
Abstract
Cancer Stem Cells/Cancer Initiating Cells (CSCs/CICs) is a rare sub-population within a tumor that is responsible for tumor formation, progression and resistance to therapies. The interaction between CSCs/CICs and tumor microenvironment (TME) can sustain “stemness” properties and promote their survival and plasticity. This cross-talk is also pivotal in regulating and modulating CSC/CIC properties. This review will provide an overview of the mechanisms underlying the mutual interaction between CSCs/CICs and TME. Particular focus will be dedicated to the immunological profile of CSCs/CICs and its role in orchestrating cancer immunosurveillance. Moreover, the available immunotherapy strategies that can target CSCs/CICs and of their possible implementation will be discussed. Overall, the dissection of the mechanisms regulating the CSC/CIC-TME interaction is warranted to understand the plasticity and immunoregulatory properties of stem-like tumor cells and to achieve complete eradications of tumors through the optimization of immunotherapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tumors are composed by heterogeneous cellular components including a rare subpopulation bearing “stemness properties” and being responsible of tumor initiation and progression. These cells have been denominated cancer stem cells (CSCs) or cancer initiating cells (CICs) [1,2,3,4,5,6]. CSCs/CICs share several characteristics with normal stem cells, such as the ability to self-renew and to give rise to differentiated progeny and the resistance to DNA damage-induced cell death [3, 5,6,7,8,9,10,11,12]. CSCs/CICs, through the cycling from proliferation to quiescence, expression of ABC drug pumps, high levels of anti-apoptotic proteins and resistance to DNA damage, are resistant to radiation and chemotherapy and play an important role in disease relapse and tumor progression [13, 14].
CSCs/CICs have been isolated from both hematological and solid tumors and they represent a rare subpopulation, comprising 0.01–10% of cells within the tumor [15]. They can be ex vivo identified based on their “stem cell-like” characteristics and the expression of certain cell surface and functional markers [16]. The identification of CSCs/CICs was first reported in leukemia, showing a hierarchical organization of tumor cells [17]. The leukemic cells were able to be engrafted upon transplantation of CD34+CD38− cells into severe combined immune-deficient (SCID) mice, which eventually led to the identification of the hierarchical organization of tumors with few cells endowed with stemness and tumorigenic properties [17]. Since then, a variety of studies highlighted the existence of “stem-like” cancer cells in solid tumors with different histological origins [5, 18,19,20,21,22,23]. Multiple molecules (e.g., ALDH-1, CD133, CD44, CD24, CBX3, ABCA5, LGR5, etc) have been identified as CSC/CIC-associated markers with differential expression depending on the tissues of derivation, highlighting the high grade of heterogeneity of these cells [16] (Table 1). Most of these molecules are over-expressed by CSCs/CICs but are also shared with either differentiated tumor cells or normal stem cells [4, 34]. As a result, detecting the presence of these cells within tumor lesions though probing for CSC/CIC- associated markers has not provided conclusive results. The xenotransplantation in immune deficient mice represents a useful tool to demonstrate in vivo the tumorigenic properties CSCs/CICs [35]. Xenograft models have contributed to prove the existence within tumor lesions of cell population endowed with stemness properties that upon serial transplantation could propagate both tumorigenic CSCs/CICs and malignant cells with differentiated phenotype without tumorigenic properties [18]. These subpopulations can be identified only through transplantation in immune deficient mice [4, 36,37,38].
Nevertheless, the available CSC/CIC-associated markers are dependent on spatial and temporal features, with their modulation occurring in relation to their inoculation in immunodeficient mice, proving the high level of plasticity of these cells and that none of the available markers can be exploited to monitor the in vivo fate of these cells [1, 39, 40] CSCs/CICs, similarly to normal stem cells, require a “niche” to allow the survival of these cells and their cycling from quiescence to proliferation and to maintain stemness and multipotency [41,42,43]. The “niche” is represented by the tumor microenvironment (TME), which is composed of multicellular and dynamic compartments that include fibroblasts, endothelial, stromal, mesenchymal and immune cells [41]. The interaction of TME with stem-like cancer cells can regulate the fate of these cells through modulating the proliferation, differentiation, immunological properties and resistance to therapies [44,45,46,47,48,49,50].
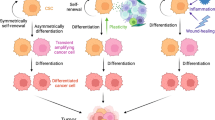
The high grade of heterogeneity and plasticity of CSCs/CICs can depend on their tissue of derivation and, importantly, on their cross-talk with TME [4, 16, 51,52,53]. Limiting the isolation and the functional characterization of CSCs/CICs to the usage of phenotypic markers is unsatisfactory and do not consider the possibility that “stemness” function of tumor cells can be reversible, as shown by Quintana et al. for melanoma [1, 39]. Moreover, xenotransplantation of these cells in immune deficient mice is lacking the important variable of the TME and its role in affecting the fate of CSCs/CICs [1]. Therefore, the lack of standardized methods to isolate CSCs/CICs and of in vivo models allowing to monitor the cross-talk of these cells with TME can lead to the high extent of variability in assessing the functional properties of these cells and in preventing to accurately determine their fate and role in the tissue of origins and in the clinical outcome of cancer patients [54, 55]. The tool of sphere forming assay to propagate in vitro CSCs/CICs is too simplified, lacking the important component of TME and of the “niche”, preventing the constant monitoring of plasticity and heterogeneity of these cells (Figs. 1, 2).

Differential immunogenic profile by CSCs/CICs vs. bulk tumor cells. CSCs/CICs can express defective levels of HLA molecules and APM components leading to low immunogenicity and escape from immune responses. In the presence of efficient expression of ligands of NK-associated activatory receptors, these cells can become susceptible to NK cell recognition. Moreover, TAAs can be expressed at suboptimal levels by CSCs/CICs. Neoantigens, generated by somatic mutation bearing tumor cells are equally expressed by both CSCs/CICs and differentiated tumor cells. The latest TAAs represent highly immunogenic target molecules, since they are not expressed by normal cells. APM: antigen processing machinery; CSCs/CICs: cancer stem cells/cancer initiating cells; NK: natural killer cells

Immunotherapy strategies to target CSCs/CICs. An overview of immunotherapy approaches including adoptive cell therapy with either 1. TCR or CAR engineered T lymphocytes; 2. Immune check point blockade with mAbs; 3. Cancer vaccination with TAAs expressed by both CSCs/CICs and differentiated tumor cells; 4. Innate immune response or 5. γδ T cell recognition of tumor cells. Combination of either multiple immunotherapy approaches or with standard therapies warrant further investigation to assess the efficacy in increasing the immunogenicity of CSCs/CICs and to implement the targeting of these cells by immune responses. CSCs/CICs: cancer stem cells/cancer initiating cells; TAA: tumor associated antigen
Therefore, the combination of deep genomic, molecular and functional profiling of CSCs/CICs could represent a relevant method to achieve a comprehensive functional characterization of these cells and, possibly, of their role in tumor outcome [56].
CSCs/CICs have been identified as the tumor components responsible for resistance to standard therapy, as well as immunotherapy [10, 57,58,59,60]. Although clinical responses in cancer patients are observed following treatments, these cells can remain in the minimal residual disease and upon changes in the environment they can exit the quiescence status and give rise to novel malignant lesion(s) or even initiate the metastatic colonization [8, 61,62,63].
The extensive molecular and immunological characterization of CSCs/CICs is warranted in order to understand the mechanisms regulating their plasticity, quiescence, interaction with the TME and resistance to therapies and to immune responses.
Immunological Profile of CSCs
HLA Molecules and APM Components
The expression of HLA class I and class II molecules and APM has been investigated in CSCs/CICs isolated from colorectal cancer (CRC) and glioblastoma multiforme (GBM) showing an overall aberrant expression of these molecules, with, in some cases, failure in their modulation by the pre-treatment with IFNs (both alpha and gamma) or DNA demethylating agent (5-Aza CdR) [64, 65]. This impairment in antigen processing and presentation by CSCs/CICs lead, upon co-culture of these cells with autologous T cells, to a preferential selection and differentiation of TH2 type T cells and failure in eliciting effector functions [64, 65]. The suboptimal expression of HLA class I molecules and APM components was also reported in CSCs/CICs isolated from different type of solid tumors [64,65,66,67,68,69]. These peculiar observations suggested that the defective expression of HLA molecules could represent a tool for the identification of CSCs/CICs [70]. On the contrary, the side population (SP) cells derived from CRC and endowed with stemness properties, showed detectable level of HLA class I molecules as well susceptibility to antigen-specific cytotoxic T lymphocytes (CTLs) [71], however this study has been performed using long-term in vitro established cell lines, that could have lost phenotypic properties of primary CSCs/CICs. Indeed, stem-like cells isolated by sphere-forming assay displayed aberrant expression of HLA class I and APM components [16, 65]. Contradictory results were obtained also in glioblastoma multiforme (GBM); CSCs/CICs isolated as sphere forming cells from this tumor have been shown to exhibit the expression of HLA class I molecules [72] while, when applying these analyses to primary GBM-derived sphere forming cells, defective expression of HLA class I and APM molecules was detected [64]. Stem-like cells expressing ABCB5 and isolated from melanoma were found to express suboptimal levels of HLA class I molecules while they were positive for HLA class II (45). APM components (e.g., LMP2, LMP7 MECL-1, TAP1 and TAP2) detected through mRNA analyses were found to be expressed in tumor sphere-models from different solid malignancies, representing a tool for in vitro enrichment of stem-like cells and for the investigation of micro-metastasis [73]. However, HLA class I and class II molecules were down-modulated in these cells as compared to differentiated tumor cells, also following their pre-treatment in vitro with IFN-γ, highlighting an impairment of antigen presentation by these cells [64]. It needs to be considered that this study did not analyze the expression at protein levels of APM molecules, therefore post-transcriptional mechanism could affect their expression. Moreover, long term in vitro established cell lines were used to isolate tumor cell spheres while in other studies reporting defective expression of APM components, primary CSCs/CICs have been investigated. These examples highlight that an overall suboptimal immunogenic potency by CSCs/CICs resulting in low or impaired susceptibility to T cell mediated immune responses (Fig. 1). This represents a mechanisms of evasion by immune responses that is shared with normal stem cells, that could represent a typical feature of cells with stemness properties [74]. Importantly, the failure in the expression of HLA molecules by tumor cells was found as one of the mechanisms of failure of the clinical activity of immune checkpoint blockade agents in cancer patients [75, 76], indicating that either CSCs/CICs can display immune evasion mechanisms shared by differentiated tumor cells or that indeed the suboptimal expression of HLA molecules of these cells and their resistance to T cell recognition can protect these cells from immunotherapy interventions, leading to tumor recurrence or progression. However, the lack of standardization in methods for both, the isolation of cells with stemness properties and to analyze HLA and APM molecules, represents a limitation in providing conclusive results. Nevertheless, detailed analysis to identify the molecular mechanisms that lead to aberrant expression of HLA molecules and APM components are warranted.
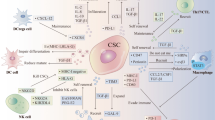
The suboptimal expression of HLA class I molecules if associated with detectable NKG2D ligands, can drive the increased susceptibility of CSCs/CICs to Natural Killer (NK) cells. This phenomenon has been observed in CSCs/CICs from glioma, melanoma, and CRC [68, 77,78,79]. However, down-modulation of NKG2D ligands on CSCs/CICs has been documented, e.g., in GBM patients [64] suggesting that the expression of low levels of NK cell activating ligands can result in the impairment of anti-CSCs/CICs innate immune responses (Fig. 1). The expression profile of molecules activating innate immune responses on CSCs/CICs can be affected by their crosstalk with TME, and thus, by their plasticity that can influence the fate in vivo of these cells.
Tumor Associated Antigens and Adaptive Immune Responses against CSCs/CICs
Tumor associated antigens (TAAs) can be recognized by T lymphocytes when exposed on the surface of tumor cells in the form of peptide/HLA complexes [80, 81]. They are categorized into three groups; (i) the overexpressed/self-antigens that are expressed at high levels by tumor cells and detectable, although at lower levels, on normal tissues (e.g., MART-1/Melan-A, hTERT, EGFR, survivin). (ii) Cancer testis (CT) antigens that are detectable on tumor cells and not on normal cells, except for testis and trophoblast (e.g., NY-ESO1, MAGE A3-A4, PRAME, CT83, SSX2). (iii) Neoantigens or mutated antigens derived by non-synonymous mutations in cancer cells (e.g., MUM-1, CDK4, ME1, ACTN4, HLA-A2) [82]. The neoantigens are higher immunogenic compared to differentiation/self TAAs since are tumor specific and do not induce tolerogenic mechanisms in immune cells [83,84,85]. Neoantigens have been shown to drive immune responses and to mediate efficient T cell recognition of tumor cells, leading to cancer eradication in patients treated with either mutanome based vaccines or adoptive cell therapy (ACT) with tumor infiltrating lymphocytes [84,85,86]. Notably, CSCs/CICs bearing a somatic mutation in the CRC-associated “driver” gene SMAD4, could elicit antigen-specific T cell responses directed to both stemness and differentiated components of tumor [87].
A transcriptome analysis of the SP cells and main population (MP) derived from CRC, breast and lung cancer revealed a preferential expression of 18 CT antigens (MAGEA2, MAGEA3, MAGEA4, MAGEA6, MAGEA12, MAGEB2, GAGE1, GAGE8, SPANXA1, SPANXB1, SPANXC, XAGE2, SPA17, BORIS, PLU-1, SGY-1, TEX15 and CT45A1) in CSCs/CICs [32]. The TAA DNAJB8, that is a member of the heat shock protein (HSP) 40 family, was found to be preferentially expressed in renal cell carcinoma (RCC); interestingly this protein played an important role in the maintenance of CSCs/CICs. DNAJB8-specific immune responses could be detected in a mice model study of DNA vaccination for RCC, rendering this molecule appealing for targeting CSCs/CICs by the immune system [32, 33] (Table 1). Recently a new antigen, Ankyrin repeat and SOCS box protein 4 (ASB4), was described as target molecule of CTLs recognizing CSCs/CICs and not the differentiated cellular components of the tumor [88]. Suboptimal expression of TAAs (MART-1, ML-IAP, NY-ESO-1, and MAGE-A) was reported in melanoma-derived CSCs/CICs (Fig. 1)[89]. Similar results were obtained in CSCs/CICs isolated from GBM and CRC (Fig. 1 and Table 1) [64, 65]. On the other hand, CD133+ CSCs/CICs isolated from melanoma were shown to express either NY-ESO-1 or DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 3, X-linked (DDX3X) representing target of tumor-specific T cells [90, 91]. Other studies have described the isolation of T lymphocytes recognizing TAAs expressed by CSCs/CICs such as IL-13Rα2, SOX2 and CD133 in GBM, CEP55 and COA-1 in CRC and EpCAM in retinoblastoma (Table 1) [28, 65, 71, 92].
Innate Immune Responses and their Relationship with CSCs/CICs
Natural killer (NK) cells are the first line of defense against cancer development and metastasis. NK cells have been described to efficiently recognize and kill in vitro CSCs/CICs isolated from CRC, melanoma and glioblastoma [68, 78, 93, 94]. The efficiency of NK cell-mediated lysis of CSCs/CICs was dependent on the expression of NCR ligands (NKp30 and NKp44), NKG2D ligands and when suboptimal or negative expression of HLA class I molecules were found on the surface of CSCs/CICs (Fig 1) [68, 78, 93,94,95]. Tallerico et al. found that CSC/CIC but not their differentiated counterpart of CRC is susceptible to NK cells [77]. Similar results have been reported in GBM and melanoma, highlighting that the amount of ligands of activatory NK receptors on CSCs/CICs was determinant for efficient innate immune responses [68, 77, 78]. In patients with acute myeloid leukemia (AML), the suboptimal expression of NKG2D ligands has been described as a mecahnisms of escape by tumor cells from NK cell recognition [96], confirming that these molecules can affect the susceptibility of cancer cells to innate responses. The observations that NKG2D ligands could represent as biomarkers for prediction of clinical responses to immune checkpoint blockade in melanoma highlight that the pattern of NKG2D ligands expression by tumor cells can affect the type and efficiency of elicited anti-tumor immune responses [97]. Therefore, the levels and pattern of expression of NKG2D ligands by tumor cells, including CSCs/CICs could be a predictive marker for the choice of the type of immunotherapy interventions.
Dendritic cells (DCs) are antigen presenting cells (APCs) that can activate either innate or adaptive immune responses [98]. In addition, they play an important role in the formation of anti-tumor T- and B cell immunologic memories [99]. Immature DCs can capture the tumor-derived antigens by phagocytosis or pinocytosis and then migrate to lymphoid organs where they present these TAAs in the form of HLA/peptide complexes to T cells, resulting in antigen-specific immune responses [100,101,102]. However, DCs depending on their morphological and phenotypic subtypes can either induce anti-tumor immune responses or promote tumor growth and progression [103]. The crosstalk of tumor with their TME is a crucial factor which results in the development of cancer [104]. Along this line, it has been described that high extent of expression of the chemokine (C-X-C motif) ligand 1 (CXCL1) by tumor and stromal cells can promote CSCs/CICs survival and proliferation and attract at tumor site DCs with suppressive functions, that could correlate tumor progression and poor survival of patients [104].
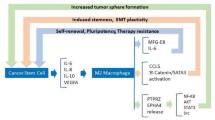
Macrophages represent important players for innate immune responses and can act as APCs similarly to DCs [105]. Based on their phenotype and functions they can be distinguished in two subpopulations: 1. The M1 subtype that are characterized by elevated pro-inflammatory cytokines, such as IL-12, IL-1β, IL-6, and tumor necrosis factor α (TNF-α), increased expression of HLA class II molecules, generation of reactive oxygen and nitrogen intermediates and ability to induce TH1-type T cell responses [106]. 2. In the presence IL-4, IL-10, and IL-13, macrophages can polarize towards M2 phenotype. These cells express scavenging, mannose and galactose receptors, IL-10, vascular endothelial growth factor (VEGF), matrix metalloproteinases (MMPs) and activation of the arginase pathway, leading to pro-tumoral effects [105,106,107, 109]. The cross-talk between CSCs/CICs and TAM is orchestrated by STAT3 signaling [102, 103]. Upon iper-modulation of STAT3 in TAM, they can promote stemness, survival and proliferation in cancer cells while the latest cells can induce the immunosuppressive properties of TAM, leading to the impairment of cancer immune-surveillance [110].
Myeloid derived suppressor cells (MDSCs) are immune cells endowed with suppressive functions that can inhibit the effector functions of immune responses [111]. The frequency of these cells either at tumor site or in the circulation has been described as a prognostic factor for patients’ survival as well as of responsiveness to immunotherapy [112]. Interestingly, STAT3 can lead the differentiation of monocytes towards MDSCs in pancreatic tumors [113] regulating also the development of CSCs/CICs [113]. The secretion of pro-inflammatory cytokines and chemokines by tumor cells can induce the differentiation and recruitment of immunosuppressive cells that can also contribute to sustain the inflammatory TME and to the interaction and reciprocal influence of CSCs/CICs and their niche [110, 113,114,115]. Another key regulator of the cross-talk between CSCs/CICs and TAM and DCs is represented by CD47 [116]. This molecule is over-expressed by CSCs/CICs of B cell malignancies. The binding of this molecule to the signal regulatory protein alpha (SIRPα), that mediates phagocytic functions in DCs and macrophages, has been shown to mediate the impairment of innate responses [116]. The cross-talk between CSCs/CICs and myeloid cells can affect both the fate and immunological profile of these cells, with implications for their susceptibilities to immune responses. Further studies should be designed to dissect the interactions of CSCs/CICs with different immune cells, although the major limitation is represented by the lack of in vivo models to monitor the interaction of these different immune cell population in the context of TME.
Immunomodulatory Properties of CSCs/CICs
The immunological profiling of CSCs/CICs has revealed that they share some characteristics with embryonic, hematopoietic and mesenchymal stem cells displaying immunoregulatory functions that render these cells invisible to immune responses and able to escape from tumor immune responses [4, 74, 117]. The principle mechanisms governing the immunomodulation of CSCs/CICs are described below.
Cytokines, Chemokines, Growth Factors & Immune Checkpoint Molecules
The observations that CSCs/CICs isolated from different tumor types can secrete soluble factors, such as Galectin-3, GDF-15, IL-10, IL-13, PGE2 and TGFb, or express immune checkpoint molecules with immunosuppressive functions, have suggested that these cells can regulate the impairment of immune responses as well as regulating a pro-tumoral TME (Table 2) [4, 51, 18–130]. These immunosuppressive factors have been described to induce the differentiation of regulatory T cells (Tregs) or MDSCs and M2 macrophages, resulting in the impairment of effector functions of innate and adaptive responses [4, 51, 130, 131]. Moreover, pro-inflammatory cytokines such as IL-6, IL-8, IL-10 and IL-13, released by CSCs/CICs can contribute to maintain an inflammatory and suppressive TME representing the “niche” sustaining cellular stemness (Table 2) [132, 133]. Indoleamine 2,3-dioxigenase (IDO), that mediates the catabolism of tryptophan, has been shown to be expressed by CSCs/CICs, contributing to the differentiation of Tregs, skewing the cytokine profiling of T cells toward TH2 -type and inhibiting the survival and proliferation of CTL (Table 2) [133, 134].
In addition, the CSC/CIC-associated expression of IL-4 and CD200, through cell-to-cell interaction, lead to the inhibition of T cell effector functions (Table 2) [65, 135]. It has been demonstrated that the over-expression of IL-4 by CRC-derived CSCs/CICs has led to inefficient TCR-mediated proliferation and antigen recognition of CTLs [65]. Interestingly, the neutralization of this cytokine by mAb could overcome the T cell mediated anti-tumor impairment and induce antigen-specific recognition of both CSCs/CICs and differentiated tumor cells [65]. This study showed that, upon up-regulation of HLA class I and APM expression through IFN-γ treatment of CSCs/CICs, T cells could specifically recognize a neoantigen, SMAD4, generated by a non-synonymous mutation bearing stem-like cells and bulk tumor cells [65]. Thus, CTL reactivity against CSCs/CICs and the TAA-specific immunosurveillance could be improved by the usage of strategies to correct the low immunogenic profile of these cells.
The immune suppressive profile of CSCs/CICs has been also confirmed by evidences describing the expression by these cells of immune checkpoint molecules (e.g. CTLA-4, PD-L1, B7-H3 or B7-H4) (Table 2) [4, 34, 64, 65, 136]. These observations highlighted the similarities between CSCs/CICs and normal stem cells in terms of the immune profile [34, 137, 138]. Moreover, altered expression of STAT3 pathway in CSCs/CICs can also affect their immune suppressive activity through inhibiting T cell proliferation and activation, inducing the differentiation of Tregs and triggering T cell apoptosis [121]. The observations reported above show that multiple mechanisms and molecular pathways are either up-regulated or aberrantly activated in CSCs/CICs resulting in their immune suppressive properties, therefore the blockade of these signaling through the combination of inhibitory agents should be considered in order to rescue the tumor-specific immune responses.
MicroRNAs
miRNAs are non-coding RNAs regulating at post-transcriptional levels, through complementary binding to target mRNA, the expression of genes [139]. The altered regulation of gene expression in tumor cells can occur by both up- or down regulation of miRNA [139]. The most common activity of miRNAs in CSCs/CICs is represented by the control of the expression of either oncogenes (e.g., MiR34a, MiR31 or MiR205) or tumor suppressor genes [139]. The aberrant expression of few miRNAs, such as miRNAs 451and 199b-5p, has been shown to affect stem-like cell properties isolated from different type of tumors (e.g., GBM, breast cancer and medulloblastoma) [139,140,141,142,143]. Of note, miRNAs displaying regulatory activity on immune-related genes (e.g, miRNA-199a that can regulate the IFN-mediated responses) can play a role in the differentiation of mammalian CSCs/CICs [144]. The level of miRNA-124, through regulating the expression of STAT3, can affect the efficiency of anti-CSC/CIC T cell responses in GBM [145]. Along this line, miR203 and miR92 can control the stemness and immunological profiles of melanoma cells [146, 147].
Immune Evasion and Tumor Dormancy
Tumor dormancy is represented by quiescent cells that can remain occult and undetectable by regular diagnostic methods for long intervals of time, even after initial clinical responses to therapies [148]. Quiescence of cells is the ability to exit cell cycle and remain in G0 phase until permissive environmental condition will lead to enter back into the cycling phase. This is considered one of the principle mechanisms underlying tumor dormancy. CSCs/CICs display the ability to cycle between quiescence and proliferation and together with their resistance to therapies represent the link between these cells and tumor dormancy [2, 5, 8, 9, 149,150,151]. Furthermore, the immune suppressive mechanisms associated with CSCs/CICs can orchestrate the evasion of these cells from immune recognition and immunosurveillance, and could be considered additional factors responsible of tumor dormancy [152].
An important mechanisms of immune-surveillance is the homing of immune cells to the tumor site, which ultimately form the immune infiltrate. Tumors arising from epithelial breast cancer are known to possess high levels MHC class I molecules and of infiltrating T effector cells and M1 macrophages. The immune infiltrate from mesenchymal like breast cancer tumors exhibit low levels of MHC class I molecules, high levels of PD-L1 and contain Tregs, M2 like macrophages as well as exhausted T cells [153]. CSCs/CICs that are considered the architects of their own microenvironment [154], as well as generated by epithelial-to-mesenchymal transition (EMT) can be potentially responsible for the type of immune infiltration depending of their pattern of immune profile.
Common gene expression patterns have been found in normal mammary stem cells and dormant tumor cells from breast cancer suggesting the possible presence of stem-like cells in dormant tumors [151]. In addition, different cell sub-populations could be isolated from relapsed AML endowed with differential tumorigenic ability depending on their up-regulation of stemness signaling [155].
A better understand of the relationship between stemness properties, immunological profile of CSCs/CICs and tumor dormancy will provide insights on the mechanisms of therapeutic resistance of these cells and will allow to identify strategies for complete tumor eradication.
Immunological Targeting of CSCs/CICs
Cancer Vaccines
The recognition of TAAs expressed by CSCs/CICs by T cells have been documented (see Table 1). These in vitro or in vivo models were based on the usage of TAAs that represented sources of antigens for the therapeutic administration of cancer vaccines in cancer patients [81]. However, the principle limiting factor of the clinical efficacy of this strategy is represented by the usage of “self”/tolerogenic TAAs, shared with normal tissues [81]. The low or negative expression of these categories of antigens and of CT-TAAs by CSCs/CICs can represent an additional reason of failure of high rate and long duration of clinical responses observed in cancer patients treated with cancer vaccines [4, 34]. In addition, the sub-optimal levels of HLA class I molecules and APM by stem-like cells can drive the failure in targeting CSCs/CICs by cancer vaccines leading to the development of tumor dormancy and tumor recurrence, although the observance of initial clinical efficacy of these therapeutic interventions [4, 34].
DC-based vaccines, exploiting these cells as APC to present TAAs to T cell-mediated responses, represent also a therapeutic strategy for cancer patients showing encouraging clinical activity [102, 156,157,158,159,160,161,162]. DCs loaded with either CSC/CIC-lysates or mRNA isolated from these cells represented source of antigens for vaccination in the context of Phase I/II clinical trials of GBM patients [163, 164]. These studies provided proof of principle of improved overall survival of cancer patients treated with CSC/CIC targeted immunotherapy [163,164,165]. Immune responses, with, in some cases increased frequency of circulating NK cells, were detected in patients showing clinical benefit from these treatments [163, 165]. Of note, these therapeutic interventions could overcome the failure of CSCs/CICs in expressing efficient levels of HLA class I molecules and in presenting TAAs to T cells, documenting for the first time, that cancer vaccine, if eliciting NK cell-mediated responses, could target stem-like tumor cells [158, 163,164,165]. Tumor cell clones expressing immunogenic neoantigens can undergo immune selection due to the recognition and elimination by T lymphocytes, leading to the survival of tumor cell clones not expressing strong immunogenic antigens and maintaining the expression of low immunogenic TAAs [166] (and see https://www.biorxiv.org/content/10.1101/536433v1). This process is also associated with immune evasions mechanisms developed by both tumor cells and TME [166].
In some tumors, the decrease in antigen presentation is a result of epigenetic silencing of the genes involved in antigen presentation machinery. The usage of demethylating agents such as 5-Aza-2′-deoxycytidine to reduce the methylation of genes involved in antigen presentation, is a potential strategy to increase the antigen presentation in these CSC/CICs. The effect of demethylation has been shown in CSCs/CICs from breast cancer, where it resulted in high expression levels of TAP1, which is involved in antigen presentation [167].
In addition, increased antigen presentation also improves the potential of discovering novel antigens, which can then be helpful in development of new anti-cancer vaccines (Fig. 2).
Immune Checkpoint Blockade
Immune checkpoints, including CTLA-4, PD-1 and PD-L1, are important physiological regulators of innate and adaptive immune responses [168]. Biological inhibitory agents have been clinically developed, revealing striking therapeutically success [169,170,171,172,173,174,175]. However, a significant proportion of cancer patients failed to benefit from these therapies.
The effectiveness of immune checkpoint blockade (ICB) is largely dependent on the tumor microenvironment [176]. Tumors such as melanoma, bladder cancer and non-small cell lung cancer (NSCLC) can be characterized as “hot” tumors due to their inflamed TME, high levels of and neo-antigen expression and of T cell infiltration and detection of PD-L1. These tumors have been reported to be associated with higher frequency of susceptibility to immune checkpoint treatments. On the other hand, prostate cancer is considered to be a “cold” tumor, due to minimal level of T cell infiltration, and limited response to single agent checkpoint inhibition [176].
Expression of immune checkpoint molecules has been observed in CSCs/CICs from different histological origins [51]. PD-L1 expression was detected at high levels in CSCs/CICs isolated from primary human head and neck squamous cell carcinoma (HNSCC), gastric and breast cancer, CRC and GBM [34, 64, 65, 177,178,179]. ,CSCs/CICs could theoretically be targeted in vivo by immune checkpoint blockade agents, enhancing the clinical efficacy of cancer vaccines, as demonstrated in a mouse model [180]. However, recent reports describing that clinical failure of these therapies was associated with defective expression of HLA class I molecules by tumor cells [75, 76], suggest that these cells might evade from the ICB-mediated unleash of immune responses (Fig. 2).
Adoptive Cell Therapy
ACT is represented by the isolation of T lymphocytes from cancer patients, their ex vivo expansion, and the infusion back into patients [181,182,183]. In addition, T lymphocytes engineered to express TCR with high affinity for a cognate TAA could be exploited for ACT studies [183]. Highly encouraging and sustained responses mediated by adoptively transferred TCR targeting the TAA NY-ESO-1 have recently been reported in different tumor types, such as breast cancer and myeloma [86, 184]. Nevertheless, the antigen choice is highly relevant to prevent severe toxicities due to “off-target” cross-reaction with normal tissues sharing the same antigens or expressing molecules mimicking the TAAs [182].
Neoantigens have been described as candidate TAAs efficiently recognized by T cells that can be exploited for ACT of cancer patients and, interestingly, CTL targeting these antigens could be isolated from tumor infiltrating lymphocytes (TILs) of melanoma and other type of malignant lesions [183, 185, 186]. Nevertheless, ACT to target neoantigens can represent a promising approach for treatment of cancer patients upon assessment of HLA expression by both CSCs/CICs and differentiated tumor cells and, in case of suboptimal levels of expression, the achievement of their up-regulation by pre-treatment with immunomodulating agents [4, 34, 51].
T cell can be genetically modified to express a chimeric antigen receptor (CAR) that is composed of epitope-specific domains isolated from mAbs linked to T cell-derived activatory/costimulatory molecules [182, 187,188,189]. CAR-T cells can recognize TAAs independently on the expression of HLA molecules and APM components [189]. CAR-T cell therapy for some subgroups of hematological malignancies represent the salvage intervention leading to stable clinical responses and improved overall survival of patients refractory to standard therapies or with recurrences [187, 189,190,191,192]. The usage of CAR-T cell therapy for solid tumors is currently under investigation, showing encouraging results in cancer patients with aggressive tumor types, including malignant mesothelioma, pancreatic cancer and GBM [193,194,195,196,197].
CAR-T cells targeting TAAs, such as CD133, EGFRvIII, EpCAM, CSPG4 and B7-H3, expressed by different type of solid tumors, including CSC/CIC components, have been developed in pre-clinical studies [196,197,198,199,200,201]. These studies have shown that the targeting of TAAs that are expressed only by tumor cells, including CSCs/CICs, and not by normal cells, could provide the rational for safe and efficient clinical development of CAR-T cells therapy for these tumors(Fig. 2) [200, 202]. Moreover, the combination of CAR-T cells targeting dual TAAs, EGFRvIII and CD133, has been used for the successful therapeutic treatment of a patient with advanced cholangiocarcinoma [203].
CAR-T cells targeting NKG2D ligands on CSCs/CICs have been investigated and tested both in vitro and in vivo [204, 205]. CSCs/CICs from glioblastoma expressing detectable NKG2D ligands could be efficiently targeted by CAR- T cells. These tools showed to efficintly eliminate also xenograft tumors [205]. Nevertheless, a major limitation associated with the use of these CAR-T cells is represented by the variability of the levels of NKG2D ligands on the surface of CSCs/CICs, depending on their origin and methodology used for their ex vivo isolation.
Nevertheless, further investigations aimed at a comprehensive genomic and immunological characterization CSCs/CICs are warranted to implement the efficiency and safety of ACT strategies.
Conclusions
Recent advances in the genomic, molecular and immunological profiling of CSCs/CICs have contributed to the identification of dysregulated molecular pathways that orchestrate stem-like cancer cells and their interaction with TME. The heterogeneity and plasticity of these cells and the mutual effect of TME and CSCs/CICs on the resulting anti-tumoral or pro-tumoral environment, represent the principle limitations in predicting the fate of these cells and their role in cancer patients’ outcome. In addition, these cells have been identified as key players in therapeutic resistance of tumors and in the development of their dormancy. The down-modulation of HLA molecules and NK activatory ligands on CSCs/CICs, through decreasing their susceptibility to T or NK cell targeting, might represent one of the principle factors leading to resistance to immunotherapy. Although, the mechanisms regulating the levels of HLA molecules and NKG2D ligands on CSCs/CICs are yet to be dissected. Due to the complexity of the cross-talk between CSCs/CICs and TME and the high plasticity of these cells, it is difficult to predict what type of immune cells could play a relevant role in targeting CSCs/CICs. Multifactorial investigations, including the immunological profile, immunomodulating molecules, the interaction with TME and the type of immune cell infiltration will allow to provide insights. Nevertheless, the available tools to isolate and characterize CSCs/CICs, such as spheroids, immunodeficient mice, antigenic profile, are unsatisfactory to dissect the cross-talk of these cells with TME. Moreover, the development of pre-clinical in vivo models engrafted with human immune system is desirable to allow the monitoring of the interaction of CSCs/CICs with TME.
The targeting of CSCs/CICs by immunotherapy could result in the complete tumor eradication and stable clinical responses in cancer patients. This goal could be achieved by the design of combination of strategies based on innate and/or antigen-specific T cell responses with immunoregulatory agents that can render CSCs/CICs susceptible to cell-mediated immunosurveillance. In cases where epigenetic factors are responsible for low antigen presentation, the usage of demethylating agents could represent a potential strategy to overcome the low expression of HLA molecules.
Molecular approaches dissecting the fate of CSCs/CICs within tumor tissues will allow to develop immune-based precision medicine approaches and to identify biomarkers predictive of patients’ responsiveness to therapies.
Abbreviations
- ALDH:
-
Aldehyde dehydrogenase
- APC:
-
Antigen presenting cells
- APM:
-
Antigen processing machinery
- CAR:
-
Chimeric antigen receptor
- CIC:
-
Cancer initiating cell
- CRC:
-
Colorectal cancer
- CT:
-
Cancer testis
- CTLA-4:
-
Cytotoxic lymphocyte antigen-4
- CSPG4:
-
Chondroitin sulphate protidoglycan 4
- HLA:
-
Human leukocyte antigen
- IDO:
-
Indoleamine 2,3-dioxygenase
- GBM:
-
Glioblastoma multiforme
- GDF-15:
-
Growth differentiation factor-15
- IFN:
-
Interferon
- IL-4:
-
Interleukin 4
- IL-10:
-
Interleukin 10
- IL-13:
-
Interleukin 13
- IL-13α2:
-
α2 chain of IL-13 receptor
- mAb:
-
Monoclonal antibody;
- MDSC:
-
Myeloid derived suppressor cell
- NSCLC:
-
Non-small cell lung cancer
- PD-1:
-
Programmed death 1
- PD-L1:
-
Programmed death ligand 1
- RCC:
-
Renal cell carcinoma
- STAT3:
-
Signal transducer and activator of transcription 3
- TGFB:
-
Transforming growth factor beta
- TAA:
-
Tumor associated antigen
- Treg:
-
T regulatory cell.
References
Shackleton M, Quintana E, Fearon ER, Morrison SJ (2009) Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 138(5):822–829
Clarke MF, Dick JE, Dirks PB et al (2006) Cancer stem cells--perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res 66(19):9339–9344
Li Y, Laterra J (2012) Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res 72(3):576–580
Maccalli C, De Maria R (2015) Cancer stem cells: perspectives for therapeutic targeting. Cancer Immunol Immunother 64(1):91–97
Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature. 414(6859):105–111
Wicha MS, Liu S, Dontu G (2006) Cancer stem cells: an old idea--a paradigm shift. Cancer Res 66(4):1883–1890 discussion 1895-1886
Tang DG (2012) Understanding cancer stem cell heterogeneity and plasticity. Cell Res 22(3):457–472
Eyler CE, Rich JN (2008) Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 26(17):2839–2845
Hadjimichael C, Chanoumidou K, Papadopoulou N, Arampatzi P, Papamatheakis J, Kretsovali A (2015) Common stemness regulators of embryonic and cancer stem cells. World J Stem Cells 7(9):1150–1184
Maugeri-Sacca M, Vigneri P, De Maria R (2011) Cancer stem cells and chemosensitivity. Clin Cancer Res 17(15):4942–4947
Soltanian S, Matin MM (2011) Cancer stem cells and cancer therapy. Tumour Biol 32(3):425–440
Steinbichler TB, Dudas J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. (2018) Therapy resistance mediated by cancer stem cells. Semin Cancer Biol
Ricci-Vitiani L, Fabrizi E, Palio E, De Maria R (2009) Colon cancer stem cells. J Mol Med (Berl) 87(11):1097–1104
Vezzoni L, Parmiani G (2008) Limitations of the cancer stem cell theory. Cytotechnology. 58(1):3–9
Morrison BJ, Steel JC, Morris JC (2018) Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 18(1):469
Maccalli C, Volonte A, Cimminiello C, Parmiani G (2014) Immunology of cancer stem cells in solid tumours. A review. Eur J Cancer 50(3):649–655
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 367(6464):645–648
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100(7):3983–3988
Clarke MF, Becker MW (2006) Stem cells: the real culprits in cancer? Sci Am 295(1):52–59
Singh SK, Clarke ID, Hide T, Dirks PB (2004) Cancer stem cells in nervous system tumors. Oncogene. 23(43):7267–7273
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature. 432(7015):396–401
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM (2007) Identification of pancreatic cancer stem cells. Cancer Res 67(3):1030–1037
Ricci-Vitiani L, Pagliuca A, Palio E, Zeuner A, De Maria R (2008) Colon cancer stem cells. Gut. 57(4):538–548
Visus C, Wang Y, Lozano-Leon A, Ferris RL, Silver S, Szczepanski MJ, Brand RE, Ferrone CR, Whiteside TL, Ferrone S, DeLeo A, Wang X (2011) Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8(+) T cells. Clin Cancer Res 17(19):6174–6184
Brown CE, Starr R, Martinez C, Aguilar B, D'Apuzzo M, Todorov I, Shih CC, Badie B, Hudecek M, Riddell SR, Jensen MC (2009) Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res 69(23):8886–8893
Glumac PM, LeBeau AM (2018) The role of CD133 in cancer: a concise review. Clin Transl Med 7(1):18
Zinzi L, Contino M, Cantore M, Capparelli E, Leopoldo M, Colabufo NA (2014) ABC transporters in CSCs membranes as a novel target for treating tumor relapse. Front Pharmacol 5:163
Mitra M, Kandalam M, Harilal A, Verma RS, Krishnan UM, Swaminathan S, Krishnakumar S (2012) EpCAM is a putative stem marker in retinoblastoma and an effective target for T-cell-mediated immunotherapy. Mol Vis 18:290–308
Choi D, Lee HW, Hur KY et al (2009) Cancer stem cell markers CD133 and CD24 correlate with invasiveness and differentiation in colorectal adenocarcinoma. World J Gastroenterol 15(18):2258–2264
Shapira S, Kazanov D, Weisblatt S, Starr A, Arber N, Kraus S (2011) The CD24 protein inducible expression system is an ideal tool to explore the potential of CD24 as an oncogene and a target for immunotherapy in vitro and in vivo. J Biol Chem 286(47):40548–40555
Schmitz M, Temme A, Senner V et al (2007) Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br J Cancer 96(8):1293–1301
Yamada R, Takahashi A, Torigoe T, Morita R, Tamura Y, Tsukahara T, Kanaseki T, Kubo T, Watarai K, Kondo T, Hirohashi Y, Sato N (2013) Preferential expression of cancer/testis genes in cancer stem-like cells: proposal of a novel sub-category, cancer/testis/stem gene. Tissue Antigens 81(6):428–434
Nishizawa S, Hirohashi Y, Torigoe T, Takahashi A, Tamura Y, Mori T, Kanaseki T, Kamiguchi K, Asanuma H, Morita R, Sokolovskaya A, Matsuzaki J, Yamada R, Fujii R, Kampinga HH, Kondo T, Hasegawa T, Hara I, Sato N (2012) HSP DNAJB8 controls tumor-initiating ability in renal cancer stem-like cells. Cancer Res 72(11):2844–2854
Maccalli C, Rasul KI, Elawad M, Ferrone S (2018) The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol.
Clevers H (2011) The cancer stem cell: premises, promises and challenges. Nat Med 17(3):313–319
Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, di Virgilio A, Conticello C, Ruco L, Peschle C, de Maria R (2008) Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 15(3):504–514
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, de Maria R (2007) Identification and expansion of human colon-cancer-initiating cells. Nature. 445(7123):111–115
Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S, Cocorullo G, Gulotta G, Dieli F, de Maria R, Stassi G (2014) CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 14(3):342–356
Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ (2008) Efficient tumour formation by single human melanoma cells. Nature. 456(7222):593–598
Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M (2010) A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 141(4):583–594
Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T (2017) Drug resistance driven by cancer stem cells and their niche. Int J Mol Sci 18(12)
Fuchs E, Tumbar T, Guasch G (2004) Socializing with the neighbors: stem cells and their niche. Cell. 116(6):769–778
Xie T, Li L (2007) Stem cells and their niche: an inseparable relationship. Development. 134(11):2001–2006
Ungefroren H, Sebens S, Seidl D, Lehnert H, Hass R (2011) Interaction of tumor cells with the microenvironment. Cell Commun Signal 9:18
Whiteside TL (2008) The tumor microenvironment and its role in promoting tumor growth. Oncogene. 27(45):5904–5912
Cabarcas SM, Mathews LA, Farrar WL (2011) The cancer stem cell niche--there goes the neighborhood? Int J Cancer 129(10):2315–2327
Cully M (2018) Tumour microenvironment: fibroblast subtype provides niche for cancer stem cells. Nat Rev Cancer 18(3):136
Cully M (2018) Cancer: fibroblast subtype provides niche for cancer stem cells. Nat Rev Drug Discov 17(3):163
Malanchi I, Santamaria-Martinez A, Susanto E et al (2012) Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 481(7379):85–89
Seidel S, Garvalov BK, Wirta V et al (2010) A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain. 133(Pt 4):983–995
Maccalli C, Parmiani G, Ferrone S (2017) Immunomodulating and Immunoresistance properties of Cancer-initiating cells: implications for the clinical success of immunotherapy. Immunol Investig 46(3):221–238
Easwaran H, Tsai HC, Baylin SB (2014) Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 54(5):716–727
Dick JE (2008) Stem cell concepts renew cancer research. Blood. 112(13):4793–4807
Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, Eberhart CG (2007) Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 25(10):2524–2533
Sakariassen PO, Prestegarden L, Wang J, Skaftnesmo KO, Mahesparan R, Molthoff C, Sminia P, Sundlisaeter E, Misra A, Tysnes BB, Chekenya M, Peters H, Lende G, Kalland KH, Øyan AM, Petersen K, Jonassen I, van der Kogel A, Feuerstein BG, Terzis AJ, Bjerkvig R, Enger PØ (2006) Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc Natl Acad Sci U S A 103(44):16466–16471
Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, Canty AJ, Danska JS, Bohlander SK, Buske C, Minden MD, Golub TR, Jurisica I, Ebert BL, Dick JE (2011) Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 17(9):1086–1093
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 444(7120):756–760
Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5(4):275–284
Diehn M, Clarke MF (2006) Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst 98(24):1755–1757
Francipane MG, Alea MP, Lombardo Y, Todaro M, Medema JP, Stassi G (2008) Crucial role of interleukin-4 in the survival of colon cancer stem cells. Cancer Res 68(11):4022–4025
Frank NY, Schatton T, Frank MH (2010) The therapeutic promise of the cancer stem cell concept. J Clin Invest 120(1):41–50
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC (2008) Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 100(9):672–679
Wilson BJ, Schatton T, Zhan Q, Gasser M, Ma J, Saab KR, Schanche R, Waaga-Gasser AM, Gold JS, Huang Q, Murphy GF, Frank MH, Frank NY (2011) ABCB5 identifies a therapy-refractory tumor cell population in colorectal cancer patients. Cancer Res 71(15):5307–5316
Di Tomaso T, Mazzoleni S, Wang E et al (2010) Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res 16(3):800–813
Volonte A, Di Tomaso T, Spinelli M et al (2014) Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J Immunol 192(1):523–532
Catalano V (2015) Resistance of cancer stem cells to cell- mediated immune responses. Vol 7: Springer
Codony-Servat J, Rosell R (2015) Cancer stem cells and immunoresistance: clinical implications and solutions. Transl Lung Cancer Res 4(6):689–703
Pietra G, Manzini C, Vitale M, Balsamo M, Ognio E, Boitano M, Queirolo P, Moretta L, Mingari MC (2009) Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int Immunol 21(7):793–801
Zhang D, Tang DG, Rycaj K (2018) Cancer stem cells: regulation programs, immunological properties and immunotherapy. Semin Cancer Biol 52(Pt 2):94–106
Grau JJ, Mesia R, de la Iglesia-Vicente M et al (2016) Enrichment of cells with Cancer stem cell-like markers in relapses of Chemoresistant patients with locally advanced head and neck squamous cell carcinoma. Oncology. 90(5):267–272
Inoda S, Hirohashi Y, Torigoe T, Morita R, Takahashi A, Asanuma H, Nakatsugawa M, Nishizawa S, Tamura Y, Tsuruma T, Terui T, Kondo T, Ishitani K, Hasegawa T, Hirata K, Sato N (2011) Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells. Am J Pathol 178(4):1805–1813
Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Sawaya R, Lang FF, Heimberger AB (2010) Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res 16(2):461–473
Busse A, Letsch A, Fusi A, Nonnenmacher A, Stather D, Ochsenreither S, Regenbrecht CR, Keilholz U (2013) Characterization of small spheres derived from various solid tumor cell lines: are they suitable targets for T cells? Clin Exp Metastasis 30(6):781–791
Ichiryu N, Fairchild PJ (2013) Immune privilege of stem cells. Methods Mol Biol 1029:1–16
Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, du VY, Schlessinger J, Goldberg SB, Chiang A, Sanmamed MF, Melero I, Agorreta J, Montuenga LM, Lifton R, Ferrone S, Kavathas P, Rimm DL, Kaech SM, Schalper K, Herbst RS, Politi K (2017) Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung Cancer. Cancer Discov 7(12):1420–1435
Chowell D, Morris LGT, Grigg CM et al (2018) Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 359(6375):582–587
Tallerico R, Todaro M, Di Franco S et al (2013) Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol 190(5):2381–2390
Castriconi R, Daga A, Dondero A et al (2009) NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol 182(6):3530–3539
Tseng HC, Arasteh A, Paranjpe A et al (2010) Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS One 5(7):e11590
Maccalli C, Rasul KI, Elawad M, Ferrone S (2018) The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol 53:189–200
Parmiani G, Russo V, Maccalli C, Parolini D, Rizzo N, Maio M (2014) Peptide-based vaccines for cancer therapy. Hum Vaccin Immunother 10(11):3175–3178
Lu YC, Robbins PF (2016) Cancer immunotherapy targeting neoantigens. Semin Immunol 28(1):22–27
Forshew T, Murtaza M, Parkinson C et al (2012) Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 4(136):136ra168
Robbins PF (2017) Tumor-infiltrating lymphocyte therapy and Neoantigens. Cancer J 23(2):138–143
Sahin U, Derhovanessian E, Miller M et al (2017) Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 547(7662):222–226
Zacharakis N, Chinnasamy H, Black M et al (2018) Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med 24(6):724–730
Mennonna D, Maccalli C, Romano MC et al (2017) T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut. 66(3):454–463
Miyamoto S, Kochin V, Kanaseki T et al (2018) The antigen ASB4 on Cancer stem cells serves as a target for CTL immunotherapy of colorectal Cancer. Cancer Immunol Res.
Schatton T, Schutte U, Frank NY et al (2010) Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res 70(2):697–708
Gedye C, Quirk J, Browning J, Svobodová S, John T, Sluka P, Dunbar PR, Corbeil D, Cebon J, Davis ID (2009) Cancer/testis antigens can be immunological targets in clonogenic CD133+ melanoma cells. Cancer Immunol Immunother 58(10):1635–1646
Koshio J, Kagamu H, Nozaki K, Saida Y, Tanaka T, Shoji S, Igarashi N, Miura S, Okajima M, Watanabe S, Yoshizawa H, Narita I (2013) DEAD/H (asp-Glu-Ala-asp/his) box polypeptide 3, X-linked is an immunogenic target of cancer stem cells. Cancer Immunol Immunother 62(10):1619–1628
Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D'Apuzzo M, Barish ME, Forman SJ, Jensen MC (2012) Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T cells. Clin Cancer Res 18(8):2199–2209
Tallerico R, Garofalo C, Carbone E (2016) A new biological feature of natural killer cells: the recognition of solid tumor-derived Cancer stem cells. Front Immunol 7:179
Parmiani G (2016) Melanoma cancer stem cells: markers and functions. Cancers (Basel) 8(3)
Ames E, Canter RJ, Grossenbacher SK et al (2015) NK cells preferentially target tumor cells with a Cancer stem cell phenotype. J Immunol 195(8):4010–4019
Paczulla AM, Rothfelder K, Raffel S et al (2019) Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 572(7768):254–259
Maccalli C, Giannarelli D, Chiarucci C, Cutaia O, Giacobini G, Hendrickx W, Amato G, Annesi D, Bedognetti D, Altomonte M, Danielli R, Calabrò L, di Giacomo AM, Marincola FM, Parmiani G, Maio M (2017) Soluble NKG2D ligands are biomarkers associated with the clinical outcome to immune checkpoint blockade therapy of metastatic melanoma patients. Oncoimmunology. 6(7):e1323618
Cella M, Sallusto F, Lanzavecchia A (1997) Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol 9(1):10–16
Keller AM, Xiao Y, Peperzak V, Naik SH, Borst J (2009) Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood. 113(21):5167–5175
Hanke N, Alizadeh D, Katsanis E, Larmonier N (2013) Dendritic cell tumor killing activity and its potential applications in cancer immunotherapy. Crit Rev Immunol 33(1):1–21
Palucka K, Banchereau J (2013) Dendritic-cell-based therapeutic cancer vaccines. Immunity. 39(1):38–48
Nouri-Shirazi M, Banchereau J, Bell D et al (2000) Dendritic cells capture killed tumor cells and present their antigens to elicit tumor-specific immune responses. J Immunol 165(7):3797–3803
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell. 140(6):883–899
Hsu YL, Chen YJ, Chang WA et al. (2018) Interaction between Tumor-Associated Dendritic Cells and Colon Cancer Cells Contributes to Tumor Progression via CXCL1. Int J Mol Sci 19(8)
Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 124(2):263–266
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122(3):787–795
Gordon S, Martinez FO (2010) Alternative activation of macrophages: mechanism and functions. Immunity. 32(5):593–604
Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, Yagita H, Takaoka A, Tahara H (2011) Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A 108(30):12425–12430
Mantovani A, Sica A, Locati M (2005) Macrophage polarization comes of age. Immunity. 23(4):344–346
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, Hewitt S, Udupi GM, Gallagher WM, Wegner C, West BL, Wang-Gillam A, Goedegebuure P, Linehan DC, DeNardo D (2013) Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73(3):1128–1141
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ (2013) The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 138(2):105–115
Filipazzi P, Huber V, Rivoltini L (2012) Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol Immunother 61(2):255–263
Panni RZ, Sanford DE, Belt BA et al (2014) Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic Cancer. Cancer Immunol Immunother 63(5):513–528
Sherry MM, Reeves A, Wu JK, Cochran BH (2009) STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 27(10):2383–2392
Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, Margolick JB, Liotta LA, Petricoin E 3rd, Zhang Y (2007) Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A 104(41):16158–16163
Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, van Rooijen N, Weissman IL (2009) CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 138(2):286–299
Ankrum JA, Ong JF, Karp JM (2014) Mesenchymal stem cells: immune evasive, not immune privileged. Nat Biotechnol 32(3):252–260
Brown MA, Hural J (2017) Functions of IL-4 and control of its expression. Crit Rev Immunol 37(2–6):181–212
Terabe M, Park JM, Berzofsky JA (2004) Role of IL-13 in regulation of anti-tumor immunity and tumor growth. Cancer Immunol Immunother 53(2):79–85
Miyazono K (2000) Positive and negative regulation of TGF-beta signaling. J Cell Sci 113(Pt 7):1101–1109
Wei J, Barr J, Kong LY et al (2010) Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther 9(1):67–78
Emmerson PJ, Duffin KL, Chintharlapalli S, Wu X (2018) GDF15 and growth control. Front Physiol 9:1712
Dumic J, Dabelic S, Flogel M (2006) Galectin-3: an open-ended story. Biochim Biophys Acta 1760(4):616–635
Colmont CS, Benketah A, Reed SH et al (2013) CD200-expressing human basal cell carcinoma cells initiate tumor growth. Proc Natl Acad Sci U S A 110(4):1434–1439
Wright GJ, Puklavec MJ, Willis AC et al (2000) Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. 13(2):233–242
Schatton T, Frank MH (2009) Antitumor immunity and cancer stem cells. Ann N Y Acad Sci 1176:154–169
Salmaninejad A, Khoramshahi V, Azani A, Soltaninejad E, Aslani S, Zamani MR, Zal M, Nesaei A, Hosseini SM (2018) PD-1 and cancer: molecular mechanisms and polymorphisms. Immunogenetics. 70(2):73–86
Yao Y, Ye H, Qi Z et al (2016) B7-H4(B7x)-mediated cross-talk between Glioma-initiating cells and macrophages via the IL6/JAK/STAT3 pathway Lead to poor prognosis in Glioma patients. Clin Cancer Res 22(11):2778–2790
Harris SG, Padilla J, Koumas L, Ray D, Phipps RP (2002) Prostaglandins as modulators of immunity. Trends Immunol 23(3):144–150
Yoshimura A, Muto G (2011) TGF-beta function in immune suppression. Curr Top Microbiol Immunol 350:127–147
Wan YY, Flavell RA (2007) 'Yin-Yang' functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol Rev 220:199–213
Shacter E, Weitzman SA (2002) Chronic inflammation and cancer. Oncology (Williston Park) 16(2):217–226 229; discussion 230-212
Parker KH, Beury DW, Ostrand-Rosenberg S (2015) Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res 128:95–139
Smith C, Chang MY, Parker KH et al (2012) IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov 2(8):722–735
Kawasaki BT, Mistree T, Hurt EM, Kalathur M, Farrar WL (2007) Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem Biophys Res Commun 364(4):778–782
Hirohashi Y, Torigoe T, Tsukahara T, Kanaseki T, Kochin V, Sato N (2016) Immune responses to human cancer stem-like cells/cancer-initiating cells. Cancer Sci 107(1):12–17
Kim SY, Cho HS, Yang SH, Shin JY, Kim JS, Lee ST, Chu K, Roh JK, Kim SU, Park CG (2009) Soluble mediators from human neural stem cells play a critical role in suppression of T-cell activation and proliferation. J Neurosci Res 87(10):2264–2272
Ljujic B, Milovanovic M, Volarevic V et al (2013) Human mesenchymal stem cells creating an immunosuppressive environment and promote breast cancer in mice. Sci Rep 3:2298
Chhabra R, Saini N (2014) MicroRNAs in cancer stem cells: current status and future directions. Tumour Biol 35(9):8395–8405
Garzia L, Andolfo I, Cusanelli E et al (2009) MicroRNA-199b-5p impairs cancer stem cells through negative regulation of HES1 in medulloblastoma. PLoS One 4(3):e4998
Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro CL, Huebner K, Croce CM (2014) Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci U S A 111(12):4536–4541
Petrelli A, Carollo R, Cargnelutti M et al (2015) By promoting cell differentiation, miR-100 sensitizes basal-like breast cancer stem cells to hormonal therapy. Oncotarget. 6(4):2315–2330
Roscigno G, Quintavalle C, Donnarumma E, Puoti I, Diaz-Lagares A, Iaboni M, Fiore D, Russo V, Todaro M, Romano G, Thomas R, Cortino G, Gaggianesi M, Esteller M, Croce CM, Condorelli G (2016) MiR-221 promotes stemness of breast cancer cells by targeting DNMT3b. Oncotarget. 7(1):580–592
Celia-Terrassa T, Liu DD, Choudhury A et al (2017) Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a-LCOR axis. Nat Cell Biol 19(6):711–723
Wei J, Wang F, Kong LY, Xu S, Doucette T, Ferguson SD, Yang Y, McEnery K, Jethwa K, Gjyshi O, Qiao W, Levine NB, Lang FF, Rao G, Fuller GN, Calin GA, Heimberger AB (2013) miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res 73(13):3913–3926
Sahranavardfard P, Firouzi J, Azimi M, et al. (2019) MicroRNA-203 reinforces stemness properties in melanoma and augments tumorigenesis in vivo. J Cell Physiol
Shidal C, Singh NP, Nagarkatti P, Nagarkatti M (2019) MicroRNA-92 expression in CD133+ melanoma stem cells regulates immunosuppression in the tumor microenvironment via integrin-dependent activation of TGF-beta. Cancer Res
Wang SH, Lin SY (2013) Tumor dormancy: potential therapeutic target in tumor recurrence and metastasis prevention. Exp Hematol Oncol 2(1):29
Costello RT, Mallet F, Gaugler B et al (2000) Human acute myeloid leukemia CD34+/CD38- progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res 60(16):4403–4411
Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, Fukata M, Miyamoto T, Lyons B, Ohshima K, Uchida N, Taniguchi S, Ohara O, Akashi K, Harada M, Shultz LD (2007) Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 25(11):1315–1321
Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG, di Fiore PP (2010) Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 140(1):62–73
Steinbichler TB, Dudas J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II (2018) Therapy resistance mediated by cancer stem cells. Semin Cancer Biol 53:156–167
Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, Weinberg RA (2017) Epithelial-to-Mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res 77(15):3982–3989
Prager BC, Xie Q, Bao S, Rich JN (2019) Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell 24(1):41–53
Shlush LI, Mitchell A, Heisler L et al (2017) Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 547(7661):104–108
Encabo A, Solves P, Mateu E, Sepulveda P, Carbonell-Uberos F, Minana MD (2004) Selective generation of different dendritic cell precursors from CD34+ cells by interleukin-6 and interleukin-3. Stem Cells 22(5):725–740
Hutten T, Thordardottir S, Hobo W, Hübel J, van der Waart A, Cany J, Dolstra H, Hangalapura BN (2014) Ex vivo generation of interstitial and Langerhans cell-like dendritic cell subset-based vaccines for hematological malignancies. J Immunother 37(5):267–277
Cornel AM, van Til NP, Boelens JJ, Nierkens S (2018) Strategies to genetically modulate dendritic cells to potentiate anti-tumor responses in hematologic malignancies. Front Immunol 9:982
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature. 392(6673):245–252
Kirk CJ, Mule JJ (2000) Gene-modified dendritic cells for use in tumor vaccines. Hum Gene Ther 11(6):797–806
Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Hüsemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Türeci Ö, Sahin U (2016) Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 534(7607):396–401
Salcedo M, Bercovici N, Taylor R, Vereecken P, Massicard S, Duriau D, Vernel-Pauillac F, Boyer A, Baron-Bodo V, Mallard E, Bartholeyns J, Goxe B, Latour N, Leroy S, Prigent D, Martiat P, Sales F, Laporte M, Bruyns C, Romet-Lemonne JL, Abastado JP, Lehmann F, Velu T (2006) Vaccination of melanoma patients using dendritic cells loaded with an allogeneic tumor cell lysate. Cancer Immunol Immunother 55(7):819–829
Finocchiaro G, Pellegatta S (2016) Immunotherapy with dendritic cells loaded with glioblastoma stem cells: from preclinical to clinical studies. Cancer Immunol Immunother 65(1):101–109
Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tønnesen P, Suso EM, Sæbøe-Larssen S, Sandberg C, Brinchmann JE, Helseth E, Rasmussen AM, Lote K, Aamdal S, Gaudernack G, Kvalheim G, Langmoen IA (2013) Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother 62(9):1499–1509
Pellegatta S, Eoli M, Frigerio S, Antozzi C, Bruzzone MG, Cantini G, Nava S, Anghileri E, Cuppini L, Cuccarini V, Ciusani E, Dossena M, Pollo B, Mantegazza R, Parati EA, Finocchiaro G (2013) The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology. 2(3):e23401
Vinay DS, Ryan EP, Pawelec G et al (2015) Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol 35(Suppl):S185–S198
Sultan M, Vidovic D, Paine AS et al (2018) Epigenetic silencing of TAP1 in Aldefluor(+) breast Cancer stem cells contributes to their enhanced immune evasion. Stem Cells 36(5):641–654
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4):252–264
Adachi K, Tamada K (2015) Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci 106(8):945–950
Haanen JB, Robert C (2015) Immune checkpoint inhibitors. Prog Tumor Res 42:55–66
Kyi C, Postow MA (2016) Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy. 8(7):821–837
Postow MA, Callahan MK, Wolchok JD (2015) Immune checkpoint blockade in Cancer therapy. J Clin Oncol 33(17):1974–1982
Rotte A, Jin JY, Lemaire V (2018) Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol 29(1):71–83
Wilden SM, Lang BM, Mohr P, Grabbe S (2016) Immune checkpoint inhibitors: a milestone in the treatment of melanoma. J Dtsch Dermatol Ges 14(7):685–695
Xia Y, Medeiros LJ, Young KH (2016) Immune checkpoint blockade: releasing the brake towards hematological malignancies. Blood Rev 30(3):189–200
Gajewski TF (2015) The next hurdle in Cancer immunotherapy: overcoming the non-T-cell-inflamed tumor microenvironment. Semin Oncol 42(4):663–671
Zhi Y, Mou Z, Chen J, He Y, Dong H, Fu X, Wu Y (2015) B7H1 expression and epithelial-to-Mesenchymal transition phenotypes on colorectal Cancer stem-like cells. PLoS One 10(8):e0135528
Yang Y, Wu KE, Zhao E et al (2015) B7-H1 enhances proliferation ability of gastric cancer stem-like cells as a receptor. Oncol Lett 9(4):1833–1838
Wu Y, Chen M, Wu P, Chen C, Xu ZP, Gu W (2017) Increased PD-L1 expression in breast and colon cancer stem cells. Clin Exp Pharmacol Physiol 44(5):602–604
Shi X, Zhang X, Li J, Mo L, Zhao H, Zhu Y, Hu Z, Gao J, Tan W (2018) PD-1 blockade enhances the antitumor efficacy of GM-CSF surface-modified bladder cancer stem cells vaccine. Int J Cancer 142(10):2106–2117
Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA (2005) Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23(10):2346–2357
Hinrichs CS, Rosenberg SA (2014) Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev 257(1):56–71
Rosenberg SA, Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 348(6230):62–68
Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J, Kulikovskaya I, Sinha SK, Kronsberg S, Gupta M, Bond S, Melchiori L, Brewer JE, Bennett AD, Gerry AB, Pumphrey NJ, Williams D, Tayton-Martin HK, Ribeiro L, Holdich T, Yanovich S, Hardy N, Yared J, Kerr N, Philip S, Westphal S, Siegel DL, Levine BL, Jakobsen BK, Kalos M, June CH (2015) NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21(8):914–921
Robbins PF, Lu YC, El-Gamil M et al (2013) Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 19(6):747–752
Tran E, Rosenberg SA (2014) T-cell therapy against cancer mutations. Oncotarget. 5(13):4579–4580
Boyiadzis MM, Dhodapkar MV, Brentjens RJ et al (2018) Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J Immunother Cancer. 6(1):137
Guedan S, Posey AD Jr., Shaw C et al. (2018) Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 3(1)
June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC (2018) CAR T cell immunotherapy for human cancer. Science. 359(6382):1361–1365
Alcantara M, Tesio M, June CH, Houot R (2018) CAR T-cells for T-cell malignancies: challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia. 32(11):2307–2315
Hay KA, Turtle CJ (2017) Chimeric antigen receptor (CAR) T cells: lessons learned from targeting of CD19 in B-cell malignancies. Drugs. 77(3):237–245
Kohn DB, Dotti G, Brentjens R, Savoldo B, Jensen M, Cooper LJ, June CH, Rosenberg S, Sadelain M, Heslop HE (2011) CARs on track in the clinic. Mol Ther 19(3):432–438
Watanabe K, Kuramitsu S, Posey AD Jr, June CH (2018) Expanding the therapeutic window for CAR T cell therapy in solid tumors: the Knowns and unknowns of CAR T cell biology. Front Immunol 9:2486
DeRenzo C, Krenciute G, Gottschalk S (2018) The landscape of CAR T cells beyond acute lymphoblastic leukemia for pediatric solid tumors. Am Soc Clin Oncol Educ Book 38:830–837
Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, Sergeeva A, Wood MS, Dotti G, Salvado B, Ruisaard K, Clise-Dwyer K, John LS, Rezvani K, Alatrash G, Shpall EJ, Molldrem JJ (2016) A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy. 18(8):985–994
Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G (2015) Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 6(1):171–184
Morgan RA, Johnson LA, Davis JL et al (2012) Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther 23(10):1043–1053
Beard RE, Zheng Z, Lagisetty KH et al (2014) Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer. 2:25
Pellegatta S, Savoldo B, Di Ianni N, et al. (2018) Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med 10(430)
Du H, Hirabayashi K, Ahn S et al (2019) Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell 35(2):221–237 e228
Deng Z, Wu Y, Ma W, Zhang S, Zhang YQ (2015) Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol 16:1
Wang Y, Geldres C, Ferrone S, Dotti G (2015) Chondroitin sulfate proteoglycan 4 as a target for chimeric antigen receptor-based T-cell immunotherapy of solid tumors. Expert Opin Ther Targets 19(10):1339–1350
Feng KC, Guo YL, Liu Y et al (2017) Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol 10(1):4
Sun B, Yang D, Dai H, et al. (2019) Eradication of hepatocellular carcinoma by NKG2D-specific CAR T-cells. Cancer Immunol Res
Yang D, Sun B, Dai H, Li W, Shi L, Zhang P, Li S, Zhao X (2019) T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J Immunother Cancer 7(1):171
Acknowledgements
This study was supported by Qatar National Research Fund, grant no. NPRP10-0129-170277.
Funding
Open Access funding provided by the Qatar National Library.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All the authors have no conflict of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ravindran, S., Rasool, S. & Maccalli, C. The Cross Talk between Cancer Stem Cells/Cancer Initiating Cells and Tumor Microenvironment: The Missing Piece of the Puzzle for the Efficient Targeting of these Cells with Immunotherapy. Cancer Microenvironment 12, 133–148 (2019). https://doi.org/10.1007/s12307-019-00233-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-019-00233-1