
Abstract
Potato viruses such as Potato virus Y (PVY) cause diseases that affect potato quality and thus damage potato production worldwide. Current tests for viral infection use double-antibody sandwich enzyme linked immunosorbent assays (DAS‑ELISA) or reverse transcription polymerase chain reaction (RT-PCR)/real-time RT-PCR. Despite many advantages, these assays have a number of drawbacks that affect cost and time of diagnosis. Reverse transcription loop-mediated isothermal amplification (RT-LAMP) allows fast detection of target RNA. Here, we developed a closed-tube real-time RT‑LAMP assay for fluorescent detection of PVY. Specific RT-LAMP primers were designed to target the conserved region of the sequence encoding the PVY coat protein. The assay was specific and facilitated sensitive PVY detection in a single tube at 65 °C. The time-to-positive values depended on the PVY concentration in tested samples. The effectiveness of RT‑LAMP in testing field-grown plants compared favorably with DAS‑ELISA and RT-PCR; under the tested conditions, RT-LAMP was about 1000-fold more sensitive than DAS‑ELISA and lateral flow assay (LFA) and about 10-fold more sensitive than RT‑PCR. Thus, this fluorescent RT-LAMP assay has great potential for routine detection of PVY.
Resumen
Los virus de la papa causan enfermedades que reducen enormemente la calidad. Entre ellos, el virus Y de la papa (PVY) es el más importante en la producción de papa en todo el mundo. En la actualidad la incidencia de la infección viral en papa se prueba mediante DAS ELISA o RT-PCR/RT-PCR de tiempo real. A pesar de muchas ventajas, estos ensayos tienen un número de dificultades que afectan el costo y el tiempo del diagnóstico viral. Una amplificación térmica de transcripción reversa mediada por enroscamiento (RT-LAMP) es un método novedoso que permite detección rápida del ARN objetivo. En este trabajo se desarrolló un ensayo en tubo cerrado de RT-LAMP de tiempo real para detección fluorescente del PVY. Se diseñaron iniciadores específicos RT-LAMP apuntando a la región conservada de la secuencia que codifica para la cubierta proteica. El ensayo fue específico y facilitó la sensibilidad de detección del PVY en tubo individual a 65 °C. Los valores del tiempo al positivo dependieron de la concentración del PVY en las muestras probadas. Bajo las condiciones de prueba, RT-LAMP fue cerca de 1000 veces más sensible que DAS ELISA y que el ensayo de flujo lateral, y como 10 veces más sensible que RT-PCR. La efectividad de RT-LAMP al probarla en plantas en el campo se comparó favorablemente con DAS ELISA y RT-PCR. En consecuencia, presenta un gran potencial para detección de rutina del PVY.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Potato virus Y, a type member of the genus Potyvirus (family Potyviridae), is considered scientifically and economically as one of the most important viruses distributed worldwide (Quenouille et al. 2013; Scholthof et al. 2011). PVY causes a major threat to potato yields and also significantly affects tobacco, pepper, and tomato (Crosslin 2012; Quenouille et al. 2013; Scholthof et al. 2011; Singh et al. 2008; Wróbel and Wąsik 2013). In nature the virus infects solanaceous and non-solanaceous weeds (Kaliciak and Syller 2009). Many aphid species transmit PVY, in a non-persistent manner, with Myzuspersicae (Sulz.) acting as its most important vector (Kostiw and Trojanowska 2011; Quenouille et al. 2013; Verbeek et al. 2010). The highly diverse genetic structures of PVY strains fall into five main clades: C1, C2, Chile, N, and O, with many recombination variants among N and O strains (Quenouille et al. 2013; Singh et al. 2008).
Seed certification systems worldwide require accurate PVY detection. The most common procedure to detect PVY involves bio-amplification of the virus by cutting eye cores from dormant tubers and growing offspring plants in a greenhouse for 3-4 weeks. The offspring are next inspected visually for virus symptoms and/or tested by the enzyme-linked immunosorbent assay (ELISA) (Zahn et al. 2010). Alternatively, tubers can be tested directly by RT‑PCR (Crosslin and Hamlin 2011) or real-time RT-PCR (Singh et al. 2012). However, the cost and complexity of immunological and molecular methods restricts their use to dedicated, specialized laboratories (Boonham et al. 2008).
Diagnostic methods using loop-mediated isothermal amplification of DNA (LAMP) facilitate fast, sensitive, and specific detection of target DNA (Notomi 2000) without the need for expensive equipment. When combined with reverse transcription (RT‑LAMP), these assays can also detect pathogens with an RNA genome (Notomi 2000). In its simplest form, the RT‑LAMP reaction can be performed in a water bath and the amplification can be detected visually or monitored by measuring turbidity caused by the formation of magnesium pyrophosphate (Mori et al. 2001). Colorimetric (Goto et al. 2009) and fluorescent (Tomita et al. 2008) variants of the assay were also developed. RT-LAMP has been used to detect various pathogens, including plant viruses such as Plum pox virus (Varga and James 2006), Potato leafroll virus (Ahmadi et al. 2013; Ju 2011), Bean pod mottle virus (Wei et al. 2012), Cassava brown streak virus (Tomlinson et al. 2013), Pepino mosaic virus (Hasiów-Jaroszewska and Borodynko 2013), Papaya leaf distortion virus (Shen et al. 2014) and Cucurbit chlorotic yellows virus (Wang et al. 2014). The agarose gel based and turbidimetric variants of the method were also adopted for detection of PVY (Nie 2005; Zhao et al. 2012). Here, we develop a closed tube, real-time fluorescent RT‑LAMP assay for fast and sensitive detection of PVY.
Materials and Methods
Source of Plants and Viruses
Tubers of cultivar Vital, infected with PVY, were obtained from the virus collection held in the Młochów Research Center of the Plant Breeding and Acclimatization Institute – National Research Institute. Tubers of cultivars Giewont, Leona, and Pungo, infected with Potato virus M (PVM), Potato virus S (PVS), and Potato virus X (PVX), respectively, and virus-free tubers of cultivar Sagitta were obtained from the Laboratory of Genetic Resources and In Vitro Cultures of the Department of Potato Protection and Seed Science at Bonin, Plant Breeding and Acclimatization Institute ‑ National Research Institute. Virus-free and virus-infected plants were obtained by planting tubers in the greenhouse. The resulting plants were tested for the viruses using DAS-ELISA kits for PVY, PVM, PVS, and PVX. DAS-ELISA was performed according to the manufacturer’s instructions (Bioreba).
RNA Isolation
Total RNA was extracted from leaves of healthy and virus-infected plants using RNA 3-zone reagent (Novazym Poland) according to the manufacturer’s instructions. RNA was dissolved in DEPC-treated water, and stored at −80 °C before use. Quantity and quality of RNA preparations was estimated using the Epoch microplate spectrophotometer (BioTek) by measuring optical densities at 260 and 280 nm.
Design of RT-LAMP Primers
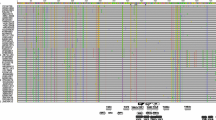
The coat protein coding sequences of forty PVY isolates were obtained from GenBank and multiple alignment analysis was performed using Genieous 5.0.4 software (Biomatters). The Gene Bank accession numbers of the sequences used in the alignment were: AM931253, AM931254, AB025416, AB270705, AB331548, AB331540, AB331546, X79305, AY601681, EF027866, HM586901, JN034579, JN034563, GU074000, EU252529, HM586906, HM586913, JN034570, HM586918, EF027868, EU885418, KC875237, GU550507, HM586924, EU161658, EF027863, EF027888, EF027865, EF027891, EF027893, HM586930, JN034576, HM586942, JN034573, HM586947, HM586948, HM586946, KC506396, FM200035, and X54611. Based on the alignment, a set of primers targeting a conserved region of the coat protein sequence was designed with LAMP Designer software (PREMIER Biosoft). The set included two outer primers (F3 and B3), two inner primers (FIP and BIP), and two loop primers (LF and LB). Primer details are listed in Table 1.
RT-LAMP
The optimized real-time RT-LAMP mixture (25 μl) contained: 1.5 μmol each of FIP and BIP, 0.75 μmol each of the loop primers (LF and LB) and 0.375 μmol each of the outer primers (F3 and B3), 15 μl Isothermal Mastermix Fluorescent Dye (IS0-001, Novazym Poland), 10 U reverse transcriptase (Roche) and 2 μl of the target RNA. The amplification was performed at 65 °C for 30 min and the specificity of the resulting products was analyzed by determination of annealing temperature (Ta) by slow annealing (0.05 °C/s) for 5 min, starting at 95 °C and ending at 80 °C. Both amplification and annealing steps, as well as a real-time monitoring of fluorescent signal, were performed in a Genie II Ultra rapid amplification instrument (OptiGene). Reactions containing total RNA from virus-free plants and mock reactions supplemented with water instead of RNA were included in each run as negative controls. Fluorescence readings recorded by the Genie II were exported to Excel (MS Office) and amplification curves were plotted using GraphPad software (GraphPad Software, Inc.).
Optimization of RT-LAMP Reaction for PVY Detection
The effect of primer concentration on reaction efficacy was tested by adding different amounts of each primer in the respective pair to the reaction: (A) 2 µmol Fip/Bip, 0.5 µmol F3/B3, 1 µmol LF/LB; (B) 1 µmol Fip/Bip, 0.25 µmol F3/B3, 0.5 µmol LF/LB; (C) 0.8 µmol Fip/Bip, 0.2 µmol F3/B3, 0.4 µmol LF/LB; (D) 0.5 µmol Fip/Bip, 0.125 µmol F3/B3, 0.25 µmol LF/LB. As a negative control (E) reaction supplemented with total RNA from a healthy plant was used. The effect of reverse transcriptase (RTase) concentration was tested by adding 20 U, 10 U, or 5 U of enzyme per reaction.
Specificity of the RT-LAMP
The specificity of RT‑LAMP was evaluated by adding 100 ng of total RNA isolated from plants infected with PVM, PVS, and PVX to the RT-LAMP reaction. As a negative control, total RNA isolated from virus-free plants was used. In the mock reaction, water control was used instead of RNA.
RT-PCR
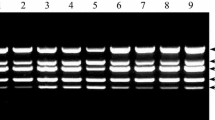
RT-PCR was carried out according to the conditions optimized previously (Zacharzewska et al. 2014), except that the primer set Y5 was replaced with the RT-LAMP outer primers (F3 and B3, Table 1) to target the PVY genome segment amplified in the RT-LAMP assay. Reverse transcription reactions were carried out with the Reverse Super Verte KIT with random primers (Novazym Poland) following the manufacturer’s protocol. The resulting cDNA was amplified by PCR using F3 as forward and B3 as reverse primer, generating a 331 bp product. The PCR mixture contained: 2.5 μl of the cDNA, 2.5 mM MgCl2, 0.2 mM dNTPs, 0.8 μM of each forward and reverse primer, and 1.25 U Go‑Taq HotStart Polymerase (Promega) in a 25 μl reaction. The temperature profile followed was: initial denaturation at 95 °C for 2 min, 35 cycles at 92 °C for 30 s, annealing at 58 °C for 40 s, extension at 72 °C for 1 min and final extension at 72 °C for five minutes. The reaction products were separated on a 1.5 % agarose gel at 140V for 50 min and stained with GelGreen dye (Biotium). Gels were excited using UV lightbox (Invitrogen) and documented with a Nikon D90 with an orange filter.
Sensitivity of RT-LAMP Compared to DAS-ELISA, LFA and RT-PCR
Comparison of sensitivity of detection methods was carried out using sap extracted from leaves of potato plants infected with PVY. Sap dilutions were prepared with DAS-ELISA sample buffer (1xPBS, 2 % PVP, 0.05 % Tween 20) and ranged from ten-fold (dilution factor = 1x101) to one million-fold (dilution factor = 1x106). LFA PVY AgriStrip Complete kit, Bioreba, Cat. No.: 112981) and DAS‑ELISA (Bioreba PVY polyclonal kit, Cat. No.: 110580) were carried out according to the manufacturer's instructions, using the prepared dilution series. To determine DAS-ELISA sensitivity, optical densities (OD) recorded at 405 nm after 60 minutes of signal development were plotted as a function of the logarithm of the dilution factor using GraphPad software (GraphPad Software, Inc.). The DAS-ELISA threshold was defined as twice the value of the mean OD value recorded under the same conditions for 10-fold diluted virus-free sap (healthy control). To quantitate the results of LFA, the relative intensities of control (C) and PVY‑specific, test (T) bands, developed on the lateral flow devices were quantitated using Phoretix 1D software (Totallab). The percent of PVY-positive signal was determined by dividing the value of T by the sum of the values of T and C. Data were fitted using GraphPad software (GraphPad Software, Inc.). To determine sensitivity of the molecular methods, total RNAs were isolated from all prepared dilutions. These RNAs were subsequently tested by RT‑PCR and RT-LAMP according to the procedures described above. Each experiment was repeated at least three times.
Results
Designing and Optimization of the Reaction Conditions for One-Step RT-LAMP Assay
Designing specific primers is a critical factor for developing RT-LAMP assays (Notomi 2000). To find a conserved sequence for primer design, we aligned 40 sequences of the coat protein from different isolates of PVY. Basing on this alignment, a specific set of primers (Table 1) was designed to amplify a conserved region of the coat protein. It was impossible to avoid mismatches between a primer and the corresponding sequence of several isolates, but most of the mismatches were located far from the 3’-end of the primers. Importantly, the 3’‑end sequences of the FIP and BIP primers fully matched all aligned isolates (Fig. 1). For amplification, we modified a commercial kit for fluorescent LAMP amplification by adding thermostable reverse transcriptase (RTase) to the reaction mix. To optimize the reaction conditions, we investigated the influence of the concentration of primers and RTase on the reaction efficacy (Fig. 2). Optimal amplification required the highest concentration of primers that we tested (Fig. 2, panel a) but we observed little or no difference between 10 and 20 U of RTase (Fig. 2, panel b). Thus, for further investigation, 10 U per reaction was used.

Comparison of the RT‑LAMP primer sequences with the corresponding genomic fragments of PVY isolates. F3, B3 – forward and reverse outer primers, F2 and F1c – segments of FIP primer, B2 and B1c –segments of BIP primer, LF and LB – loop primers

Optimization of RT-LAMP for PVY detection. a Effect of primer concentration (FIP-BIP/F3-B3/LF-BF) on the reaction efficacy. A – 2/0.5/1, B -1/0.25/0.5, C – 0.8/0.2/0.4, D – 0.5/0.125/0.25 µM of each primer in respective combination, E – reaction supplemented with total RNA from healthy plant (negative control). b Effect of reverse transcriptase (RTase) concentration. Amount of added RTase (in Units of enzyme) is indicated. Reaction supplemented with total RNA from healthy plant (negative control) – healthy, reaction supplemented with water instead of RNA – mock
Specificity of Detection of PVY by One-Step RT-LAMP
The specificity of the designed primers was confirmed by amplification of RNA isolated from leaves of virus-free plants, plants infected with PVY, and with other viruses commonly infecting potato, including Potato virus M (PVM), Potato virus S (PVS), and Potato virus X (PVX). As expected, only PVY was detected (Fig. 3, panel a). The annealing temperature of the amplified RT-LAMP product was equal to the predicted Ta = 86.5 °C (Fig. 3, panel b).

Specificity of PVY detection. a Amplification plots for reactions supplemented with total RNA extracted from plants infected with PVY, PVX, PVM, and PVS. Reaction supplemented with total RNA from a healthy plant (negative control) – healthy, reaction supplemented with water instead of RNA – mock. b Annealing curves recorded for amplicons in reactions described in panel A
Sensitivity of RT-LAMP Assay Compared to DAS-ELISA, LFA and RT-PCR
To compare the sensitivities of different assays, we tested the same dilution series with LFA (Fig. 4, panels a and b), DAS-ELISA (Fig. 4, panel c), RT‑LAMP (Fig. 4, panels d and e), and RT‑PCR (Fig. 4, panel f). A similar limit of detection was observed for LFA and DAS‑ELISA (5x102, Fig. 4, panels b and c). Detection of PVY by LFA in samples diluted from 10-fold to 50-fold required an incubation time of about five minutes, but detection of PVY in samples diluted 500-fold (the lowest detectable PVY concentration), required an incubation time of up to 20 minutes. The limit of detection of the RT-PCR was observed to be at dilutions of 5x104. In the RT-LAMP assay, the PVY‑specific signal was detected up to a dilution factor of 5x105. However, the lower the PVY concentration, the longer the amplification time required to detect the virus (Fig. 4, panels d and e). PVY in sap diluted 10-fold was detected after 11 minutes, while the time-to-positive value (Tp) for sap diluted 5x105-fold was 22 minutes (Fig. 4, panel d). The relation between Tp values and the logarithm of the dilution factor was linear over several orders of magnitude (Fig. 4, panel e).

Comparison of sensitivity of LFA, DAS‑ELISA, RT-LAMP, and RT‑PCR.a Determination of the sensitivity of the PVY detection by LFA using Bioreba AgriStrip lateral flow devices. Lateral flow devices were placed into tubes filled with 0.2 ml of sap extracted from PVY-infected plants and diluted 1x101 – 1, 2.5x101 – 2, 5x101 – 3, 1x102 – 4, 2x102 – 5, 5x102 – 6, 1x103 – 7, 2x103 – 8. Sap from a healthy plant, diluted 10-fold, was used as a negative control – 9. Virus positive zone – T, test control zone – T. Three independent repetitions are shown. b Quantitative analysis of results obtained in A. Lateral flow devices were photographed after completion of the assay and intensities of the C and T spots were quantitated as described in Materials and Methods. c Determination of the sensitivity of PVY detection by DAS‑ELISA. d Determination of sensitivity of the RT‑LAMP assay. Amplification plots for specific dilution factors are indicated. Negative control reaction supplemented with total RNA from a healthy plant – healthy. e Mean time to positive (Tp) values plotted as a function of logarithm of dilution factor. f Determination of sensitivity of PVY detection by RT‑PCR in sap from a PVY-infected plant diluted 1x101 – 2, 2.5x101 – 3, 5x101 – 4, 1x102 – 5, 2x102 – 6, 5x102 – 7, 1x103 – 8, 2x103 – 9, 5x103 – 10, 1x104 – 11, 1.5x104 – 12, 2.5x104 – 13, 5x104 – 14, 5x105 – 15, 1x106 – 16. Negative controls:reverse transcription reaction supplemented with water instead of RNA – 17, PCR reaction supplemented with water instead of cDNA – 18, RT-PCR reaction supplemented with RNA from virus-free plant – 19. Positive control: RT-PCR reaction supplemented with PVY RNA
Comparison of Detection Methods for Field Samples Evaluation
Certified, virus-free tubers of Irys and Rosalind cultivars were planted in experimental fields during the spring. Leaves of 30 offspring plants of each cultivar were collected in September. The leaves were tested by DAS-ELISA, RT-PCR, and RT-LAMP as described above (Table 2). Thirty Rosalind samples tested PVY‑free by all applied methods (Table 2). Twenty-eight Irys samples tested positive for PVY by RT-PCR and RT-LAMP, and twenty-four of these tested positive by DAS‑ELISA. Four plants that were negative by DAS-ELISA but positive in both molecular tests (plants 5, 10, 11 and 29) produced very faint bands in RT-PCR and required longer amplification time in RT‑LAMP (results not shown). Two plants (2 and 27) tested PVY-free by all detection methods (Table 2).
Discussion
In this study, we report a novel test for the fluorescent detection of PVY, using a closed-tube, one-step real‑time RT-LAMP assay carried out in a Genie II Ultra rapid amplificator. Detection of PVY by RT-LAMP does not require specialized equipment (Nie 2005), but use of an amplificator, capable of real-time monitoring of fluorescence, enabled us to use this method for quick, sensitive, and routine detection of PVY. Also, due to the availability of commercial kits optimized for fluorescent detection, little optimization is required (Fig. 2). The closed-tube format of the assay eliminates the risk of carry-over contamination. These elements enabled us to develop a practical assay, which is very promising for routine detection of PVY.
The PVY genomic sequence shows high variability in different isolates (Crosslin 2012; Gray et al. 2010; Nie et al. 2012; Quenouille et al. 2013; Singh et al. 2008); the ideal diagnostic test should detect both known and emerging PVY isolates. To allow this as much as possible, we designed RT-LAMP primers based on an alignment of the coat protein coding sequence of forty PVY isolates. The designed primers matched well to the sequences of the aligned isolates, especially in the 3’-end regions of FIP and BIP primers (Fig. 1). Thus, the primer sets should amplify different PVY isolates; however this needs to be confirmed experimentally.
One of advantages of RT‑LAMP is its high specificity, provided by six primers targeting eight different regions in the amplified sequence (Nagamine et al. 2002; Goto et al. 2007). Indeed, the primer set designed in this work promoted amplification only in the reactions with RNA from PVY-infected plants (Fig. 3, panel a) and the resulting amplicon had the annealing temperature expected for PVY (Fig. 3, panel b). The ability to determine the annealing temperature allowed us to assess the specificity of the amplification products without separation a gel, similar to the analysis of the melting temperature of the reaction products of real-time PCR.
The developed RT-LAMP assay facilitates very fast detection of PVY, in 11-30 minutes, depending on the virus concentration (Fig. 4, panel d). Thus this method has the potential to be applied as point-of-care method (Boonham et al. 2008; De Boer and Lopez 2012). However, PVY detection by RT-LAMP requires prior isolation of RNA from tested samples, which extends the time required for the assay by at least half an hour, depending on the method of RNA isolation (Zacharzewska et al. 2014) and makes the assay difficult under field conditions. RT-LAMP was reported to be less sensitive to inhibitory compounds than RT-PCR (Kaneko et al. 2007); thus it is possible that in the RT-LAMP workflow, RNA isolation may be omitted or significantly simplified. However, our attempts to detect PVY directly in diluted sap have failed so far, mainly due to the high fluorescence of sap components (Zacharzewska B., Chołuj J. and Treder K., unpublished). At the moment, the only true point-of-care method of virus detection is the lateral flow assay (LFA), a test based on immunochromatography (Danks and Barker 2000; Boonham et al. 2008; De Boer and Lopez, 2012). The LFA test is very simple, requires no training or laboratory equipment to perform, operates directly on diluted plant sap, and produces results in several minutes, with sensitivity equal to that of DAS-ELISA (compare Fig. 4, panel b vs panel c). The test, in the form of lateral flow devices dedicated to specific plant viruses, is sold by multiple commercial companies and offers a useful tool for plant inspection in the field. However in Poland, LFA is more than 10‑fold more expensive than DAS‑ELISA and about twice as expensive as RT-PCR or RT-LAMP. Moreover, the sensitivity level offered by LFA, and similar to that of DAS‑ELISA, is not sufficient to detect potato viruses directly in samples from field-grown plants or harvested tubers (Hill and Jackson 1984). Reliable detection of potato viruses by immunological methods requires prior virus bio-amplification in plants originating from tested tubers in a grow‑out test (Hill and Jackson 1984, Zahn et al. 2010). Also, LFA is not well suited for testing tubers in a grow-out assay, due to its poor performance in high-throughput screening, where DAS‑ELISA is still the gold standard. Under the conditions used in this work, the RT‑LAMP assay was 1000-fold more sensitive than both of the immunological assays, and 10-fold more sensitive than RT‑PCR (Fig. 4). This corresponds well to the RT-LAMP sensitivity reported for other plant viruses (Hasiów-Jaroszewska and Borodynko 2013; Shen et al. 2014; Tomlinson et al. 2013; Wang et al. 2014), as well as for detection of PVY in tobacco (Zhao et al. 2012). The RT‑LAMP assay was also characterized by a close to linear relationship between the logarithm of the virus dilution and the time to positive signal accumulation (Fig. 4, panel e). Therefore, this assay may be applied for quantitative or semi-quantitative estimation of viral RNA, similarly to real‑time RT‑PCR (Tomlinson et al. 2013). Also the effectiveness of RT‑LAMP in testing field-grown plants compared favorably with DAS‑ELISA and RT-PCR (Table 2). RT‑LAMP is significantly faster than real-time RT‑PCR, even including the RNA isolation step; PVY detection can be completed in less than 1 hour. Thus, RT‑LAMP can potentially combine the high-throughput advantage of DAS‑ELISA with the robustness of LFA, while offering exceptional sensitivity (Fig. 4, panels d and e) at the real‑time RT‑PCR level (Tomlinson et al. 2013), depending on simplification of the sample preparation step, as discussed above.
In conclusion, we have established a fluorescent real-time RT-LAMP assay for the sensitive detection of PVY. Including RNA isolation, the RT‑LAMP assay can be accomplished in less than 1 hour. This assay may be a promising alternative to methods currently used for routine detection of PVY.
References
Ahmadi, S., M.A. Almasi, F. Fatehi, P.C. Struik, and A. Moradi. 2013. Visual detection of potato leafroll virus by one-step reverse transcription loop-mediated isothermal amplification of DNA with hydroxynaphthol blue dye. Journal of Phytopathology 161: 120–124.
Boonham, N., R. Glover, J. Tomlinson, and R. Mumford. 2008. Exploiting generic platform technologies for the detection and identification of plant pathogens. European Journal of Plant Pathology 121: 355–363.
Crosslin, J.M. 2012. PVY: an old enemy and a continuing challenge. American Journal of Potato Research 90: 2–6.
Crosslin, J.M., and L.L. Hamlin. 2011. Standardized RT-PCR conditions for detection and identification of eleven viruses of potato and potato spindle tuber viroid. American Journal of Potato Research 88: 333–338.
Danks, C., and I. Barker. 2000. On-site detection of plant pathogens using lateral-flow devices. EPPO Bulletin 30: 421–426.
De Boer, S.H., and M.M. Lopez. 2012. New grower-friendly methods for plant pathogen monitoring. Annual Review of Phytopathology 50: 197–218.
Goto, M., H. Hayashidani, K. Takatori, and Y.H. Kudo. 2007. Rapid detection of enterotoxigenic Staphylococcus aureus harbouring genes for four classical enterotoxins, SEA, SEB, SEC and SED, by loop-mediatedisothermal amplification assay. Letters in Applied Microbiology 45: 100–107.
Goto, M., E. Honda, A. Ogura, A. Nomoto, and K.-I. Hanaki. 2009. Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. Biotechniques 46: 167–172.
Gray, S., S. De Boer, J. Lorenzen, A. Karasev, J. Whitworth, P. Nolte, R. Singh, A. Boucher, and H. Xu. 2010. Potato virus Y: an evolving concern for potato crops in the United States and Canada. Plant Disease 94: 1384–1397.
Hasiów-Jaroszewska, B., and N. Borodynko. 2013. Detection of Pepino mosaic virus isolates from tomato by one-step reverse transcription loop-mediated isothermal amplification. Archives of Virology 158: 2153–2156.
Hill, S.A., and E.A. Jackson. 1984. An investigation of thereliability of ELISA as a practical test for detecting potatoleaf roll virus and potato virus Y in tubers. Plant Pathology 33: 21–26.
Ju, H. 2011. Simple and rapid detection of potato leafroll virus by reverse transcription loop-mediated isothermal amplification. Plant Pathology Journal 27: 385–389.
Kaliciak, A., and J. Syller. 2009. New hosts of potato virus Y (PVY) among common wild plants in Europe. European Journal of Plant Pathology 124: 707–713.
Kaneko, H., T. Kawana, E. Fukushima, and T. Suzutani. 2007. Tolerance of loop-mediatedisothermal amplification to a culture mediumand biological substances. Journal of Biochemical and Biophysical Methods 70: 499–501.
Kostiw, M., and E. Trojanowska. 2011. Impact of feeding time on PVYN and PVYNTN transmission by Myzus Persicae (SULZ.). Journal of Plant Protection Research 51: 429–434.
Mori, Y., K. Nagamine, N. Tomita, and T. Notomi. 2001. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochemical and Biophysical Research Communications 289: 150–154.
Nagamine, K., T. Hase, and T. Notomi. 2002. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Molecular and Cellular Probes 16: 223–229.
Nie, X. 2005. Reverse transcription loop-mediated isothermal amplification of DNA for detection of potato virus Y. Plant Disease 89: 605–610.
Nie, X., M. Singh, Y. Pelletier, and D. McLaren. 2012. Recent advances on potato virus Y research in Canada. American Journal of Potato Research 90: 14–20.
Notomi, T. 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Research 28: 63e–63.
Quenouille, J., N. Vassilakos, and B. Moury. 2013. Potato virus Y: a major crop pathogen that has provided major insights into the evolution of viral pathogenicity. Molecular Plant Pathology 14: 439–452.
Scholthof, K.-B.G., S. Adkins, H. Czosnek, P. Palukaitis, E. Jacquot, T. Hohn, B. Hohn, K. Saunders, T. Candresse, P. Ahlquist, C. Hemenway, and G.D. Foster. 2011. Top 10 plant viruses in molecular plant pathology. Molecular Plant Pathology 12: 938–954.
Shen, W., D. Tuo, P. Yan, X. Li, and P. Zhou. 2014. Detection of Papaya leaf distortion mosaic virus by reverse-transcription loop-mediated isothermal amplification. Journal of Virological Methods 195: 174–179.
Singh, R.P., J.P.T. Valkonen, S.M. Gray, N. Boonham, R.A.C. Jones, C. Kerlan, and J. Schubert. 2008. Discussion paper: the naming of potato virus Y strains infecting potato. Archives of Virology 153: 1–13.
Singh, M., R.P. Singh, M.S. Fageria, X. Nie, R. Coffin, and G. Hawkins. 2012. Optimization of a real-time RT-PCR assay and its comparison with ELISA, conventional RT-PCR and the grow-out test for large scale diagnosis of potato virus Y in dormant potato tubers. American Journal of Potato Research 90: 43–50.
Tomita, N., Y. Mori, H. Kanda, and T. Notomi. 2008. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nature Protocols 3: 877–882.
Tomlinson, J.A., S. Ostoja-Starzewska, I.P. Adams, D.W. Miano, P. Abidrabo, Z. Kinyua, T. Alicai, M.J. Dickinson, D. Peters, N. Boonham, and J. Smith. 2013. Loop-mediated isothermal amplification for rapid detection of the causal agents of cassava brown streak disease. Journal of Virological Methods 191: 148–154.
Varga, A., and D. James. 2006. Use of reverse transcription loop-mediated isothermal amplification for the detection of Plum pox virus. Journal of Virological Methods 138: 184–190.
Verbeek, M., P.G.M. Piron, A.M. Dullemans, C. Cuperus, and R.A.A. Van Der Vlugt. 2010. Determination of aphid transmission efficiencies for N, NTN and Wilga strains of potato virus Y. Annals of Applied Biology 156: 39–49.
Wang, Z., Q. Gu, H. Sun, H. Li, B. Sun, X. Liang, Y. Yuan, R. Liu, and Y. Shi. 2014. One-step reverse transcription loop mediated isothermal amplification assay for sensitive and rapid detection of Cucurbit chlorotic yellows virus. Journal of Virological Methods 195: 63–66.
Wei, Q.-W., C. Yu, S.-Y. Zhang, C.-Y. Yang, K. Miriam, W.-N. Zhang, D.-L. Dou, and X.-R. Tao. 2012. One-step detection of Bean pod mottle virus in soybean seeds by the reverse-transcription loop-mediated isothermal amplification. Virology Journal 9: 187.
Wróbel, S., and A. Wąsik. 2013. Seed potato production in Poland. American Journal of Potato Research 3: 260–268.
Zacharzewska, B., A. Przewodowska, and K. Treder. 2014. The adaptation of silica capture RT-PCR for the detection of potato virus Y. American Journal of Potato Research. doi:10.1007/s12230-014-9383-y.
Zahn, V., J. Dahle, and K.H. Pastrik. 2010. Visual inspection of potato seedlings in routine virus diagnosis: validation of a rating method. EPPO Bulletin 40: 429–434.
Zhao, L., J. Cheng, X. Hao, X. Tian, and Y. Wu. 2012. Rapid detection of tobacco viruses by reverse transcription loop-mediated isothermal amplification. Archives of Virology 157: 2291–2298.
Acknowledgments
The authors would like to thank Prof. Ewa Zimnoch-Guzowska for providing tubers infected with PVY and Mgr. Danuta Sekrecka for providing virus-free plants and sources of PVM, PVS and PVX. We also want to thank Dr. Betty Chung for critical review of the manuscript and helpful comments. This study was supported financially by the Ministry of Agriculture and Rural Development of the project: “The development of the procedures for the detection of viral infections in potato tubers directly after harvesting or during dormancy” (4‑3‑00‑7-03) and by National Science Centre (Grant N N310 728540).
Author information
Authors and Affiliations
Corresponding author
Additional information
Agnieszka Przewodowska and Bogumiła Zacharzewska contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Przewodowska, A., Zacharzewska, B., Chołuj, J. et al. A One-Step, Real-Time Reverse Transcription Loopmediated Isothermal Amplification Assay to Detect Potato Virus Y. Am. J. Potato Res. 92, 303–311 (2015). https://doi.org/10.1007/s12230-015-9430-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12230-015-9430-3