
Abstract
Hydrogenative rearrangement of biomass-derived furfurals (furfural and 5-hydroxymethyl furfural) to C5 cyclic compounds (such as cyclopentanones and cyclopentanols) offers an expedient reaction route for acquiring O-containing value-added chemicals thereby replacing the traditional petroleum-based approaches. The scope for developing efficient bifunctional catalysts and establishing mild reaction conditions for upgrading furfurals to cyclic compounds has stimulated immense deliberation in recent years. Extensive efforts have been made toward developing catalysts for multiple tandem conversions, including those with various metals and supports. In this scientific review, we aim to summarize the research progress on the synergistic effect of the metal–acid sites, including simple metal–supported acidic supports, adjacent metal acid sites–supported catalysts, and in situ H2-modified bifunctional catalysts. Distinctively, the catalytic performance, catalytic mechanism, and future challenges for the hydrogenative rearrangement are elaborated in detail. The methods highlighted in this review promote the development of C5 cyclic compound synthesis and provide insights to regulate bifunctional catalysis for other applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
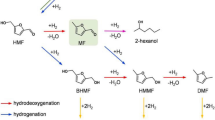
Lignocellulose is a renewable resource of carbon, yielding an annual production of ~ 170 billion tons of materials, fuels, and chemicals [1, 2]. Catalytic upgradation of biomass to oxygen-containing value-added chemicals demonstrates a providential route for reducing carbon emissions and developing green chemical industries. Lignocellulose-derived furfurals (i.e., furfural and 5-hydroxymethyl furfural) can be efficiently generated from the dehydration of carbohydrates (i.e., pentoses and hexoses) [3,4,5]. These have been employed as the most critical biobased platform compounds to upgrade various fine chemicals, such as furan alcohols (i.e., furan alcohol and 2,5-bishydroxymethyl furan), furancarboxylic acids (i.e., furancarboxylic acid and 2,5-furandicarboxylic acid) [6, 7], methylfurans (i.e., 2-methylfuran and 2,5-dimethylfuran) [8, 9], linear polyols (i.e., 1,2-pentanediol, 1,4-pentanediol, 1,5-pentanediol, 2,5-hexanediol, and 1,2,6-hexanetriol) [10, 11], cyclopentanones (i.e., cyclopentanone and 3-hydroxymethyl cyclopentanone), and cyclopentanols (i.e., cyclopentanol and 3-hydroxymethyl cyclopentanol) [12,13,14]. Among these, C5 cyclic compounds, including cyclopentanones and cyclopentanols, are highly desirable because of their comprehensive applications. For example, they are pivotal building blocks for various drug molecules, perfumes, polymers, and natural products with biological activities because these compounds concurrently constitute active carbonyl and hydroxyl groups in a small C5 cyclic molecule. Furthermore, they can be employed as reactants to synthesize high-density fuels through alkali-catalyzed aldol condensation reaction. Traditionally, cyclopentanone and cyclopentanol are derived through the decarboxylation–cyclization of petroleum-based 1,6-adipic acid and hydration of cyclopentene, respectively. Hydrogenative rearrangement provides an alternative synthesis route for cyclopentanones/cyclopentanols and proliferates the utilization potential of biomass.
Several research groups have reported the conversion of furfurals to cyclic compounds over metal–acid bifunctional catalysts [15,16,17,18,19,20]. Conventionally, furfural (FFA) requires performing distinct types of cascade reaction steps, such as C=O hydrogenation to furan alcohol (FA) over metal sites, rearrangement to form 4-hydroxymethyl-2-cyclopentenone (HCP) over acid sites, hydrogenation and dehydration to cyclopentanone (CPO) or further hydrogenation to cyclopentanol (CPL) (Scheme 1a). Similarly, 5-hydroxymethyl furfural (HMF) can be converted to 3-hydroxymethyl cyclopentanone (HCPN) or 3-hydroxymethyl cyclopentanol (HCPL) via the same route using the C=O-hydrogenated intermediate 2,5-bishydroxymethyl furan (BHF) and rearranged intermediate 4-hydroxy-4-hydroxymethyl-2-cyclopentanone (HHCPN) (Scheme 1b). However, some over-hydrogenated byproducts [i.e., tetrahydrofurfuryl alcohol (THFA) and 2,5-bishydroxymethyl tetrahydrofuran (THBHF)] are easily produced via furan ring hydrogenation, thus reducing the selectivity of these target cyclic compounds [21]. The efficient and selective preparation of cyclic compounds prevail a monumental challenge and inevitably requires delicate regulation of the metal and acid sites for the hydrogenation and acid-catalyzed steps. Furthermore, the cooperative effect, which plays an indispensable role in modulating catalytic activity and selectivity, requires comprehensive investigation.

Hydrogenative rearrangement pathway of furfurals to cyclic compounds
Reports on the synthesis of CPOs from FFAs have been well-reviewed. In particular, Dutta et al. [22] reviewed the catalytic performance of distinct types of heterogeneous catalysts and reaction conditions. However, comprehensive reviews on synergistic catalysis for promoting hydrogenative rearrangement to cyclic compounds are lacking. Herein, we review the research progress of concerted catalytic mechanisms in the hydrogenative rearrangement cascade reaction. We present the synergistic catalytic mechanism for regulating the acid support as well as for adjusting the distance between metal and acid sites and in situ hydrogen-modified active sites. Subsequently, we discuss the relation between the active sites and catalytic performance on substrate adsorption, hydrogenation activity, and acid-catalyzed activity. This review is expected to enhance the understanding of the synergetic cooperation of hydrogenation and acidic sites in advancing the progression of FFAs. Furthermore, this review can stimulate substantial deliberation in the development of comparatively efficient and/or new catalytic transformations enabled by selective hydrogenation and the acid-catalyzed synergetic mechanism, thereby providing accession for biorefineries.
Hydrogenative Rearrangement of Furfurals to C5 Cyclic Compounds
Synergistic Catalysis by Regulating Acid Support
The aforementioned reaction process indicates that the hydrogenative rearrangement of FFAs requires a metal site for accomplishing the hydrogenation step. Hronec et al. [23] introduced the green and environmentally friendly path of converting FFA to CPO. Employing water as the solvent and noble or non-noble metals supported on carbon as catalysts, they performed the reaction under a H2 pressure of 8.0 MPa at temperatures of 160–180 °C. The authors proposed the following reaction mechanism: First, FFA and H2 are absorbed and activated on the surface of the metal catalyst. Second, the C=O of FFA is hydrogenated to form the intermediate FA, and the C–OH of FA is cleaved to produce carbocation catalyzed by the H+ formed via the dissociation of water. Third, CPO is formed via the rearrangement of the carbocation and subsequent hydrogenation steps. Accompanied by the primary reaction, furanic polymers are formed on the catalyst surface via the partial polymerization of FFA (Fig. 1a). However, the reaction conditions are abrasive, and the synthesis efficiency of CPO is unsatisfactory owing to the lack of an acidic site on the carbon support. The neutral support affects the rearrangement step and causes FA accumulation, an intermolecular C–C coupling reaction that produces humins. Considering these drawbacks of the partial polymerization of FFA, reducing the reaction temperature has been the objective of many research groups who have reported various acidic metal–organic frameworks (MOFs) (such as Cu–BTC, MIL-101, MIL-100, UIO-66, and FeZn–PBA), metal oxides (ZrO2, Y2(Sn0.7Ce0.3)2O7-δ, Y2(Sn0.65Al0.35)2O7-δ/Al2O3, La2TiO7, NiMoO4, γ-Al2O3, and Ti3AlOx), layered double hydroxides (LDH), zeolites (such as Al–MCM-41, SBA-15, HY, and ZSM-5), and supported reduced metals (such as Pt, Pd, Au, Ru, Co, Ni, Cu, and Ni2P) as bifunctional catalysts for accomplishing reactions under mild reaction conditions: temperature of 120–160 °C and H2 pressure of 2.0–4.0 MPa (Table 1) [12, 13, 15, 16, 18, 23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70].

a Proposed reaction mechanism for the rearrangement of FFA to CPO; b Hydrogenative rearrangement performance of various catalysts for the conversion of HMF to HCPN; c Selective synthesis of CPO from FFA over Pd/Prussian blue. (Reproduced with permission from Ref. [18, 38]. Copyright © 2012,2019, Elsevier ScienceDirect)
Unlike FFA, regulating catalytic sites for HMF is relatively more challenging because the electron-withdrawing hydroxymethyl group of C=O-hydrogenated BHF escalates the difficulty in accomplishing the rearrangement. To illustrate, the predominant product of HMF over noble metal/MIL-101 is intermediate BHF because the weak Lewis acidity of the unsaturated coordination of Cr3+ ions is incapable of further opening the furan ring to produce HCPN [35]. Ohyama et al. [71, 72] reported that an Au/strong acidic support (such as Ta2O5 and Nb2O5) strengthens bifunctional catalysis, leading to the rearrangement of BHF and affording HCPN in 86% yield (Table 2). Designing a universal catalyst that solves the requirements for active sites is essential to drive the conversion of various FFAs. Li et al. and Deng et al. [36,37,38] have reported that noble metals supported on moderate acid MOF material catalysts (such as Fe-MIL-100, Pd/Cu–BTC, and Pd/FeZn Prussian blue (PBA)) exhibit a selective hydrogenative rearrangement for FFA and HMF at a temperature of 150 °C, a H2 pressure of 4.0 MPa, and a reaction time 6–12 h. The yield of CPO and HCPN is > 85%. However, weak Lewis acidic Pd/FeNi Prussian blue can only catalyze the synthesis of furfuryl alcohols (FA and BHF) (Fig. 1b, c). These experimental results demonstrate that acid strength plays a pivotal role in the synthesis of CPOs. To better comprehend the acidic sites on the catalyst surface, a few characterization methods were employed for further analyses (namely, NH3-temperature-programmed desorption, in situ pyridine-adsorbed FTIR, 31P magic angle spin nuclear magnetic resonance) [38, 62, 70].
Synergistic Catalysis Through Distance Modification Between Metal and Acid Site
The precise control of nanoscale features in bifunctional catalysts has been exploited to enhance synergistic catalytic performance. FFAs have unsaturated C=C and C=O bonds; however, C=C hydrogenation is often thermodynamically favored over C=O hydrogenation. A pivotal factor for high selectivity toward CPOs/CPLs is the rational design of heterogeneous catalysts to enhance C=O hydrogenation selectivity. An efficient strategy for selective C=O hydrogenation of furan aldehydes was deployed through structural engineering of active sites such as O vacancies. Deng et al. [56] reported a series of pyrochlore-based (A2B2O7, A2B2O6O') bifunctional catalysts for the synthesis of CPOs. The pyrochlore support possesses natural O vacancies in accordance with the principle of electroneutrality. The B-site cations are easily substituted by a C-site metal cation with a lower valence, further strengthening the O vacancy concentration of the support. Bare metal ions near O vacancies provide a pure Lewis acidity to pyrochlore, and the acidity of these catalysts can be regulated by adjusting the type of metal cations [56,57,58]. In comparison to metal/traditional acid support (Pd/Hβ, Pd/TiO2, and Pd/Nb2O5), the Pd/pyrochlore exhibits evident advantages, such as acceleration of the C=O hydrogenation and rearrangement step, inhibition of furan ring hydrogenation reaction (Fig. 2a, b). Among Pd/pyrochlore catalysts (La2Sn2O7, Y2Sn2O7 and Y2(Sn0.7Ce0.3)2O7-δ), Pd/Y2(Sn0.7Ce0.3)2O7-δ exhibits the apex catalytic performance with CPO yields of > 92%. To comprehend the catalytic effect of O vacancies, in situ attenuated total reflection infrared (ATR-IR) spectra were incorporated to evaluate the molecular level interaction of FFA with the catalyst (Fig. 2c). The signal for C=O is redshifted in comparison with that for pristine FFA; however, the position of the C=O signal for the furan ring bands remains unchanged during interaction with Pd/pyrochlore, indicating that the O vacancy of catalysts can selectively absorb the O atom of C=O bond via a vertical configuration. This selective C=O adsorption guarantees selective C=O hydrogenation and inhibits the furan hydrogenation, thus reducing the byproduct yield of THFA. The adsorption kinetics data illustrate that Pd/pyrochlore possesses enhanced adsorption quality of FFA than traditional Pd/TiO2, further proving the selective adsorption of O vacancy (Fig. 2d). The reaction kinetics studies regarding the C=O hydrogenation step of FFA and the rearrangement step of FA are also performed. These studies found that the hydrogenation and rearrangement steps follow pseudo-first-order kinetics, and the concentration of oxygen vacancies strongly influences reaction rate constants (Fig. 2e, f). Oxygen vacancies not only provide the adsorption sites for C=O activation but also afford the Lewis acidic sites for the rearrangement steps. Moreover, the catalyst exhibits a stable recycling performance and generality for various FFAs, including FFA and HMF. Furthermore, Gao et al. [58] also demonstrated the universality of oxygen vacancy for tandem reactions using other pyrochlore-based catalysts (Pd/Y2(Sn0.65Al0.35)2O7-δ/Al2O3, Pd/La2TiO7, Pd/La2Zr2O7, Pd/La2Ce2O7). This study provides an effective biological preparation route for CPOs via the synergistic catalysis of metal sites and O vacancy of the support.

a Hydrogenative rearrangement performance of various catalysts for the conversion of FFA to CPO. b Synthesis of products from FFA over Pd/acidic supports and pyrochlore; c ATR-IR spectra of FFA adsorbed on Pd/Y2Sn2O7 and Pd/TiO2. d Adsorption kinetics of FFA under Y2Sn2O7 and TiO2. e Relation between FFA hydrogenation rate and number of oxygen vacancies. f Relation between FA hydrolysis rate and Lewis acid concentration. (Reproduced with permission from Ref. [56]. Copyright © 2020, American Chemical Society)
On the surface of the aforementioned metal nanoparticles, which are supported through acidic support catalysts, the metal sites (metal nanoparticles) and acid sites (acidic support) are spatially separated, not only factoring a considerable accumulation of C=O-hydrogenated intermediates (i.e., FA, BHF) in the cascade reaction system but also limiting the cooperative catalytic performance of distinct active sites. Tong et al. [69] prepared a class of metal phosphide nanoparticles (Ni2P, CoP) for hydrogenative rearrangement reactions. Metal cations and metal atoms coexist on the nanoparticle surface. The former plays the role of a Lewis acid site, whereas the latter can be engaged as a hydrogenation site. The hydrogen activation ability and acidic performance of these catalysts can be regulated by adjusting the kind of metal cations and the topological structure (Fig. 3a, b). Interestingly, for the furfural reaction, Ni2P exhibits a 62.8% yield of CPL by the hydrogenative rearrangement route. In contrast, CoP displays a novel hydrogenative hydrolysis route with an 80.2% yield of 1,2,5-pententriol under 150 °C. According to the ATR-IR results, Ni2P nanoparticles are capable of adsorbing C=O of FFA selectively through vertical adsorption (Fig. 3c). The adsorption configuration ensures selective C=O hydrogenation over the metal sites, and the in situ-generated FA can be rapidly transferred to the adjacent metal cations for the rearrangement permitting efficient synthesis of CPL. CoP can adsorb both C=O as well as the furan ring of FFA via parallel adsorption. FFA can be converted to dihydrofuran alcohol by C=O hydrogenation and furan ring semi-hydrogenation over the hydrogenation site. The dihydrofuran alcohol can be hydrolyzed over the nearby Lewis acidic sites, preventing over-hydrogenation to THFA. The close metal site (Coδ+ and Niδ+) and acidic site (Co2+ and Ni2+) over metal phosphide provide an efficient synthesis method for CPL and demonstrate a novel synthesis route for 1,2,5-pententriol (Fig. 3d).

a Pyridine-adsorbed FTIR spectra of metal phosphides. b MS signals of HD generation over metal phosphides during H2–D2 exchange reaction. c ATR-IR spectra of FFA adsorbed over various metal phosphides. d Proposed reaction mechanism over various metal phosphides. e FFA conversion to CPO over Ni/Al2O3 modifying with phosphorus. f H2-FTIR spectra on Sr2P2O7. g Isotope-label tracing experiment over Sr2P2O7/Ni2P. (Reproduced with permission from Ref. [60, 61, 69]. Copyright © 2021, 2023, 2021, Royal Society of Chemistry, Elsevier ScienceDirect, American Chemical Society)
Gao et al. [60] investigated the synthesis of CPO from FFA over different nickel phosphides (Ni2P, Ni3P, and Ni12P5) supported Al2O3 (Fig. 3e). Nickel nanoparticles are common hydrogenation sites, and the introduction of phosphorus regulates the selectivity of hydrogenation products. Under optimal reaction conditions, the total yield of CPO and CPL can reach more than 90% following reaction at 150–190 °C, 3.0 MPa H2 for 2 h. As the hydrogenation site, nickel phosphides can selectively adsorb the C=O group of FFA and activate hydrogen, further catalyzing the formation of FA. The introduction of additional acidic sites further promotes the furfuryl alcohol rearrangement step and finally leads to the formation of CPO and CPL. Cheng et al. [61] constructed a Sr2P2O7/Ni2P composite catalyst for the hydrogenative rearrangement of FFAs to CPOs, proving the universality of various FFAs. The nanoscale activity of Sr2P2O7/Ni2P was crucial for the construction of the catalytic interface with modulated hydrogen activation and acid sites. They found that Sr2P2O7 can activate H2 to participate in the reaction. The H2-FTIR experiments display the formation of Sr–H and O–H bonds after H2 activation, implying that H2 could be heterolyzed on Sr2P2O2 (Fig. 3f) [76]. The mechanism was further investigated by isotope-label tracing experiments (H218O, D2O, or D2), which confirmed that both H2O and H2 participated in the transformation (Fig. 3g).
Synergistic Catalysis by Hydrogen Spillover and Heterolysis
To further enhance the intimacy of metal sites and acidic sites, Li et al. [59] and Yuan et al. [55] have constructed an intimate H+–H– pair by the hydrogen spillover mechanism. Some reducible metal oxides (such as NiMoO4 and CeO2) were used as supports to load noble metal nanoparticles. H2 can be homolyzed to hydrogen atoms on the nanoparticles, migrate to the support, and form proton H+ accompanied by the reduction of the support valence state (Fig. 4a, b). Also, the H atom on the Pd nanoparticles can be polarized into H– because of the greater electronegativity of H than Pd. The in situ-generated H+–H– pair with asymmetry can selectively adsorb and activate the C=O group of FFA to ensure the selective C=O hydrogenation and inhibit the furan hydrogenation. In addition, the generation of the H––H+ pair triggers the transformation of the original Lewis acidity to Brønsted acidity, as evidenced by in situ pyridine-adsorbed FTIR spectra (Fig. 4c). To better investigate the relation between acidity and acid catalytic ability, kinetic studies of the rearrangement of FA were conducted under H2 atmosphere using pyridine as an in situ blocking reagent for occupying both Lewis and Brønsted acid sites or 2,6-dimethylpyridine for poisoning Brønsted acid sites [77]. These results indicate that both Lewis and Brønsted acidity can catalyze the rearrangement reaction. However, the in situ-generated Brønsted acids essentially accelerated the rearrangement steps (Fig. 4d). In addition, Deng et al. [62] reported that H+–H– pairs can be generated over partial oxidized Pd/Ti3AlC2 by in situ hydrogen spillover mechanism. Similarly, the H+–H– pairs not only function as the hydrogenation sites for selective C=O hydrogenation but also provide acid sites for the acid-catalyzed step in the conversion of FFAs. As a result, the natural bifunctional properties of H+–H– pairs exhibit an 81.6% yield of CPO from FFA under 120 °C. Pd/Ti3AlC2 demonstrates excellent catalytic performance at the lowest temperature reported to date, substantially reducing the reported minimum reaction temperature for FFA conversion to about 30 °C.

a Proposed catalytic reaction mechanism. b Pd 3d, Mo 3d and O 1 s XPS spectra. c Pyridine-adsorbed FTIR spectra. d Rearrangement rate constants of various catalysts after the in situ titration by pyridine and 2,6-dimethylpyridine. (Reproduced with permission from Ref. [59]. Copyright © 2022, Elsevier Science Direct)
Additionally, Deng et al. [70] reported a new perspective for strengthening bifunctional hydrogenation and acid-catalyzed reactions without classical acid hosts. A series of Co-embedded N-doped carbon (Co@Co–NCs) as catalysts were exploited for the conversion of FFA to CPL. The Co nanoparticle core plays a vital role in C=O hydrogenation. Intriguingly, it promotes H2 heterolysis over the Co–N pair and a subsequent water-mediated transformation of Hδ––Co–N–Hδ+ to OHδ––Co–N–Hδ+ accompanied by the generation of H2 (Fig. 5a). Namely, H2 acts as a catalyst to induce the acid–base transformation of the Co–NC shell from Lewis acid–base pairs (Co–N) into Brønsted acid − base pairs (OHδ––Co–N–Hδ+). The Brønsted acid sites of in situ-generated OHδ––Co–N–Hδ+ were confirmed by 31P magic angle spin nuclear magnetic resonance measurements, and the H+ species formed on Co–N via hydrogen heterolysis was characterized by.

a Proposed reaction mechanism over Co@Co-NC. b.13P with a probe of TMPO. c In situ IR spectra for the subsequent D2 and D2O activation at 150 °C. d MS signals of HD generation during D2–H2O treatment. (Reproduced with permission from Ref. [70]. Copyright © 2022, American Chemical Society)
ATR-IR. Substantial HD generation is detected via mass spectrometry (MS) when Co–NC is exposed to a mixture of D2 and H2O (Fig. 5b–d). The OHδ––Co–N–Hδ+ construction selectively adsorbs and reacts with the electronegative O atom of the asymmetric C=O, promoting selective hydrogenation. The Brønsted acidity of OHδ––Co–N–Hδ+ fundamentally promotes the acid-catalyzed step. Furthermore, they demonstrated the directed synthesis of 3-methyl cyclopentanol from 5-methyl-furfural, demonstrating the remarkable universality of the selective hydrogenation rearrangement route.
In another work, multishell hollow materials were synthesized from ZIFs-MOFs with controllable nanostructures. This strategy is in accordance with the pyrolysis of multilayer solid ZIFs. The number of carbon shells and the synthesized hollow Co@Co–NC can be precisely controlled by a step-by-step crystal growth method. Due to the three-dimensional hierarchical structure, highly dispersed Co nanoparticles and nitrogen-doped carbon Co@Co–NCs can be engaged as efficient catalysts for this reaction. Multishell Co@Co–NC catalyzes the production of CPL in high yield. Under the reaction conditions of 160 °C and 2.0 MPa H2, the CPL yield reached 97.0% after 8 h reaction and demonstrated excellent cyclic stability. The optimal performance of the multishell Co@Co–NC catalyst was attributed to its structural advantages: (1) the abundant stratified holes in the hollow shell can enhance the mass diffusion capacity; (2) the high dispersion of Co nanoparticles in the hollow shell can increase the density of highly exposed active sites [65].
Summary and Perspective
This review presented a mechanistic insight into synergistic hydrogenative rearrangement of biomass-based FFAs to comprehend how the bifunctional sites (hydrogenation and acidic sites) selectively fix and activate the C=O group and cooperatively catalyze the rearrangement. (1) The medium-strength acid support–loaded metal sites are designed as bifunctional synergistic catalysts to enhance the intermediate acid-catalyzed rearrangement reaction, proving the universality of the catalytic conversion of FFAs. (2) Catalyst structure engineering of active sites (such as O vacancies) is applied for selective C=O hydrogenation of furan aldehydes and increasing the strength of acid carriers to enhance the synergistic catalysis. Reducing the distance between the hydrogenation and acid sites advances the mass transfer rate of the intermediate between these bifunctional sites. (3) In situ-generated hydrogen species via the hydrogen spillover or heterolysis mechanism results in the adsorption and activation of the O atom of the C=O group and leads to selective hydrogenation. Meanwhile, hydrogen species can also be engaged as acidic sites for the rearrangement steps. In summary, synergistic catalysis performs a vital role in the hydrogenative rearrangement reaction of biobased FFAs. This sheds light on the potential application of this strategy for the rational design of catalysts. This mechanism provides theoretical guidance for bifunctional synergistic catalysis of other highvalue-added fine chemicals derived from FFAs. Therefore, precise regulation of the reaction path of biomass resources and elaborate reaction mechanisms are contemplated to proliferate the scope for biomass refining. Future research can be focused on the following aspects.
-
(1)
Current reaction systems use biomass platform compounds, such as FFAs, as raw materials. Synthesis of CPO from feedstock with a higher priority (such as pentoses, rhamnose, glucose, and fructose) demonstrates better scientific and industrial prospects. This cascade of reaction strategy can avoid intermediate separation and purification and minimize energy costs by integrating several steps into a single reaction system. The reaction process would, therefore, require designing a multifunctional synergistic catalyst (with metal and acid sites) for the tandem sequence of dehydration, hydrogenation, and rearrangement steps.
-
(2)
Integrating biomass into the organic nitrogen-containing chemicals will reduce the carbon footprint and enhance the economic competitiveness of biorefining. The near hydrogenation and acid sites over metal phosphides can be engaged as catalysts to regulate the vertical adsorption of FA and enhance the mass transfer rate of intermediates on the catalyst surface [78]. On account of the synergic effect of efficient hydrogenation and acid catalysis, the reaction routes (FA hydrolysis, nitro hydrogenation, and the Paal–Knorr reaction) are proposed to provide an efficient method for pyrrole synthesis. Although significant progress has been made in inhibiting competitive adsorption (such as furan and nitro groups on metal sites, hydroxyl, and hydrogenated amino groups on acidic sites) over metal phosphide at tandem reaction, there are some challenges in using FFAs as reaction substrates due to strong reactivity of C=O in FFA.
-
(3)
Linear polyols (e.g., 1,2-pentanediol, 1,4-pentanediol, 1,5-pentanediol, 1,6-hexanediol, and 1,2,6-hexanetriol) are widely used in spices, cosmetics, medical disinfectants, and cleaning industries. Thus, further research can be performed on the “oxygen-bearing strategy” to establish the method of synthesizing polyols from FFAs. Additionally, the hydrogenative ring-opening of FFAs proceeds via a “one-pot” tandem process, in which the multifunctional catalytic sites contribute to diverse reactions and ultimately lead to overall hydrogenative ring-opening transformation. It is worth noting that the high stability of these intermetallic catalysts provides the potential to solve the catalyst deactivation caused by the leaching of metal oxide in conventional metal/metal oxide catalysts.
References
He MY, Sun YH, Han BX (2022) Green carbon science: efficient carbon resource processing, utilization, and recycling towards carbon neutrality. Angew Chem Int Ed 61(15):e202112835
Resasco DE, Wang B, Sabatini D (2018) Distributed processes for biomass conversion could aid UN Sustainable Development Goals. Nat Catal 1(10):731–735
Huber GW, Chheda JN, Barrett CJ et al (2005) Production of liquid alkanes by aqueous-phase processing of biomass-derived carbohydrates. Science 308(5727):1446–1450
Feng YC, Li Z, Long SS et al (2020) Direct conversion of biomass derived l-rhamnose to 5-methylfurfural in water in high yield. Green Chem 22(18):5984–5988
Zhao HB, Holladay JE, Brown H et al (2007) Metal chlorides in ionic liquid solvents convert sugars to 5-hydroxymethylfurfural. Science 316(5831):1597–1600
Guan W, Zhang YL, Yan CH et al (2022) Base-free aerobic oxidation of furfuralcohols and furfurals to furancarboxylic acids over nitrogen-doped carbon-supported AuPd bowl-like catalyst. Chemsuschem 15(16):e202201041
Barwe S, Weidner J, Cychy S et al (2018) Electrocatalytic oxidation of 5-(hydroxymethyl)furfural using high-surface-area nickel boride. Angew Chem Int Ed 57(35):11460–11464
Gilkey MJ, Panagiotopoulou P, Mironenko AV et al (2015) Mechanistic insights into metal lewis acid-mediated catalytic transfer hydrogenation of furfural to 2-methylfuran. ACS Catal 5(7):3988–3994
Li X, Zhang LK, Deng Q et al (2022) Promoted hydrogenolysis of furan aldehydes to 2, 5-dimethylfuran by defect engineering on Pd/NiCo2O4. Chemsuschem 15(13):e202201012
Ma RF, Wu XP, Tong T et al (2017) The critical role of water in the ring opening of furfural alcohol to 1, 2-pentanediol. ACS Catal 7(1):333–337
Zhao ZL, Yang C, Sun P et al (2023) Synergistic catalysis for promoting ring-opening hydrogenation of biomass-derived cyclic oxygenates. ACS Catal 13(8):5170–5193
Zhang GS, Zhu MM, Zhang Q et al (2016) Towards quantitative and scalable transformation of furfural to cyclopentanone with supported gold catalysts. Green Chem 18(7):2155–2164
Zhou MH, Zhu HY, Niu L et al (2014) Catalytic hydroprocessing of furfural to cyclopentanol over Ni/CNTs catalysts: Model reaction for upgrading of bio-oil. Catal Lett 144(2):235–241
Ramos R, Grigoropoulos A, Perret N et al (2017) Selective conversion of 5-hydroxymethylfurfural to cyclopentanone derivatives over Cu–Al2O3 and Co–Al2O3 catalysts in water. Green Chem 19(7):1701–1713
Hronec M, Fulajtárová K, Vávra I et al (2016) Carbon supported Pd–Cu catalysts for highly selective rearrangement of furfural to cyclopentanone. Appl Catal B 181:210–219
Li XL, Deng J, Shi J et al (2015) Selective conversion of furfural to cyclopentanone or cyclopentanol using different preparation methods of Cu–Co catalysts. Green Chem 17(2):1038–1046
Renz M (2005) Ketonization of carboxylic acids by decarboxylation: mechanism and scope. Eur J Org Chem 2005(6):979–988
Hronec M, Fulajtarová K, Liptaj T (2012) Effect of catalyst and solvent on the furan ring rearrangement to cyclopentanone. Appl Catal A 437–438:104–111
Hronec M, Fulajtárova K, Soták T (2014) Highly selective rearrangement of furfuryl alcohol to cyclopentanone. Appl Catal B 154–155:294–300
Duan Y, Zheng M, Li DM et al (2017) Conversion of HMF to methyl cyclopentenolone using Pd/Nb2O5 and Ca–Al catalysts via a two-step procedure. Green Chem 19(21):5103–5113
Zhang SJ, Ma H, Sun YX et al (2019) Catalytic selective hydrogenation and rearrangement of 5-hydroxymethylfurfural to 3-hydroxymethyl-cyclopentone over a bimetallic nickel–copper catalyst in water. Green Chem 21(7):1702–1709
Dutta S, Bhat NS (2021) Catalytic transformation of biomass-derived furfurals to cyclopentanones and their derivatives: A review. ACS Omega 6(51):35145–35172
Hronec M, Fulajtárova K, Mičušik M (2013) Influence of furanic polymers on selectivity of furfural rearrangement to cyclopentanone. Appl Catal A 468:426–431
Liu YH, Chen ZH, Wang XF et al (2017) Highly selective and efficient rearrangement of biomass-derived furfural to cyclopentanone over interface-active Ru/carbon nanotubes catalyst in water. ACS Sustain Chem Eng 5(1):744–751
Shen T, Hu RJ, Zhu CJ et al (2018) Production of cyclopentanone from furfural over Ru/C with Al11.6PO23.7 and application in the synthesis of diesel range alkanes. RSC Adv 8(66):37993–38001
Wang DD, Al-Mamun M, Gong WB et al (2021) Converting Co2+-impregnated g-C3N4 into N-doped CNTs-confined Co nanoparticles for efficient hydrogenation rearrangement reactions of furanic aldehydes. Nano Res 14(8):2846–2852
Huang L, Hao F, Lv Y et al (2021) MOF-derived well-structured bimetallic catalyst for highly selective conversion of furfural. Fuel 289:119910
Mironenko RM, Belskaya OB, Talsi VP et al (2020) Mechanism of Pd/C-catalyzed hydrogenation of furfural under hydrothermal conditions. J Catal 389:721–734
Liu XY, Zhang B, Fei BH et al (2017) Tunable and selective hydrogenation of furfural to furfuryl alcohol and cyclopentanone over Pt supported on biomass-derived porous heteroatom doped carbon. Faraday Discuss 202:79–98
Hu Z, Han MM, Chen C et al (2022) Hollow carbon sphere encapsulated nickel nanoreactor for aqueous-phase hydrogenation-rearrangement tandem reaction with enhanced catalytic performance. Appl Catal B 306:121140
Zhou MH, Li J, Wang K et al (2017) Selective conversion of furfural to cyclopentanone over CNT-supported Cu based catalysts: model reaction for upgrading of bio-oil. Fuel 202:1–11
Gong WB, Chen C, Zhang HM et al (2018) Highly dispersed Co and Ni nanoparticles encapsulated in N-doped carbon nanotubes as efficient catalysts for the reduction of unsaturated oxygen compounds in aqueous phase. Catal Sci Technol 8(21):5506–5514
Wang Y, Sang SY, Zhu W et al (2016) CuNi@C catalysts with high activity derived from metal–organic frameworks precursor for conversion of furfural to cyclopentanone. Chem Eng J 299:104–111
Advani JH, Khan NUH, Bajaj HC et al (2019) Stabilization of palladium nanoparticles on chitosan derived N-doped carbon for hydrogenation of various functional groups. Appl Surf Sci 487:1307–1315
Fang RQ, Liu HL, Luque R et al (2015) Efficient and selective hydrogenation of biomass-derived furfural to cyclopentanone using Ru catalysts. Green Chem 17(8):4183–4188
Li X, Deng Q, Zhang LK et al (2019) Highly efficient hydrogenative ring-rearrangement of furanic aldehydes to cyclopentanone compounds catalyzed by noble metals/MIL-MOFs. Appl Catal A 575:152–158
Deng Q, Wen XH, Zhang P (2019) Pd/Cu-MOF as a highly efficient catalyst for synthesis of cyclopentanone compounds from biomass-derived furanic aldehydes. Catal Commun 126:5–9
Li X, Deng Q, Zhou SH et al (2019) Double-metal cyanide-supported Pd catalysts for highly efficient hydrogenative ring-rearrangement of biomass-derived furanic aldehydes to cyclopentanone compounds. J Catal 378:201–208
Wang CH, Yu ZQ, Yang YH et al (2021) Hydrogenative ring-rearrangement of furfural to cyclopentanone over Pd/UiO-66-NO2 with tunable missing-linker defects. Molecules 26(19):5736
Wang YL, Liu C, Zhang XF (2020) One-step encapsulation of bimetallic Pd–Co nanoparticles within UiO-66 for selective conversion of furfural to cyclopentanone. Catal Lett 150(8):2158–2166
Zhang SJ, Ma H, Sun YX et al (2021) Selective tandem hydrogenation and rearrangement of furfural to cyclopentanone over CuNi bimetallic catalyst in water. Chin J Catal 42(12):2216–2224
Liu CY, Wei RP, Geng GL et al (2015) Aqueous-phase catalytic hydrogenation of furfural over Ni-bearing hierarchical Y zeolite catalysts synthesized by a facile route. Fuel Process Technol 134:168–174
Yang YL, Du ZT, Huang YZ et al (2013) Conversion of furfural into cyclopentanone over Ni–Cu bimetallic catalysts. Green Chem 15(7):1932–1940
Jia P, Lan XC, Li XD et al (2019) Highly selective hydrogenation of furfural to cyclopentanone over a NiFe bimetallic catalyst in a methanol/water solution with a solvent effect. ACS Sustainable Chem Eng 7(18):15221–15229
Gao X, Ding YY, Peng LL et al (2022) On the effect of zeolite acid property and reaction pathway in Pd–catalyzed hydrogenation of furfural to cyclopentanone. Fuel 314:123074
Date NS, Kondawar SE, Chikate RC et al (2018) Single-pot reductive rearrangement of furfural to cyclopentanone over silica-supported Pd catalysts. ACS Omega 3(8):9860–9871
Tian HL, Gao GM, Xu Q et al (2021) Facilitating selective conversion of furfural to cyclopentanone via reducing availability of metallic nickel sites. Mol Catal 510:111697
Zhang YF, Fan GL, Yang L et al (2018) Efficient conversion of furfural into cyclopentanone over high performing and stable Cu/ZrO2 catalysts. Appl Catal A 561:117–126
Ren BJ, Zhao C, Yang L et al (2020) Robust structured Cu-based film catalysts with greatly enhanced catalytic hydrogenation property. Appl Surf Sci 504:144364
Pan P, Xu WY, Pu TJ et al (2019) Selective conversion of furfural to cyclopentanone and cyclopentanol by magnetic Cu-Fe3O4 NPs catalyst. ChemistrySelect 4(19):5845–5852
Kumar A, Shivhare A, Bal R et al (2021) Metal and solvent-dependent activity of spinel-based catalysts for the selective hydrogenation and rearrangement of furfural. Sustain Energy Fuels 5(12):3191–3204
Liu MR, Yuan LY, Fan GL et al (2020) NiCu nanoparticles for catalytic hydrogenation of biomass-derived carbonyl compounds. ACS Appl Nano Mater 3(9):9226–9237
Chen S, Qian TT, Ling LL et al (2020) Hydrogenation of furfural to cyclopentanone under mild conditions by a structure-optimized Ni−NiO/TiO2 heterojunction catalyst. Chemsuschem 13(20):5507–5515
Lin W, Zhang YX, Ma ZX et al (2024) Synergy between Ni3Sn2 alloy and Lewis acidic ReOx enables selectivity control of furfural hydrogenation to cyclopentanone. Appl Catal B 340:123191
Yuan EX, Wang CL, Wu C et al (2023) Constructing hierarchical structures of Pd catalysts to realize reaction pathway regulation of furfural hydroconversion. J Catal 421:30–44
Deng QA, Gao R, Li XA et al (2020) Hydrogenative ring-rearrangement of biobased furanic aldehydes to cyclopentanone compounds over Pd/pyrochlore by introducing oxygen vacancies. ACS Catal 10(13):7355–7366
Tong ZK, Gao R, Li X et al (2021) Highly controllable hydrogenative ring rearrangement and complete hydrogenation of biobased furfurals over Pd/La2B2O7 (B=Ti, Zr, Ce). ChemCatChem 13(21):4549–4556
Gao R, Li X, Guo LY et al (2021) Pyrochlore/Al2O3 composites supported Pd for the selective synthesis of cyclopentanones from biobased furfurals. Appl Catal A 612:117985
Li X, Tong ZK, Zhu S et al (2022) Water-mediated hydrogen spillover accelerates hydrogenative ring-rearrangement of furfurals to cyclic compounds. J Catal 405:363–372
Gao GM, Shao YW, Gao Y et al (2021) Synergetic effects of hydrogenation and acidic sites in phosphorus-modified nickel catalysts for the selective conversion of furfural to cyclopentanone. Catal Sci Technol 11(2):575–593
Cheng C, Zhao CS, Zhao D et al (2023) The importance of constructing riple-functional Sr2P2O7/Ni2P catalysts for smoothing hydrogenation Ring-rearrangement of Biomass-derived Furfural compounds in water. J Catal 421:117–133
Deng Q, Zhou R, Zhang YC et al (2023) H+−H− pairs in partially oxidized MAX phases for bifunctional catalytic conversion of furfurals into linear ketones. Angew Chem Int Ed 62(9):e202211461
Zhou XY, Feng ZP, Guo WW et al (2019) Hydrogenation and hydrolysis of furfural to furfuryl alcohol, cyclopentanone, and cyclopentanol with a heterogeneous copper catalyst in water. Ind Eng Chem Res 58(10):3988–3993
Wang LL, Weng YJ, Wang XL et al (2019) Synergistic bimetallic RuMo catalysts for selective rearrangement of furfural to cyclopentanol in aqueous phase. Catal Commun 129:105745
Chen HR, Shen K, Tan YP et al (2019) Multishell hollow metal/nitrogen/carbon dodecahedrons with precisely controlled architectures and synergistically enhanced catalytic properties. ACS Nano 13(7):7800–7810
Ma YF, Wang H, Xu GY et al (2017) Selective conversion of furfural to cyclopentanol over cobalt catalysts in one step. Chin Chem Lett 28(6):1153–1158
Wang Y, Zhou MH, Wang TZ et al (2015) Conversion of furfural to cyclopentanol on Cu/Zn/Al catalysts derived from hydrotalcite-like materials. Catal Lett 145(8):1557–1565
Zhou MH, Zeng Z, Zhu HY et al (2014) Aqueous-phase catalytic hydrogenation of furfural to cyclopentanol over Cu–Mg–Al hydrotalcites derived catalysts: model reaction for upgrading of bio-oil. J Energy Chem 23(1):91–96
Tong ZK, Li XA, Dong JY et al (2021) Adsorption configuration-determined selective hydrogenative ring opening and ring rearrangement of furfural over metal phosphate. ACS Catal 11(11):6406–6415
Li X, Zhang LK, Zhou R et al (2022) Bifunctional role of hydrogen in aqueous hydrogenative ring rearrangement of furfurals over Co@Co-NC. ACS Sustain Chem Eng 10(22):7321–7329
Ohyama J, Kanao R, Ohira Y et al (2016) The effect of heterogeneous acid–base catalysis on conversion of 5-hydroxymethylfurfural into a cyclopentanone derivative. Green Chem 18(3):676–680
Ohyama J, Kanao R, Esaki A et al (2014) Conversion of 5-hydroxymethylfurfural to a cyclopentanone derivative by ring rearrangement over supported Au nanoparticles. Chem Commun 50(42):5633–5636
Perret N, Grigoropoulos A, Zanella M et al (2016) Catalytic response and stability of nickel/alumina for the hydrogenation of 5-hydroxymethylfurfural in water. Chemsuschem 9(5):521–531
Li JC, Feng YC, Wang HQ et al (2021) Highly selective ring rearrangement of 5-hydroxymethylfurfural to 3-hydroxymethylcyclopentanon catalyzed by non-noble Ni–Fe/Al2O3. Mol Catal 505:111505
Ohyama J, Ohira Y, Satsuma A (2017) Hydrogenative ring-rearrangement of biomass derived 5-(hydroxymethyl)furfural to 3-(hydroxymethyl)cyclopentanol using combination catalyst systems of Pt/SiO2 and lanthanoid oxides. Catal Sci Technol 7(14):2947–2953
Zhang S, Huang ZQ, Ma YY et al (2017) Solid frustrated-Lewis-pair catalysts constructed by regulations on surface defects of porous nanorods of CeO2. Nat Commun 8:15266
Hall JN, Bollini P (2020) Metal–organic framework MIL-100 catalyzed acetalization of benzaldehyde with methanol: Lewis or Brønsted acid catalysis? ACS Catal 10(6):3750–3763
Li X, Zhang LK, Wu ZL et al (2022) Breaking binary competitive adsorption in the domino synthesis of pyrroles from furan alcohols and nitroarenes over metal phosphide. Appl Catal B 316:121665
Acknowledgements
The authors appreciate the support from the National Natural Science Foundation of China (Nos. 22178158, 52162014 and 22065024), Science and Technology Project of Education Department of Jiangxi Province (No. GJJ2200402), Jiangxi Provincial Natural Science Foundation (No. 20224BAB213023), the Outstanding Youth Science Fund Project of Jiangxi Province (No. 20224ACB213008), the Jiangxi Provincial Double Thousand Talents Plan-Youth Program (No. S2021GDQN0947), and Natural Science Foundation of Chongqing (No. 2023NSCQ-MSX0052).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that there is no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Deng, Q. Review on Metal–Acid Tandem Catalysis for Hydrogenative Rearrangement of Furfurals to C5 Cyclic Compounds. Trans. Tianjin Univ. 29, 347–359 (2023). https://doi.org/10.1007/s12209-023-00367-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12209-023-00367-w