
Summary
In malignant diseases, targeting of immune checkpoints successfully changed the therapeutic landscape and helped to unleash anti-tumor T cell responses, resulting in durable clinical outcomes, but only in up to 50% of patients. The success of these therapies and the need to overcome intrinsic and acquired therapy resistance stimulated research to identify new pathways and targets. Numerous clinical trials are currently evaluating novel checkpoint inhibitors or recently developed strategies like modulating the tumor microenvironment, mostly in combination with approved therapies. This short review briefly discusses promising therapeutic targets, currently still under investigation, with the chance to realize clinical application in the foreseeable future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
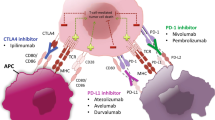
Over the past decade, immune checkpoint inhibitors (ICIs) successfully shaped the therapeutic landscape of malignant tumors. The most broadly studied and first immune checkpoint targets were cytotoxic T lymphocyte-associated antigen‑4 (CTLA-4), programmed cell death protein‑1 (PD-1) and its ligand (PD-L1). The binding of CTLA‑4 (CD152) to the ligands CD80 (B7-1) and CD86 (B7-2) delivers a negative signal to T cell activation [1], whereas the binding of PD‑1 (CD279) to its ligands PD-L1 and PD-L2 (CD273, B7-DC) suppresses the activation and function of T cells, thereby downregulating adaptive immune response [2, 3]. Ipilimumab, a monoclonal antibody (mAb) against CTLA‑4, was the first ICI approved by the United States Food and Drug Administration (FDA) in 2011 after demonstrating a survival benefit for patients with advanced melanoma over the chemotherapeutic dacarbazine. Pembrolizumab, the first humanized mAb against PD‑1, gained initial global approval for patients with unresectable or metastatic melanoma by the FDA in 2014 [4]. Since then, the indication of those mAbs to several other tumor entities, and the list of approved ICIs against PD-1/PD-L1 or CTLA‑4 have expanded [5]. The current benchmark for efficacy in melanoma therapy is the combinational therapy of anti-CTLA‑4 and anti-PD‑1 agents [6]. However, roughly half of all patients will not benefit from ICIs, and therefore identification of predictive markers allowing patient stratification regarding first- and second-line treatment strategies and avoiding toxicity of ineffective therapy is of immense clinical interest [7, 8]. The search for new potential targets and pathways has already resulted in a new portfolio of targets for novel treatment options, mostly tested in combination with PD‑1 inhibitors. Molecules targeting inhibitory pathways such as the type I transmembrane glycoproteins lymphocyte activation gene 3 protein (LAG-3), T cell immunoglobulin mucin receptor 3 (TIM-3), T cell immunoglobulin mucin receptor 3 (TIGIT) or B7 homolog 3 (B7-H3) are being investigated, as well as agonists of stimulatory checkpoint pathways, such as OX-40, the inducible T cell co-stimulator (ICOS), the glucocorticoid-induced TNFR-related protein (GITR), 4‑1BB and CD40 (Fig. 1, Table 1).

Overview of discussed immune checkpoints and their respective ligands on a tumor cell and/or antigen presenting cell (APC)
Targeting inhibitory pathways
After CTLA‑4 and PD‑1, lymphocyte activation gene 3 protein (LAG-3) was the third inhibitory receptor targeted with mAbs in clinical trials. LAG‑3 is a T cell-associated inhibitory checkpoint protein and member of the immunoglobin (Ig) superfamily, co-expressed with PD‑1 and usually present on T cells, B cells, dendritic cells (DCs) and natural killer cells (NK cells) [9]. It is responsible for regulating immune tolerance and T cell homeostasis by its inhibitory effect on effector T cell proliferation and enhancing regulatory T cell function [9, 10]. Pre-clinical studies have shown that dual PD‑1 and LAG‑3 blockade synergistically stimulate T cell responses and decrease tumor burden more than either agent alone [11]. In addition, the efficiency of the LAG‑3 antibody relatlimab seems to be increased in tumors with higher LAG‑3 expression, indicating that it might be used as a biomarker [6, 12].
The T cell immunoglobulin mucin receptor 3 (TIM‑3, CD366) is a type I transmembrane protein that can be found on a variety of immune cells and its expression was also demonstrated on melanoma tumor-infiltrating lymphocytes (TILs) [13,14,15]. Animal models of advanced melanoma demonstrated that blocking TIM‑3 reverses T cell exhaustion and dysfunction [10, 14,15,16]. Anti-TIM‑3 antagonist antibodies, like cobolimab, are currently under investigation in phase II clinical studies in combination with other checkpoint inhibitors or as a bispecific antibody (anti-PD‑1 and anti-TIM-3) in a phase I multiple-ascending dose study.
Another inhibitory receptor is the T‑cell immunoreceptor with Ig and ITIM domains (TIGIT). This transmembrane glycoprotein receptor is expressed not only on T cells, regulatory T cells (Tregs) and NK cells, but also highly expressed on melanoma cells, DCs and monocytes within the melanoma tumor microenvironment (TME) [15, 17, 18]. TIGITs immunosuppressive effects are mediated through a decreased release of pro-inflammatory cytokines and an increased release of IL-10 [19]. Vibostolimab, an anti-TIGIT antibody, is currently under investigation in a number of sub-studies to an umbrella study, testing experimental treatments for melanoma.
The B7 homolog 3 (B7-H3, CD276) protein is a type I transmembrane protein commonly expressed on antigen presenting cells (APCs), NK cells, tumor cells and tumor endothelial cells, belonging to the B7-CD28 pathway family. Its overexpression is frequent in multiple malignancies including melanoma, correlating with poor prognosis [20, 21]. Little is known about the molecular mechanisms underlying B7-H3 functions and its receptor(s) have not yet been identified. Research demonstrates that the B7-H3 pathway promotes cancer aggressiveness, while exerting inhibitory function on T cell activation, proliferation and cytokine production [20, 21]. This indicates that besides enhancing innate immunological responses against malignancies, B7-H3 blockade might also directly affect tumor behavior. The safety of the mAb enoblituzumab in combination with pembrolizumab on B7-H3 expressing melanomas and other cancers is currently evaluated. When combined with chemotherapy or other ICIs, it appears to have a synergistic effect [20, 21].
The B and T cell lymphocyte attenuator (BTLA, CD272) expressed by the majority of lymphocytes is an inhibitory receptor, structurally and functionally related to CTLA‑4 and PD‑1. Binding of BTLA to its ligand herpes virus entry mediator (HVEM) leads to an inhibition of T and B cell activation, proliferation and cytokine production [20, 22]. By expressing HVEM, melanoma cells have been shown to exploit this pathway and high levels of BTLA/HVEM correlate with progression and poor prognosis, making this pathway a promising target for checkpoint blockade [23]. The first anti-BTLA mAb approved for clinical trials is currently being assessed regarding its safety and tolerability as monotherapy in advanced malignancies.
The V‑domain Ig suppressor of T cell activation (VISTA) is a type I transmembrane protein and B7 family member. It is constitutively expressed on multiple immune cell types, mainly on myeloid cells including neutrophils, monocytes, macrophages and DCs, and can also be found on TILs [24, 25]. Structural analysis suggests that VISTA has the potential to function as a receptor and a ligand [26]. Furthermore, evidence indicates that VISTA could exert a dual role, stimulatory for APCs on the one side and inhibitory for T cells on the other [20]. Recently, VSIG‑3 was reported as a binding partner of VISTA [27]. In pre-clinical studies, VISTA blockade has demonstrated improved infiltration, proliferation and effector function of TILs within the TME, thereby altering the suppressive properties of the TME [28, 29].
Activating co-stimulatory pathways
For optimal cancer control it may not be sufficient to target negative regulatory pathways alone, but may require the activation of co-stimulatory pathways either alone or in combination with checkpoint blockade to enhance the immune response.
OX-40 (CD134, TNFRSF4) is a T cell co-stimulatory protein and member of the tumor necrosis factor receptor superfamily (TNFRSF). It is primarily expressed on activated T cells and APCs, but it is also expressed at high levels on tumor resident Tregs [30]. OX-40 agonism leads to an increase in the number of activated T cells and those cells gaining effector function, while the induction of Tregs in the periphery is suppressed [31].
Interactions of OX-40 and its ligand on activated T cells increase proliferation, effector and cytotoxic function and cytokine production of those T cells, among other features [32]. Several OX-40 agonist antibodies are investigated in clinical phase I and II trials. Increased OX-40 expression on TILs in cutaneous melanoma is associated with improved prognosis [33].
Another promising immunotherapy target is the glucocorticoid-induced TNFR-related protein (GITR), stimulating the acquired and innate immunity. It is highly expressed on Tregs and activated upon binding to its ligand GITRL, mainly expressed on APCs and endothelial cells. This exerts dual effects, down-regulation of Tregs and up-regulation of CD8+ effector cells while extending their survival [15, 34, 35]. Anti-tumor activity of agonistic anti-GITR antibodies has already been demonstrated in mouse models and is currently evaluated in phase I and II clinical trials in melanoma and other tumor types [36].
The inducible T cell co-stimulator, short ICOS (CD278), is an immune checkpoint protein structurally and functionally related to CD28, a T cell specific cell-surface receptor like CTLA‑4, and an important regulator of the immune system. ICOS is expressed on activated cytotoxic T cells, Tregs, NK cells and other types of T cells. It enhances all basic T cell functions to a foreign antigen like proliferation, secretion of cytokines and mediators up-regulating cell–cell interaction and supporting antibody secretion by B cells [37]. Up-regulation of ICOS can be detected in activated T cells, especially after anti-CTLA‑4 therapy, where it can serve as a biomarker indicating the binding of anti-CTLA‑4 antibodies to their targets. Increased expression on circulating T cells after ipilimumab therapy has been associated with improved clinical outcomes [38, 39]. Due to the upregulation of ICOS after anti-CTLA‑4 therapy, the combination of those targets can achieve potent synergistic effects [20, 37].
Another important regulator of immune response and member of the TNFRSF is 4‑1BB (CD137, TNFRSF9). This co-stimulatory molecule is expressed on innate and adaptive immune cells and triggers proliferation and prolonged survival of CD8+ effector T cells and NK cells upon binding to its ligand 4‑1BBL [20, 40]. Anti-4-1BB antibody blockade has been shown to induce potent anti-tumor T cell responses by promoting CD8+ T cell proliferation, enhancing T cell receptor (TCR) signaling and inducing immunologic memory [41, 42]. Several pre-clinical agonistic antibodies are currently under investigation, like utomilumab, in a phase II study in melanoma [41].
An additional member of the TNF family and co-stimulatory immune checkpoint receptor is the glycoprotein CD27 expressed on T cells, NK cells and Tregs. Its ligand is CD70, which is expressed on DCs, activated B and T cells [43]. When CD27 is bound by CD70, CD8+ T cell activation, survival and effector function are enhanced. To prevent unwanted stimulation of T cells, CD70 is usually not available. In this case, varlilumab or other mAbs can substitute and activate T cells receiving TCR stimulation [44].
Immune co-stimulatory receptor CD40 (TNFRSF5) is also part of the TNFRSF and expressed on APCs, including DCs, B cells, macrophages and monocytes. It plays a key role in the activation of the immune system, while binding to its ligand CD40L (CD154) on T cells [20]. Binding of CD40 leads to increased priming and activation of CD8+ T cells, mediated through increased major histocompatibility complex (MHC) surface expression on DCs, production of pro-inflammatory cytokines and B cell proliferation [15, 45]. Healthy tissue exhibits comparatively low to no CD40 expression, indicating strong potential as a cancer-specific immunological target [46]. Several clinical trials are studying the effects of CD40 as monotherapy or in combination.
Other pathways and novel agents
Besides targeting immune checkpoints and thereby inducing inhibitory or co-stimulatory immune responses, further research interest is aimed at other promising pathways and mechanisms.
Modulating the TME through indoleamine 2,3-dioxygenase 1 (IDO1) is one of those approaches. IDO1 is a tryptophan catabolizing and IFN-inducible enzyme, promoting tumor-mediated immunosuppression. It thereby inhibits effector T cells and NK cells and activates Tregs and myeloid-derived suppressor cells [15, 47]. IDO1 is overexpressed in several malignancies including melanoma, while inhibition shifts the TME from a tumor-promoting inflammatory to an immune stimulating state [48]. Especially in melanoma, previous research established a relationship between CTLA‑4, PD‑1 and IDO1 associated with poor prognosis, independent of disease stage, making IDO1 a potential target for further investigation [15, 49]. However, a phase III study showed no survival benefit from enhancing anti-PD‑1 therapy by IDO1 inhibition [50].
Another major factor in the immunosuppressive TME is the adenosine pathway, which is mediated by ectonucleotidases, like CD39 and CD73, and adenosine receptors, like A2AR. In melanoma, increased CD73 (ecto-5′-nucleotidase) expression correlates with a more aggressive, invasive phenotype and can be detected in over 50% of the metastases [51], while CD39 is overexpressed earlier in tumor development, potentially influencing the differentiation of melanocytes to melanoma cells [15]. In pre-clinical models, CD73 blockade showed inhibition of metastasis formation and improved anti-tumor immunity [52]. A first-in-human study is currently evaluating the safety of a small molecule inhibitor targeting CD73 as monotherapy or in combination with pembrolizumab in patients with melanoma and other advanced solid malignancies.
Targeting TLRs aims at a family of specialized receptors, stimulating immune responses to pathogen-associated molecular patterns (PAMPs). Among those, TLR9 has been shown to induce potent anti-tumor responses by stimulating innate and adaptive immune responses [53], ultimately leading to strong CD4+ and CD8+ T cell responses that may intensify the efficacy of ICIs [54]. Clinical activity of TLR9 has been shown in advanced melanoma patients unresponsive to PD-(L)1 inhibition.
Oncolytic peptides are cytotoxic chemotherapeutic peptides, injected intratumorally and thereby limiting systemic toxicities as well as their application to disseminated malignancies [20]. Injection of the lactoferrin-derived lytic peptide ruxotemitide leads to tumor antigen release followed by increased TIL activity and CTLA‑4 expression, suggesting administration in conjunction with anti-CTLA‑4 agents [55].
IL‑2 is nothing new and is considered the first effective immunotherapy in cancer [56]. Due to severe toxicities, IL-2R agonists have been developed to potentiate and prolong IL‑2 anti-tumor effects, thereby allowing lower doses. Bempegaldesleukin, an engineered cytokine specifically stimulating through IL-2Rβ (CD122), is currently being investigated in combination with nivolumab in a phase III clinical trial in melanoma. Pegilodecakin, the PEGylated form of IL-10, is under investigation in regard to safety and tolerability in a phase I study. Also for IL-10, a combination with PD‑1 seems reasonable due to both receptors being upregulated on TILs [20].
Promising results have also been shown with histone deacetylase (HDAC) inhibitors and pembrolizumab in patients with unresectable or metastatic melanoma that progressed during or after anti-PD‑1 therapy. Histone acetylation and deacetylation play a key role in regulating gene transcription, and inhibition of this process has emerged as a potential anticancer therapeutic in various malignancies [15].
Another emerging field of research focusses on intra-tumoral agents, like the stimulator of interferon genes (STING). STING is a transmembrane protein that is activated by cyclic dinucleotides and plays an important role in innate immunity by stimulating type 1 IFN‑1 and DC activation [15]. In mouse models, intra-tumoral injection of the dinucleotide caused regression of the injected and untreated lesions. Pre-clinical data demonstrates that tumor antigen recognition in melanoma can be enhanced by restoring MHC class I surface expression through agonist-induced activation of STING signaling [57]. This suggests that synthetic cyclic dinucleotides activating the STING pathway should be considered as a therapeutic intervention and further investigated. Phase I trials are currently testing stimulators of STING.
Discussion
Understanding the immunobiology of tumors and therapeutic resistance will remain a major challenge for the future. The aim is to develop effective immunotherapies tailored to individual subgroups of patients, especially those not achieving long-term clinical benefit. Omics-based strategies utilizing the respective bioinformatics could deliver a potential breakthrough in this ongoing (re)search in precision medicine (Fig. 2). To understand the complexity of the tumor-immune microenvironment consisting of interactions between multiple cell types, omics-guided approaches can help in assessing for instance the TME, immune system and cancer cells. Microbiome strategies, representing the next wave of treatment approaches in immuno-oncology, will likely enrich this dynamic therapeutic landscape even further. The results from pre-clinical research and subsequently clinical studies will provide potential predictive biomarkers, novel treatment options and pharmacodynamic markers to guide treatment decisions for each individual patient. Ultimately, a combination of immunotherapies or integration of immunotherapy with non-immunotherapies will likely succeed over single agent approaches, given the complexity of the immune response.

General algorithm for the optimal treatment choice
Take home message
At present, it seems to be a difficult challenge to surpass the combination of ipilimumab and nivolumab regarding efficacy. Nevertheless, novel immune checkpoints have the potential to improve clinical outcome for patients with advanced melanoma and other malignancies, by broadening the therapeutic spectrum and increasing the number of therapy options.
References
Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T‑cell immunity. Nat Rev Immunol. 2004;4:336–47.
Berger KN, Pu JJ. PD‑1 pathway and its clinical application: A 20 year journey after discovery of the complete human PD‑1 gene. Gene. 2018;638:20–5.
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD‑1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8.
Kwok G, Yau TCC, Chiu JW, Tse E, Kwong Y‑L. Pembrolizumab (Keytruda). Hum Vaccin Immunother. 2016;12:2777–89.
Li Z, Song W, Rubinstein M, Liu D. Recent updates in cancer immunotherapy: a comprehensive review and perspective of the 2018 China cancer immunotherapy workshop in Beijing. J Hematol Oncol. 2018;11:142.
Hoeller C. The future of combination therapies in advanced melanoma. memo. 2020;13:309–13.
Dummer R, Goldinger SM, Paulitschke V, Levesque MP. Curing advanced melanoma by 2025. Curr Opin Oncol. 2015;27:125–7.
Hogan SA, Levesque MP, Cheng PF. Melanoma immunotherapy: next-generation biomarkers. Front Oncol. 2018;8:178.
He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, et al. Lymphocyte-activation gene‑3, an important immune checkpoint in cancer. Cancer Sci. 2016;107:1193–7.
Anderson AC, Joller N, Kuchroo VK. Lag‑3, Tim‑3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004.
Lichtenegger FS, Rothe M, Schnorfeil FM, Deiser K, Krupka C, Augsberger C, et al. Targeting LAG‑3 and PD‑1 to enhance T cell activation by antigen-presenting cells. Front Immunol. 2018;9:385.
Ascierto P, et al. ESMO 2017, abstract LBA18. 2017.
Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim‑3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–41.
Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim‑3 and PD‑1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–86.
Ambrosi L, Khan S, Carvajal RD, Yang J. Novel targets for the treatment of melanoma. Curr Oncol Rep. 2019;21:97.
Sakuishi K, Ngiow SF, Sullivan JM, Teng MWL, Kuchroo VK, Smyth MJ, et al. TIM3(+)FOXP3(+) regulatory T cells are tissue-specific promoters of T‑cell dysfunction in cancer. OncoImmunology. 2013;2:e23849.
Chauvin J‑M, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD‑1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125:2046–58.
Johnston RJ, Yu X, Grogan JL. The checkpoint inhibitor TIGIT limits antitumor and antiviral CD8+ T cell responses. OncoImmunology. 2015;4:e1036214.
Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim‑3 ligand galectin‑9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–52.
Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11:39.
Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. 2016;22:3425–31.
Paulos CM, June CH. Putting the brakes on BTLA in T cell-mediated cancer immunotherapy. J Clin Invest. 2010;120:76–80.
Lan X, Li S, Gao H, Nanding A, Quan L, Yang C, et al. Increased BTLA and HVEM in gastric cancer are associated with progression and poor prognosis. Onco Targets Ther. 2017;10:919–26.
Lines JL, Sempere LF, Broughton T, Wang L, Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. 2014;2:510–7.
Choi JW, Kim YJ, Yun KA, Won CH, Lee MW, Choi JH, et al. The prognostic significance of VISTA and CD33-positive myeloid cells in cutaneous melanoma and their relationship with PD‑1 expression. Sci Rep. 2020;10:14372.
Flies DB, Wang S, Xu H, Chen L. Cutting edge: a monoclonal antibody specific for the programmed death‑1 homolog prevents graft-versus-host disease in mouse models. J Immunol. 2011;187:1537–41.
Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG‑3 as a ligand of VISTA inhibits human T‑cell function. Immunology. 2019;156:74–85.
Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD‑1 and CTLA‑4. Mol Cancer. 2019;18:155.
Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74:1933–44.
Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: structure and function—what questions remain? Mol Immunol. 2017;83:13–22.
Zhang X, Xiao X, Lan P, Li J, Dou Y, Chen W, et al. OX40 costimulation inhibits Foxp3 expression and Treg induction via BATF3-dependent and independent mechanisms. Cell Rep. 2018;24:607–18.
Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria J‑C, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50–66.
Ladányi A, Somlai B, Gilde K, Fejös Z, Gaudi I, Tímár J. T‑cell activation marker expression on tumor-infiltrating lymphocytes as prognostic factor in cutaneous malignant melanoma. Clin Cancer Res. 2004;10:521–30.
Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second- and third-generation drugs for immuno-oncology treatment - the more the better? Eur J Cancer. 2017;74:55–72.
Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer. 2016;67:1–10.
Zhu MMT, Burugu S, Gao D, Yu J, Kos Z, Leung S, et al. Evaluation of glucocorticoid-induced TNF receptor (GITR) expression in breast cancer and across multiple tumor types. Mod Pathol. 2020;33:1753–63.
Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42:640–55.
Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA‑4 blockade in cancer immunotherapy. J Exp Med. 2014;211:715–25.
Harvey C, Elpek K, Duong E, Simpson T, Shu CJ, Shallberg L, et al. Efficacy of anti-ICOS agonist monoclonal antibodies in preclinical tumor models provides a rationale for clinical development as cancer immunotherapeutics. J Immunother Cancer. 2015;3:O9.
Makkouk A, Chester C, Kohrt HE. Rationale for anti-CD137 cancer immunotherapy. Eur J Cancer. 2016;54:112–9.
Takeda K, Kojima Y, Uno T, Hayakawa Y, Teng MWL, Yoshizawa H, et al. Combination therapy of established tumors by antibodies targeting immune activating and suppressing molecules. J Immunol. 2010;184:5493–501.
Bartkowiak T, Curran MA. 4‑1BB agonists: multi-potent potentiators of tumor immunity. Front Oncol. 2015;5:117.
Kroemer A, Xiao X, Vu MD, Gao W, Minamimura K, Chen M, et al. OX40 controls functionally different T cell subsets and their resistance to depletion therapy. J Immunol. 2007;179:5584–91.
Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood. 2018;131:39–48.
Rakhmilevich AL, Alderson KL, Sondel PM. T‑cell-independent antitumor effects of CD40 ligation. Int Rev Immunol. 2012;31:267–78.
Piechutta M, Berghoff AS. New emerging targets in cancer immunotherapy: the role of cluster of differentiation 40 (CD40/TNFR5). ESMO Open. 2019;4:e510.
Mellor AL, Keskin DB, Johnson T, Chandler P, Munn DH. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J Immunol. 2002;168:3771–6.
Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017;77:6795–811.
Chevolet I, Speeckaert R, Schreuer M, Neyns B, Krysko O, Bachert C, et al. Characterization of the in vivo immune network of IDO, tryptophan metabolism, PD-L1, and CTLA‑4 in circulating immune cells in melanoma. OncoImmunology. 2015;4:e982382.
Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20:1083–97.
Monteiro I, Vigano S, Faouzi M, Treilleux I, Michielin O, Ménétrier-Caux C, et al. CD73 expression and clinical significance in human metastatic melanoma. Oncotarget. 2018;9:26659–69.
Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, et al. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol. 2013;191:4165–73.
Lu H. TLR agonists for cancer immunotherapy: tipping the balance between the immune stimulatory and inhibitory effects. Front Immunol. 2014;5:83.
Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117:1184–94.
Yamazaki T, Pitt JM, Vétizou M, Marabelle A, Flores C, Rekdal Ø, et al. The oncolytic peptide LTX-315 overcomes resistance of cancers to immunotherapy with CTLA4 checkpoint blockade. Cell Death Differ. 2016;23:1004–15.
Tomala J, Kovar M. IL-2/anti-IL‑2 mAb immunocomplexes: a renascence of IL‑2 in cancer immunotherapy? OncoImmunology. 2016;5:e1102829.
Falahat R, Perez-Villarroel P, Mailloux AW, Zhu G, Pilon-Thomas S, Barber GN, et al. STING signaling in melanoma cells shapes antigenicity and can promote antitumor T‑cell activity. Cancer Immunol Res. 2019;7:1837–48.
Funding
Open access funding provided by the Medical University of Vienna.
Funding
Open access funding provided by Medical University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
C. Hoeller declares the following: Speaker for Amgen, BMS, MSD, Novartis and Roche. Consultancy for Amgen, Astra Zeneca, BMS, Inzyte, MSD, Novartis, Pierre Fabre and Roche. Research support from Amgen. N. Zila and V. Paulitschke declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zila, N., Hoeller, C. & Paulitschke, V. Novel immune checkpoints beyond PD-1 in advanced melanoma. memo 14, 135–142 (2021). https://doi.org/10.1007/s12254-021-00699-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12254-021-00699-0