
Abstract
The virus “acute respiratory syndrome coronavirus 2” (SARS-CoV-2) is the etiologic agent of coronavirus disease 2019 (COVID-19), initially responsible for an outbreak of pneumonia in Wuhan, China, which, due to the high level of contagion and dissemination, has become a pandemic. The clinical picture varies from mild to critical cases; however, all of these signs already show neurological problems, from sensory loss to neurological diseases. Thus, patients with multiple sclerosis (MS) infected with the new coronavirus are more likely to develop severe conditions; in addition to worsening the disease, this is due to the high level of pro-inflammatory cytokines, which is closely associated with increased mortality both in COVID-19 and MS. This increase is uncontrolled and exaggerated, characterizing the cytokine storm, so a possible therapy for this neuronal inflammation is the modulation of the cholinergic anti-inflammatory pathway, since acetylcholine (ACh) acts to reduce pro-inflammatory cytokines and acts directly on the brain for being released by cholinergic neurons, as well as acting on other cells such as immune and blood cells. In addition, due to tissue damage, there is an exacerbated release of adenosine triphosphate (ATP), potentiating the inflammatory process and activating purinergic receptors which act directly on neuroinflammation and positively modulate the inflammatory cycle. Associated with this, in neurological pathologies, there is greater expression of P2X7 in the cells of the microglia, which positively activates the immune inflammatory response. Thus, the administration of blockers of this receptor can act in conjunction with the action of ACh in the anticholinergic inflammatory pathway. Finally, there will be a reduction in the cytokine storm and triggered hyperinflammation, as well as the level of mortality in patients with multiple sclerosis infected with SARS-CoV-2 and the development of possible neurological damage.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The emergence of a new virus from the coronavirus family, SARS-CoV-2, the etiological agent of COVID-19, has resulted in a mass spread and a pandemic that has already exceeded 1 year in duration. In addition, it has brought a new challenge for neurologists, especially those who treat patients with MS. This is due to the drastic alteration of clinical management, requiring its alteration and adaptation to the new scenario that the world is in. COVID-19 usually presents with mild symptoms; about 15% of affected individuals can develop a serious disease, which presents with respiratory failure, generalized systemic inflammation, and lethality, which may require mechanical ventilation and intensive care [1].
Therefore, the reality of patients with MS is still unclear in relation to the risk factors for the disease and it is not known whether these people are at higher risk for severe conditions or infection with COVID-19 [2]. However, the fact that MS therapy is based on immunosuppressants, it is believed that the reduction of immune activity is directly related to the increased risk of serious infections compared to the general population. Thus, the guidelines of the Multiple Sclerosis International Federation (MSIF) are recommending extra care among patients with MS in order to minimize their exposure to the virus [3].
Thus, the modulation of the anti-cholinergic inflammatory pathway and purinergic receptors can be analyzed as potential treatments. In this light, considering that the side effects are much less, the stimulation of the vagus nerve and these receptors has been strongly used for therapeutic purposes, being able to improve sepsis, lung injury, rheumatoid arthritis, stroke, traumatic brain injury, obesity, diabetes, cardiovascular control, and pain control [4].
In addition, this therapy has been initiated to reduce respiratory symptoms in patients infected with COVID-19, with excellent results [5]. The benefits of this modulation range from reducing the use of nebulizers and corticosteroids, a fact highly recommended by the World Health Organization, in view of concerns about the spread of SARS-CoV-2 [6], to immediate symptomatic relief [7]. Thus, the use of targeting neural pathways for the treatment of inflammatory diseases is a new scope for exploring this pathway.
Pathophysiology of MS
MS an autoimmune and demyelinating disease of the central nervous system, involving neuroinflammatory processes, demyelination, and axonal degeneration [8], leading to a chronic degenerative and immune-mediated condition of the central nervous system (CNS) [9]. In this inflammatory process, there is an accumulation of macrophages and inflammatory infiltrates in the CNS, contributing to demyelination and among its etiological causes are environmental factors and the involvement of susceptible genes [10].
The manifestations of MS are very variable; however, in most patients, it occurs with the appearance of clinical symptoms on a recurrent basis, followed by partial or total recovery. This form is called Relapsing–Remitting Multiple Sclerosis (RRMS). After 10 to 15 years of illness, half of the untreated patients progress to a progressive pattern, in which there is a progressive deterioration over the years, being classified as Secondary Progressive Multiple Sclerosis (SPMS). Still, there is a form that occurs in approximately 15% of patients, in which MS has been relentless from the beginning, being called Primary Progressive Multiple Sclerosis (PPMS) [11], yet the Progressive Relapsing Multiple Sclerosis (PRMS) represents 5% of the cases and the patients present progressive disability from the beginning [12].
When assessing the pathophysiology of the disease, it appears that the autoimmune condition is triggered primarily by T lymphocytes, with increasing evidence of significant involvement of B lymphocytes. Self-reactivity is triggered by peripheral lymphocyte activation after a change in myelin self-tolerance and to CNS antigens [13]. The triggering factor can be an environmental antigen (such as a virus, for example), which leads to lymphocyte activation, or through molecular mimicry, in which there is a cross reaction between host proteins (such as the basic myelin protein) and an exogenous protein pathogenic (as a viral antigen) [14, 15].
Sequentially, the activated autoreactive T lymphocytes migrate to the CNS, crossing the blood–brain barrier, and can be reactivated by cells presenting local antigens (dendritic cells and macrophages), leading to an intense inflammatory cascade, marked by the release of cytokines and chemokines and recruitment of additional inflammatory cells, which lead to oligodendrocytes loss and damage to myelin [16]. This leads to “spreading of the epitope,” causing a greater exposure of targets to T lymphocytes, amplifying and continuing the local inflammatory response [17]. In turn, the axonal lesion occurs both by the initial inflammatory process, and by the exhaustion of the repair mechanisms by the constant activation of the microglia and the complement, as well as by the indirect action of the pro-inflammatory cytokines, such as the tumor necrosis factor α (TNF-α), nitric oxide, and matrix metalloproteinases [8].
Regarding B lymphocytes, its first indication of involvement with MS was in the identification of ectopic follicles of B lymphocytes present in the meninges of patients with SPMS [18]. This cell proliferation is consistent with the sustained inflammation of the meninges, mainly in the progressive forms, supporting the hypothesis that cytotoxic factors diffused by the meninges favor the subpial cortical lesions and lead to the worsening of the clinical condition [19, 20]. Going further, successful B-cell-based immunotherapies corroborated the participation of these cells in the pathogenesis of MS [21].
Correlation Between COVID-19 and MS
SARS-CoV-2 is known mainly to affect the respiratory system; however, several studies and case reports have demonstrated the potential neurotropism of this virus [22]. Neurological symptoms that have been observed in patients with COVID-19 include dizziness, headache, impaired consciousness, acute cerebrovascular disease, encephalopathy, ataxia, seizures, nerve pain, changes in smell, and taste and visual impairment (optic neuritis and Guillain-Barré syndrome) [23,24,25,26,27].
So far, there are different possibilities that support the SARS-CoV-2 to reach the central nervous system (CNS). Among the hypotheses, there is that the hematogenous or lymphatic distribution of this virus through the systemic blood stream to the brain, in which the flow is slower and can favor the damage to the endothelial cells, as well as offering access to the brain [28]. Contributing to this, SARS-CoV-2 particles were found in the capillary endothelium and in autopsied neurons from a frontal lobe sample [29]. Such fact could be explained by the interaction of angiotensin-converting enzyme 2 (ACE2), present in these cells, with the virus present in the blood and lymphatic circulation, favoring its replication and its damage to the vascular and neuronal tissue [28].
Another possibility is that the virus is able to spread through the cribriform plaque, reaching the olfactory bulb [30]. After reaching the cranial nerves, such as the olfactory nerve, several infectious agents, including coronaviruses, are able to perform retrograde axonal transport [28], transiting through microtubules and reaching neuronal cell bodies [31]. Once established in the brain, the virus is able to spread through neurotransmitter pathways, such as serotonergic in the dorsal raphe system and through hematogenous means [32].
It is also believed that the “Trojan horse mechanism” can occur with SARS-CoV-2. In this, infected leukocytes cross the blood–brain barrier and distribute the virus in the CNS. In the context of systemic inflammation, the blood–brain barrier permeability is increased and, for this reason, immune cells that express ACE2, such as monocytes and lymphocytes, which may be infected, have easy access to the CNS [33]. Finally, pericellular dissemination to adjacent cells can occur, in which the virus is able to pass through the space between olfactory neurons and reach neighboring cells. This mechanism also occurs in the cerebrospinal fluid and in the mucous layer of the respiratory tract [34].
When looking for a correlation between COVID-19 and demyelinating diseases in addition to MS, it is possible to find several reports involving the peripheral nervous system, mainly under the spectrum of Guillain-Barré Syndrome [35]. In addition, it is also possible to analyze the involvement of the central nervous system, having in the literature reports of acute disseminated encephalomyelitis [36], meningo-encephalitis [37], and optic neuritis [38].
Although there are incipient reports correlating COVID-19 and the demyelinating processes in the CNS, some studies help to better understand the relationship between viral infections and central demyelination. For example, there is an increased risk for the development of MS when an infection with the Epstein-Barr virus occurs [39]. In addition, viruses such as varicella-zoster, influenza, or adenovirus are associated with a higher frequency and severity of relapses in patients with MS [40, 41]. In addition, the study by Marrodan et al. [42] demonstrated that viral respiratory tract infections can be related to a greater exacerbation of MS (Fig. 1).

Pathophysiology of multiple sclerosis triggered by SARS-CoV-2 infection. SARS-CoV-2 can reach the CNS by different routes, including hematogenous and dissemination through the cribriform plaque and the olfactory bulb, in addition to being able to perform retrograde axonal transport and propagate by neurotransmitter pathways. When the blood–brain barrier is compromised, both the virus and viral particles are able to reach the cells that make up the CNS. The virus can remain dormant in astrocytes and oligodendrocytes and, due to molecular mimicry between viral particles and the basic myelin protein, trigger a cross reaction and cause autoimmunity. The innate and adaptive immune response triggered includes the activation of microglia and B and T lymphocytes. These will lead to the destruction of myelin, culminating in the neurodegenerative process related to MS
Inflammation and Attack of Neuronal Cells
MS is a chronic, immune-mediated neurodegenerative disease that can involve environmental and genetic factors [43]. The hallmark of MS is the presence of sclerotic plaques, which indicate the final stage of the pathological process that includes inflammation, demyelination, and remyelination, as well as oligodendrocyte depletion, astrocytosis, and neuronal and axonal degeneration [44, 45]. As a consequence of these processes, there are symptoms, sensory, visual, and motor deficiencies, in addition to fatigue, pain, and cognitive deficits [45].
As factors that trigger and maintain the pathogenesis of MS, there is the complex interaction between the dysregulation of the innate and adaptive immune system, genetic susceptibility, and the influence of environmental factors [46]. The different populations of T cells, such as the CD8 + cytotoxic T lymphocyte (TCD8 +), regulatory T (Treg), helper T type 1 (TH1), and T-helper 17 (TH17) [47], B cells, as well as other cells of the peripheral innate immune system (monocytes, dendritic cells), astrocytes and microglia are recognized as the central components of MS immunopathology [48]. In addition, MS is associated with a cytokine storm that generates an increase in proinflammatory cytokines such as interferon-γ (IFN-γ), interleukin-12 (IL-12), tumor necrosis factor alpha (TNF-α), and beta (TNF-β), in addition to decreasing anti-inflammatory responses by reducing interleukin-10 (IL-10) and transforming growth factor beta (TGF-β). Both types of cytokines are produced by astrocytes and microglia and interact with the complex peripheral immune system [49, 50].
The initial lesion of MS begins with the passage of autoreactive T lymphocytes through the blood–brain barrier [51, 52], with the subsequent activation of the microglia and other immune cells that will initiate a high secretion of pro-inflammatory cytokines [45] and that will be responsible for the initial lesion and demyelination of neuronal axons [53]. The dysregulation of the immune response will lead to an inflammatory process in some specific sites, mediated by the perivascular accumulation of TCD8 + .
In addition, plaques will be established around the corpus callosum, lateral ventricles, cortex, and white matter, as well as in the optic nerve and spinal cord [45]. The presence of cells active in innate and adaptive immunity, in addition to the high release of pro-inflammatory cytokines, will intensify the inflammatory process through the activation of the naive microglia [54, 55]. Thus, the production of the humoral response by B lymphocytes in the meninges can cause even more damage to the cortex [56].
When there is stimulation of astrocytes in the face of injuries and injuries, as when viral infections occur, a cellular response called astrogliosis is triggered. This, in turn, significantly changes cell biology and aims to improve neuronal survival, restore integrity to the blood–brain barrier, and isolate the injured region, creating a glial scar [57]. However, in some conditions that probably involve the proinflammatory activation of microglia and signaling pathways related to toll-like/factor nuclear kappa B/(activator of transcription 3) (toll-like/NFκB/STAT3), astrocytes initiate proinflammatory gene expression that favors neurodegeneration [58, 59]. The microglia, after a viral infection, remains activated and initiate the release of pro-inflammatory molecules and reactive oxygen species, recruiting cells from the peripheral immune system and promoting the activation of macrophages [60].
Cytokines and growth factors that are released by microglia and activated astrocytes help in the process of remyelination. However, over time, astrocytes cause a sealing of the lesion and gliosis hinders the process of additional remyelination, causing the ability to accommodate cumulative deficits to be impaired and marking the moment when permanent deficits begin [45]. Approximately 85% of patients have the relapsing–remitting form of MS, in which there is a partial or complete recovery after an acute exacerbation of the demyelinating process [48].
In turn, in COVID-19, an immune response can be observed characterized by a hyperactivity of innate immunity, followed by an immunosuppression [61]. In more severe cases, the development of the cytokine storm, which occurs after 2 to 3 weeks of infection, is related to severe acute respiratory syndrome (SARS), with disseminated intravascular coagulation, multiple organ failure [62], and death [63]. The main cytokines involved are IL-1β, IL-6, IL-18, IL-33, IFNγ, and TNF-α [64, 65], which are secreted by innate immunity cells and adaptive and cause local and systemic effects [66].
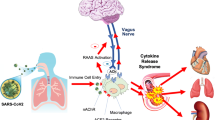
Using different mechanisms, SARS-CoV-2 can reach the CNS and, even if there is no invasion of the CNS by the virus, viral proteins present in the circulation and molecular complexes of damaged cells can reach the brain, if the blood–brain barrier is compromised [1]. After these molecules enter the brain, they can act as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), leading to innate immune responses through toll receptors present in pericytes, macrophages residing in the brain and in the brain. microglia. In the case of SARS-CoV, toll-like receiver 2 (TLR2) mediates proinflammatory responses after recognition of the protein spikes by macrophages, activating the nuclear factor κB (NF-κB) pathway [67]. Also, if the virus reaches the CNS, the damage will be caused by direct viral cytotoxicity or by immune-mediated attacks [68]. Neuroinflammation can generate more DAMPS and autoantigens, causing an increase in inflammatory responses and favoring autoimmune processes [69], as observed in MS [70] (Fig. 1).
Cytokine Storm
The moment SARS-CoV-2 reaches the CNS, invasion of astrocytes, macrophages, and microglia cells occurs, inducing a pro-inflammatory state with high amounts of IL-1β, IL-2, IL-4, IL-6, IL-10, IL-18, IFN-γ, TNF-α, among other molecules involved in the inflammatory process [71]. In addition, in vitro studies have found that SARS-CoV-2 induces the expression of IL-6, IL-12, IL-15, and TNF-α in glial cells [72]. The cytokine storm at the CNS level may occur due to the local production of cytokines or the ability of cytokines and immune cells to cross the blood–brain barrier, due to impairments in their integrity [73]. This storm of cytokines will lead to neuronal death, activation of microglia, impairment of synaptic plasticity, and damage to neurotransmitter signaling [74].
When analyzing the immunological characteristics of COVID-19 and the pathogenic process of MS, it is clear that the cytokine storm triggered by SARS-CoV-2 infection can be related to neuroinflammation and rupture of the blood–brain barrier, favoring greater relapse of MS [75]. In addition to the cytokine storm, COVID-19 is able to promote changes in cell populations of T and B lymphocytes and in natural killers cells (NK), and the initiation of the response of these innate and adaptive immune cells may favor the relapse of MS [76].
Corroborating what has been discussed, large amounts of pro-inflammatory cytokines, such as TNF-α, IL-6, IFN-γ, and IL-1β, were found in the cerebrospinal fluid of MS patients suffering from lesions in the gray or white substance [77]. It is known that TNF-α has already been isolated postmortem in active areas of degeneration in the brains of patients with MS. This cytokine promotes the apoptosis of oligodendrocytes and the infiltration of immune cells in the cerebrospinal fluid, while the injection of TNF-α monoclonal antibodies prevents the development of MS in animal models [78].
In turn, high levels of IL-6 inhibit the differentiation of T lymphocytes into Treg, creating an inflammatory and demyelinating condition in mice. Furthermore, IL-6 deficient mice are resistant to MS and the administration of antibodies to IL-6 receptors (IL-6R) promotes the inhibition of myelin degeneration in animal models of autoimmune encephalomyelitis. In addition, IL-6 favors compromising the integrity of the blood–brain barrier, facilitating the entry of cytotoxic T cells [78]. Finally, it is also known that antibodies to the IL-1β receptor antagonize the demyelination process in animal models of autoimmune encephalomyelitis, suggesting possible routes of action for IL-1β in the pathogenesis of MS [79] (Fig. 2).

Neurodegeneration process triggered by the cytokine storm in COVID-19. After the entry of SARS-CoV-2 in the CNS, infection of astrocytes and microglia cells occurs, leading to the release of several pro-inflammatory cytokines, such as IL-1β, IL-2, IL-4, IL-6, TNF-α, and IFN-γ. The impairment of the blood–brain barrier by the systemic pro-inflammatory state also allows the entry of more pro-inflammatory cytokines and a greater number of viruses in the CNS. The storm of cytokines at the level of the nervous system can lead to neuronal death, activation of microglia, impairment of synaptic plasticity, and impairment of neurotransmission. The cytokine storm and the rupture of the blood–brain barrier can promote the relapse of MS, in addition to (A) promoting the activation of B and T lymphocytes and NK cells, favoring the neurodegenerative process of MS. (B) In addition, studies have found that TNF-α, isolated in the brains of patients with MS, is able to promote the apoptosis of oligodendrocytes and the infiltration of immune cells, corroborating the destruction of the myelin sheath. (C) Going further, high concentrations of IL-6 are able to inhibit the differentiation of T lymphocytes into Treg, establishing an inflammatory and demyelination state in animal models
Cross-Reaction and Autoimmune Response
Going beyond the cytokine storm, there is another hypothesis that relates the autoimmune process due to the cross reaction between the viral particles and the basic myelin protein, this being the point of origin for the neural demyelination process. To support this hypothesis, the genome of other coronaviruses (CoV-OC43 and CoV-229E) and their antibodies have already been isolated from the CNS of patients with MS, and particles similar to the coronavirus can be found in perivascular regions of the human brain with MS [32]. In addition, the virus may be dormant in astrocytes and oligodendrocytes and, due to molecular mimicry, trigger autoimmunity. Strengthening this idea, studies have been found that the intracranial inoculation of murine coronavirus stimulated optic neuritis and focal encephalitis, causing chronic demyelination in animal models [80]. In these cases, the demyelinating process would be caused by adaptive and innate immune responses, including the activation of microglia, B and T lymphocytes (CD4 and CD8), together with direct viral toxicity [81, 82].
Cellular Receivers
In addition to the aspects already discussed, toll-like receptors (TLRs) are involved simultaneously in the recognition of pathogens and in the pathophysiology of MS. It has been found that when viral particles are recognized by these receptors, there is a change in the immune response in patients with MS. Therefore, through this route, a relationship could be observed between viral infections, including the coronavirus, and the development of demyelinating diseases [83].
Experimental studies carried out on mouse strains susceptible to JHMV coronavirus lineage verified an acute encephalomyelitis followed by a chronic demyelinating process [84]. In addition, multiple models of murine coronaviruses have led to demyelinating processes in rodents [85], either by causing a persistent infection in oligodendrocytes or astrocytes or by promoting an autoimmune response against myelin basic protein [86]. Thus, the expression of ACE2 receptors in the CNS favors viral tropism for SARS-CoV-2 [87] and it is known that this virus can favor neuronal loss [72], thus being able to, in the brains of people with MS, favor the neurodegenerative process [88] through a mechanism of viral toxicity that could cause direct damage to infected neuronal cells [20].
Therefore, considering the pro-inflammatory context resulting from SARS-CoV-2 infection, the persistence of this scenario can spread myelin degeneration and lead to the spread of MS [89] or even lead to first manifestation of the disease [38]. In mild cases of COVID-19, the exacerbation of MS can be temporary; however, the risk of relapse or disease progression can be significant. Still, treatment with DMD, immunosuppressants, and corticosteroids for long periods can influence the immune response, and can lead to complicated cases of COVID-19 [90]. Thus, the development of new therapies is necessary, one of which is the modulation of the cholinergic and purinergic system that are often associated with inflammatory processes.
Current MS Therapies in the Context of COVID-19
Interferon-Beta (Interferon-β1a and Interferon-β1b)
The first approved agent for the treatment of MS, available in subcutaneous and intramuscular formulations, modestly protecting against relapses and exacerbated brain injuries [91]. Because it is an immunomodulator, it acts directly on the significant increase in anti-inflammatory cells, as well as on the reduction of pro-inflammatory cytokines [92]. Also responsible for reducing the passage of inflammatory cells through the blood–brain barrier, increasing the production of nerve growth factor (NGF) and NK CD56 by IFN-β. These cells are able to release anti-inflammatory mediators and limit neuronal inflammation [91].
However, conditions such as rare leukopenia, lymphopenia, and lymphadenopathy are included as side effects of using this drug. In addition, upper respiratory tract infections are closely associated with the administration of IFN-β1a (14% vs 12% placebo), although interferon-B1b has a lower incidence [91]. It is worth noting that IFN-β1b formulations contain albumin, which is a factor capable of increasing the risk of viral transmission, even if interferons are not associated with opportunistic or serious infections.
In the context of the SARS-CoV-2 pandemic, IFN-β does not act mainly on lymphocyte depletion, and presenting lymphopenia is rare and mild, the immune response is unlikely to be affected early or late against SARS-CoV-2, not altering susceptibility to infection. In fact, a recent clinical trial has indicated IFN-β as a beneficial antiviral against coronavirus [93], and it is likely to be safe to continue treatment during the pandemic, as well as to start treatment in newly diagnosed patients. However, even though it is safe, its potency is low in relation to other agents (DMTs), not being the best choice for patients with highly active MS or with several predictors of negative results [94].
Glatiramer Acetate
Acting in a modest reduction in the annual recurrence rate, glatiramer acetate (GA) is a subcutaneous injection made up of a synthetic polypeptide structurally similar to the basic myelin protein. The mechanism of action is based on the competition with the myelin antigens on the interaction with the main molecules of the type 2 histocompatibility complex in the antigen-presenting cells [91]. Thus, preventive action is generated regarding an attack response of T cells against myelin, added to activation of type 2 helper T cells, which secrete anti-inflammatory cytokines and act to reduce inflammatory demyelination of the CNS [95].
In a situation of general infection, compared to placebo, GA had a slightly higher incidence (30% vs 28%), in bronchitis (6% vs 5%), in influenza (14% vs 13%), in nasopharyngitis (11% vs 9%), and viral upper respiratory tract infections (URTIs) (3% vs 2%) [91], although it is not recognized as a cause of opportunistic or serious infections. Thus, because lymphocyte depletion does not occur, a negative impact on the immune response against SARS-CoV-2 or even an increased propensity to infection is unlikely. It is safe to continue the treatment, as well as its beginning, regardless of the contact with the virus; however, similarly to the previous medication, its use should only be indicated for patients with mild MS, given its low potency [96].
Teriflunomide
Oral agent of modest efficacy in the preventive action against relapses and accumulation of brain lesions in MS, teriflunomide had its use approved in 2012 and is an inhibitor of dihydroorotate dehydrogenase. This is a fundamental mitochondrial enzyme in the synthesis of pyrimidine and necessary for cell division; thus, its inhibition is capable of negatively modulating the replication of auto-reactive lymphocytes [95]. In addition, its immunological side effects include neutropenia (16%) and rare lymphopenia (12%) [91].
Likewise, this medication had a slightly higher incidence in infections such as URTIs (9% vs 7%), bronchitis (8% vs 6%), and sinusitis (6% vs 4%) [91], as well as rare cases of tuberculosis and reports of reactivation of hepatitis by cytomegalovirus. After commercialization, there were fatal cases of Klebsiella, Pneumocystis jiroveci, and Aspergillus pneumonia; however, the patients had underlying comorbidities and were being co-treated with other immunosuppressants [91]. Thus, by reducing the proliferation of reactive lymphocytes, increased susceptibility to coronavirus is possible.
Thus, observing the profile of the infectious side effects and the slight change in the infectious propensity, one should evaluate the beginning or the continuation of the treatment, although it is considered relatively safe due to the beneficial antiviral effects [97]. In the same way that this treatment is likely to remain asymptomatic or mildly symptomatic, a fatal case of COVID-19 infection that was taking teriflunomide, which had advanced secondary progressive MS and comorbid myotonic dystrophy [98], has also been reported. In addition, other cases of COVID-19 infection were self-limiting [99,100,101] and there was a self-reported case of full recovery at home [102].
Fumarates
First approved in 2013 for recurrent forms of MS, dimethyl fumarate (DMF) has been replaced by diroximel fumarate, approved in 2019, which has similar bioavailability and efficacy, with reduced gastrointestinal adverse effects [103]. It is believed that the mechanism of action, although still uncertain, is based on immunomodulation through the inhibition of the Nrf-2 protein, capable of inhibiting the inflammatory cascades [95]. Lymphopenia is one of the main idiosyncratic side effects, which occurs in 37% of patients, while severe lymphopenia (< 500/mm3) occurs in only 8% [96].
The incidences of general and severe infections were very similar between the groups of patients and the control group [91]. There are cases of progressive multifocal leukoencephalopathy (PML) associated with foot-and-mouth disease (FMD), especially when prolonged severe moderate lymphopenia (< 800/mm3) is present [104]. Thus, it is possible that there is an increased susceptibility to SARS-CoV-2 infection in these patients, differently from patients without lymphopenia or with its mild version.
Thus, it is advisable to interrupt the treatment of patients with persistent severe lymphopenia and constant monitoring of cutaneous lymphocyte-associated antigen (CLA); however, in mild cases, it is safe to continue and start this therapy. Two infected patients who did not show lymphopenia achieved complete recovery, despite the continuation of DMF [98]. Another seven with suspicion [105] and two self-reported [102] also obtained favorable results, with normal or slightly decreased lymphocyte counts, illustrating that the fumarates do not reduce the immune response.
Sphingosine-1-Phosphate Modulators
Approved in 2010, the first oral agent approved for recurrent MS and the first sphingosine-1-phosphate (S1P) modulator, fingolimod, has been replaced by siponimod and ozanimod, approved in 2019 and 2020, respectively [106, 107]. This is because these drugs are more selective for the S1P1 and S1P5 subtypes, as well as lesser bradycardic side effects. The mechanism of action consists in reducing the output of T cells from the lymph nodes, decreasing the lymphocytes available for the CNS, and the central inflammation [95].
Acting directly on the availability of peripheral lymphocytes, all three drugs can increase the risk of infection [91]. In the general incidence of infections in studies with fingolimod 72% of the patients presented it and 2% in a severe incidence, approaching the values of the placebo group [108]. Influenza (13% vs 10%) and herpes viral infections (9% vs 8%) had higher values, in addition to fatal cases of herpetic infections with fingolimod. Siponimod and ozanimod had similar infectious side effects, and multifocal leukoencephalopathy becomes a risk for this class of drugs [91].
As evidenced by the increased susceptibility to viral infections, S1P modulators can, by reducing peripheral lymphocytes, increase the risk of SARS-CoV-2 infection. However, in more serious situations in COVID-19 patients with a cytokine storm, it appears to have a potential beneficial effect that is being analyzed in an ongoing clinical trial (ClinicalTrials.gov Identifier: NCT04280588) [109]. As for treatment, its continuation is likely to be safe when absolute lymphocyte count (ALC) is greater than 200/mm3 and is potentially beneficial even in patients infected with COVID-19, since two cases of worsening of the condition have been reported after interruption of fingolimod [110].
In addition, six other cases of COVID-19 infection in patients with MS and following treatment with fingolimod have been reported, in addition to four self-reported cases. In all cases, the patients had the mild version of the disease and full recovery occurred, although some infected patients stopped treatment temporarily and others continued without interruption. Patients treated with siponimod or ozanimod and infected with SARS-CoV-2 have not yet been reported [91].
Natalizumab
In recurrent MS, the first mononuclear antibody approved as monotherapy and combined therapy with IFN-β1a in 2004 was natalizumab, as it has high potency against recurrences and accumulation of lesions [95]. The mechanism of action consists of response similar to the alpha-4 integrin antagonist in the leukocyte membrane, capable of blocking the interaction of this cell with the vascular cell adhesion molecules and preventing its migration and infiltration in the CNS [95]. Thus, because it is a non-depleting immunomodulator, it was not associated with lymphopenia; however, it is known that its use increases the risk of PML.
Thus, monitoring for antibodies to the John Cunningham virus (JCV) is mandatory during treatment, given that high levels indicate viral exposure and greater chances of PML. Thus, URTIs (22% vs 16% placebo), influenza (12% vs 5% placebo), and sinusitis (8% vs 4% placebo) were recurrently seen in these patients, and fatal cases of herpetic encephalitis and meningitis were notified after marketing [91]. An unlikely increase in susceptibility to viral infections by natalizumab is illustrated by being a non-destructive cell agent; however, its side effects seem to be related to this increase in the scenario of COVID-19.
Beneficial effects of this therapy are being studied in patients infected with SARS-CoV-2 due to its action on integrin receptors [111]. Thus, it is understood that the permanence or initiation of therapy, in general, is safe, although theoretical questions raise doubts. Experts claim that this drug can reduce the movement of lymphocytes in the lungs and mucosa, potentiating viral elimination [112]; similarly, with reduced immune surveillance in the CNS, there is concern about a neuronal invasion [113] and the potential development of SARS-CoV-2 encephalitis. To date, two cases of MS patients following therapy and infected have been reported, both have had mild strokes and complete recovery, with no neurological symptoms or suspected encephalitis.
Ocrelizumab
In 2017, ocrelizumab was approved by the FDA for both recurrent and PPMS, showing high efficacy against brain injuries and relapses when compared to IFN-β1a [114] and modest efficacy when compared to placebo analyzing the progression of disability [94]. Because it is a humanized monoclonal antibody (MAB), it targets CD20 in the B cell membrane, which can result in prolonged selective lymphopenia [95] and a reduction in total ALC in a quarter of patients [96]. In addition, prolonged depletion of memory B cells can result in hypogammaglobulinemia, even if the drug does not reach plasma cells, as a result of similar long-term treatments [115].
Thus, patients following this treatment line had a higher incidence of infections, experiencing 58%, compared to 52% in patients with RRMS treated with IFN-β1a, this study with PPMS, presented 70% vs 68% for the control group. Likewise, the incidences, in RRMS studies, of URTIs (40% vs 33%), lower respiratory tract infection (LRTI) (8% vs 5%), herpes zoster (2.1% vs 1.0%), and herpes simplex (0.7% vs 0.1%) remained higher in this treatment [116], while in PPMS studies, incidences of URTI (49% vs 43%), LRTI (10% vs 9%), and herpes oral (2.7% vs 0.8%) were also high in the comparison. Thus, it is possible to increase susceptibility to SARS-CoV-2 as demonstrated in recent cases [117].
In this line of reasoning, the direct impact of ocrelizumab would not be on the initial immune response against the virus, as it mainly targets B cells. However, considering the role of these cells in humoral immunity, as well as their role as antigen-presenting cells for B cells, there is a direct link to infection control. In addition, B cell depletion can still act in the long term in an immunological impairment, increasing the risk of reinfection [118], and a failure in the development of anti-SARS-CoV-2 antibodies has also been demonstrated in patients treated with this medication after infection, not directly influencing a more serious infection [119].
Thus, the treatment response to COVID-19 infection is quite variable and discrepant. This occurs in anti-CD20 therapies due to the double effect that B cells can provide a beneficial effect for the production of neutralizing antiviral antibodies, but also a harmful effect mediated by the production of IL-6 and contribution to the cytokine storm [120]. Thus, to mitigate the risks, it may be interesting to space the infusions guided by CD19 counts, which may delay the reduction of ocrelizumab in infected patients or with sustained CD19 depletion. For newly diagnosed patients, the decision for this therapy should be cautious due to its impact on the risk of infection and future vaccination [91].
Alemtuzumab
Highly potent intravenous immunosuppressant approved for RRMS in 2014, alemtuzumab, reduces relapses of MS and the accumulation of neuronal lesions. Because it is a humanized MAB, it targets CD52 receptors on the surface of mature lymphocytes [95] and can cause generalized and prolonged lymphopenia in T cells. Thus, due to its strong effect on immune cells, the absence of T lymphocytes demands a reconstitution precocious B cells, which can trigger autoimmune disorders, such as thyroid diseases, idiopathic thrombocytopenia, and glomerulonephritis [91]. In addition, it can promote neutropenia and pancytopenia, by altering innate immunity by modifying CD52-positive macrophages and dendritic cells [121].
Thus, treatment with alemtuzumab provides a greater risk of infection, being illustrated in clinical studies by a 71% chance of incidence, compared to 53% in patients who took IFN-β1a [91]. In addition, this difference can be seen in infections such as nasopharyngitis (25% vs 19%), URTIs (16% vs 13%), sinusitis (11% vs 8%), herpetic infections (16% vs 3%), influenza (8% vs 6%), and bronchitis (7% vs 4%), plus severe infections (3% vs 1%) and including cases of pneumonia and herpes zoster [91]. Representing a strong association with viral infections and may increase susceptibility and or reinfection by SARS-CoV-2.
Considering its effect on T and B cells, as well as in innate cells, the impact of alemtuzumab on initial and long-term immunity against SARS-CoV-2 is understandable. This response is expected in the first 6 months of treatment, in which maximum lymphopenia is observed, which can significantly reduce the humoral and cellular responses against the viral protein [122]. Thus, the continuation or the beginning of the treatment must be rethought, since, in two reports of infection, both patients developed the infection after receiving the second cycle of treatment and presented lymphopenia, one severe (grade 4) and the other moderate (grade 2) [123]. Even if both have recovered and the connection between treatment and the severity of the disease is not known, attention should be paid to the use of this drug and possible consequences.
Cladribine
Published in 2010, the potent oral immunosuppressant proposed against MS relapses and the accumulation of brain lesions is a purine nucleoside analog that inhibits DNA synthesis and repair. Thus, cladribine acts to induce apoptosis of B and T cells, although its effect on T cells is shorter and less pronounced [112]. Lymphopenia occurs in the depletion phase, followed by a return to normal counting and the effect on innate immune cells is not so significant, and may rarely lead to cases of neutropenia and pancytopenia [91].
As for the incidence of infections, clinical trials showed 49% in patients with this treatment, compared to 44% in the placebo group. In addition, a higher percentage is observed in the incidence of URTI (38% vs 32%) and in severe infections (6% vs 2%) in the placebo group. When severe lymphopenia, less than 500/mm3, there was a higher incidence of herpes zoster and severe and fatal tuberculosis, and hepatitis B infections also occurred. Thus, by triggering non-selective lymphopenia in the first semester of treatment, a reduction in initial and long-term immunity may occur, justifying your caution in the pandemic scenario of COVID-19 [91].
The increased susceptibility to infection or reinfection by SARS-CoV-2 is understandable, although its oral administration and the lesser involvement of T cells reduce these effects. During the cell depletion phase, humoral and cellular responses should be reduced, altering the response against the SARS-CoV-2 viral protein, although there are no confirmed cases of this infection yet. Even so, one study showed a tendency towards higher symptoms suggestive of COVID-19 among these patients, reinforcing the need to maintain preventive measures and social isolation [124].
Thus, it is understood that the use of immunosuppressive therapies or that act to directly reduce this activity in MS, when considering a scenario of a viral infectious pandemic, is able to impair the immune response against the agent, or even significantly increase susceptibility. infection. Therefore, the study and application of alternative therapies become increasingly necessary in this pathology.
Purinergic and Cholinergic Modulation in Neuroinflammation
The purinergic system consists of extracellular nucleotides and nucleosides, specific receptors, and degradation enzymes, being closely associated with neurodegenerative and/or neuroinflammatory diseases such as MS [125]. One of these nucleotides is ATP, initially seen only as an energetic intracellular molecule, until the introduction of the concept of purinergic cotransmission in 1976, characterizing extracellular ATP as a cotransmitter in the peripheral and central nervous systems [126]. In this way, extracellular nucleotides can be released by several cells in physiological response to injury or stress, in pathological situations or innately [127].
From this point of view, the fundamental role of extracellular ATP as a signal of alert and cellular danger is visible, especially in the brain in which harmful stimuli significantly raise the level of this molecule [128]. This increase, when persistent, acts as an immunostimulatory signal and provides a neuroinflammatory environment, which can cause excitotoxicity of oligodendrocytes [19]. However, another molecule of this system, the adenosine nucleoside, a product of ATP degradation, exhibits a potent immunosuppressant and potent anti-inflammatory, acting in a manner contrary to the pro-inflammatory actions of extracellular ATP [129].
Thus, it is understood the need for a balance between ATP and adenosine for an immunological homeostasis, in order to prevent further tissue damage. The modulation and control of these molecules are mediated by a chain of ectonucleotidase enzymes, which are expressed in different tissues. In addition, the modulating role of adenosine in neuronal activity in regulating the excitability of nerve cells has neuroprotective functions through the activation of purinoreceptors in pathological conditions [130]. As a practical example, in general, changes in ectonucleotidase enzyme activities in the vascular system directly imply vascular tone, lymphocyte, and macrophage responses, as well as platelet aggregation, functions regulated by adenosine and its nucleotides [131].
Among the classification of purinergic receptors, there is the type 1 (P1) receptor, subdivided into four subtypes of adenosine receptors, while the adenosine type 2 (P2) nucleotide receptors are divided into two main families P2X (ionotropic) and P2Y (metabotropic) [126]. The expression of the P2 receptor occurs in practically all cells, and the regulated release of ATP and previous studies have shown a high expression in astrocytes, microglia, and immune cells. These are controlled by ectonucleotidases and trigger the secretion of pro-inflammatory cytokines and chemokines, which promote cell migration and proliferation, contributing to demyelination and axonal damage [132].
About the cholinergic system, the central mechanism of action is related to a greater stimulation that inflammatory agents can generate in the vagus nerve in its afferent branch. This greater activation is accompanied by greater activity in the vagus efferent nerve that is able to increase the release of acetylcholine through direct or indirect pathways and mediated by T lymphocytes. Thus, the components of this pathway act in a way to reduce production and release of pro-inflammatory cytokines, such as TNF-α and IL-6, of the immune system. In this perspective, when the severity of the cytokine storm in the pathological scenario of COVID-19 and its involvement with MS is understood, the activation of the anti-inflammatory reflex mechanism can modulate the exaggeration of the effects of pro-inflammatory cytokines and reduce the symptom level [4, 133, 134].
Thus, involving acetylcholine (ACh) as the main neurotransmitter that modulates the pathway, the cholinergic system acts on the central nervous system (CNS) and the peripheral (SNP) by means of cholinergic neurons, as well as their interaction with non-neuronal cells that regulate several physiological functions [135]. Among its components, we highlight the following: ACh, cholinacetyltransferase (ChAT), choline transporter (ChT), vesicular cholinergic transporter (vAChT), nicotinic receptors (nAChRs), muscarinic receptors (mAChRs), acetylcholinesterase (AChE), and butyrylcholinesterase (BuChE), the latter two known as cholinesterases (ChEs) [136]. These enzymes act directly in the regulation of ACh volume and activity, contributing to its homeostatic control, since they are able to hydrolyze ACh quickly and effectively, with emphasis on AChE which has a high and specific affinity for the neurotransmitter [137].
The nervous environment has high concentrations of AChE, a characteristic shared with neuromuscular junctions, red blood cells, and lymphocytes [138]. The nonspecific BuChE hydrolyzing cholinesterase can hydrolyze several esters and is also present in white matter and in certain populations of neurons and glial cells, as well as found in the blood serum, pancreas, liver, and CNS [139]. In addition, there are two classes of pre- and post-synaptic cholinergic receptors: metabotropic mAChRs and ionotropic nAChRs [140], of which the latter are widely expressed in both neuronal and non-neuronal tissues and are of great importance for the immune system and others peripheral tissues [141].
Thus, the different functions possible by this higher order system in the brain are understood, as well as the essential role in immune cells and in the maintenance of homeostasis [136]. Dendritic cells, macrophages, and B and T cells express most of the components for functional performance of the cholinergic system, providing an independent system of cholinergic nerves involved in the metabolism of ACh produced by cells of the immune system [142]. Thus, it is understood how the proliferation of immune cells, production of cytokines, auxiliary differentiation, and presentation of antigen can affect cholinergic transmission [143].
ACh acts as a classic primary excitatory neurotransmitter and its responses depending on the location, which varies between the brain and the periphery. In addition, contributing to synaptic plasticity, ACh acts as a neuromodulator in many areas of the brain. As it is also related to the modulation of lymphocyte actions, it can be considered an immunomodulator [142]. Thus, it is known of the synthetic action of ChAT in the metabolism of choline and acetate, as well as starting substrates in ACh that will be mediated by the nAChRs and mAChRs receptors [144]. However, the way in which storage occurs or even its release is still uncertain, and it can be stored in vesicles and synthesized in T cells according to demand [145].
Association Between Cholinergic and Purinergic Systems
Thus, it is clear that both components of the cholinergic system, as well as the purinergic system, can be affected due to diseases or secondary events. Constituting important signaling extracellular molecules, ACh and purines trigger cellular effects in different cells by the expression of their receptors, including in the neuronal environment. Thus, the potential modulating role for activation and inhibition of receptors in resident brain cells and peripheral cells may act in new therapeutic opportunities in MS [135].
In addition, the interaction between these systems is strongly highlighted in the literature. Evidence indicates a co-release of ATP and ACh in the brain cholinergic nerve terminals [146], being confirmed after hypoxia in the carotid body of animal models. Thus, it is perceived that ATP receptors are able to control the release of ACh, although an opposite effect has also been observed in areas of the brain and motor terminals of rats [147, 148], providing a presynaptic inhibition of release of ACh in the cerebral cortex [147]. However, with the activation of presynaptic receptors, it facilitates the release of this neurotransmitter, added to an activation of the presynaptic nAChR, justifying a direct interaction between the receptors [149].
In addition to the coexistence of these receptors at the cholinergic terminals, there is an evoked release of ACh in phrenic nerve endings, since, when stimulated, A2R facilitates this release, while A1R has inhibitory effects [150]. Coupled with this, Cunha et al. [149] analyzed this release modulated by adenosine in three areas of the hippocampus (CA1, CA3, and dentate gyrus) and noticed differences between the excitatory and inhibitory effects of A1R and A2AR [149], indicating direct action by purinergic signaling in the control of ACh centrally and peripherally.
Another aspect to be highlighted is the relationship between nACh receptors and P2X, since their simultaneous activation generates negative modulation. Thus, there is an inhibitory interaction between nicotinic receptors and P2X, as well as the independence of the submucosal neurons of these channels [151]. In an animal model, Rodrigues et al. [152] also demonstrated a strong functional interaction in the hippocampus between the P2X and nACh receptors in the presynaptic phase in controlling the release of noradrenaline, firmly evidencing the interaction between these receptors. In the rat striatum, the dopamine release in the dopaminergic terminals had their functional interaction controlled by pre-synaptic A2AR and nAChRs, and the activation of the first reduces the release of dopamine stimulated by nicotine from the isolated nerve of the terminals, being essential for sensitization motor [153].
Thus, the interaction of A2AR and nAChE is visible, and how purinoreceptors seem to control nicotinic responses. Confirmed that the standard stimulation causes changes in the presynaptic inhibition of A1R and A2AR, given the changes in the amount of extracellular adenosine, interferences in the levels of ACh in the neuromuscular junctions of rats are perceived by tonic activation of M1 and autoreceptive muscarinic facilitator M2 inhibitors [154], justifying the control of adenosine receptors over muscarinic receptors and providing new therapeutic opportunities for the treatment of MS.
Cholinergic System and Patients with MS
Expressed by macrophages and other cytokine-producing cells, nicotinic receptors, when interacting with ACh, can act in the regulation of pro-inflammatory cytokine synthesis [155]. In this perspective, the cholinergic dysfunction that occurs in diseases such as MSRR can be controlled by the activation of nAChR, which selectively modulates inflammatory responses. This fact is reinforced by previous investigations that reported low levels of ACh in the CSF and in the serum of these patients, contrasting with high levels of pro-inflammatory cytokines such as IL-1β and IL-17 in relation to the control group, justifying a probable relationship between the cytokine levels and ACh, given its inversely proportional activity [135].
Huang et al. [156] in their studies affirm the contribution of IL-17 in autoimmune diseases such as MS. Thus, reports reinforce this reasoning by demonstrating that the activity of AChE in lymphocytes and whole blood, added to the activity of BuChE in serum, was increased in patients with RRMS, highlighting an increase in pro-inflammatory cytokines [135]. Analyzing this counterpoint, it can be expected that the increase in enzyme activity contributes to the reduction of ACh concentrations, a fact confirmed by the study by Di Bari et al. [157] when reporting the dysfunction in patients with MS. These results directly associate the reduction in ACh levels caused by the overactivation of cholinesterases and the consequent reduction in cholinergic entry to the cells’ immune system [135].
BuChE activity has been involved in the activation of microglia and, in states of neurodegenerative diseases, contributing to the pro-inflammatory state since the increase in enzyme activity has been found in neuropathological studies in the brain tissue of patients with MS [158]. Another piece of evidence was the report of reduced activity and expression of the ChAT protein in the hippocampus of patients with MS, which is responsible for the synthesis of ACh, although no changes were found in the activity and expression of AChE [159].
This reduction in cholinergic innervation in the patients’ hippocampus suggests why there are so many memory problems faced by patients with MS. The cholinergic neurotransmitter system has a fundamental role in the learning function and in memory [160]. Thus, this cholinergic imbalance in the hippocampus, associated with a reduction in the synthesis of ACh, is able to exacerbate cognitive impairment in neurodegenerative diseases [145] and generate impairment in memory and loss of cognitive function, common clinical manifestations in patients with MS [135].
It is also worth mentioning the importance of the protein activity of the complement system, which makes up an important part of the innate immune system and is activated in conditions of neuroinflammation, in addition to contributing to the formation of the brain's synaptic circuits [161]. Studies have shown a correlation between CSF and C3 (complement system protein) levels and BuChE, suggesting a cholinergic influence on complement expression [162]. These elevated levels in patients with MS may be associated with MRI injuries, clinical disability, and light levels of the neurofilament chain (NfL), indicative of nerve damage [162].
Thus, there is a potential link between intrathecal cholinergic activity and complement activation. In addition, the analysis of the soluble complement 2 receptor (sCR2), which can act as an inhibitor instead of an activator in the regulation of C3 function, was found to be increased in the CSF of patients with MSRR and SPMS, suggesting a modulatory function in complement activation downstream of C3 [161].
In research on animal models, the inhibition of AChE activity was applied with a positive effect in the treatment of MS [163], a fact confirmed by another study that indicated the activity of ACh as anti-inflammatory [164]. In addition, the use of acetylcholinesterase inhibitors (AChEI) led to a reduction in lymphocyte proliferation and the secretion of pro-inflammatory cytokines in experimental autoimmune encephalomyelitis (EAE), and treatment with ACh resulted in a reduction in the levels of cytokines such as TNF-α, IL-1β, and IL-6 in the blood of mice [165], confirming the interaction of ACh and α7nAChR in monocytes, neutrophils, and macrophages in the mechanism of inflammation reduction.
Confirming this hypothesis, in rats with neuroinflammation induced by intracerebroventricular injection of lipopolysaccharide (LPS), the action of the α7nAChR receptor in anti-inflammatory reactions is visible, and may be a therapeutic target for the control of this scenario [166]. In addition, Nizri and Brenner [143] reported the effects of cholinergic agents in the therapy of EAE disease, which was dependent on the action of the receptor to reduce clinical severity, relating clinical treatment to modulation of the cholinergic system.
Several findings prove this relationship in neurological diseases and associate different variables. The presence of IFN-β in rats with the demyelinating disease was able to reduce the activities of AChE [167] and the use of immunosuppressants such as cyclosporine A reduced the enzymatic action in the cerebral cortex, striatum, hippocampus, cerebellum, and point [168]. Thus, the natural AChE inhibitor Huperzine A was used in rats with cerebral hypoperfusion (hypoxia) in order to reduce the existing chronic inflammation, also capable of acting on myelin repairs and memory deficits. Confirming that the interruption of the anti-inflammatory effects triggered by ACh via stimulation of α7nAChR can exacerbate the uncontrolled and persistent inflammation of demyelinating diseases, such as MS [169].
Purinergic System and Pathophysiology of MS
The study by Burnstock [170] reported the involvement of the P2X7 (P2X7R) and P2Y (P2YR) receptors in the pathology of MS, given their role in the remyelination processes in neurodegenerative diseases. In addition, there is a wide expression of the P2X7R receptor in the brain and a close association with other inflammatory and demyelinating pathological changes that affect the CNS [171]. Thus, acting in an integrated manner with P2X7R, P2Y1R, A2AR, ATP seems to contribute to the neurotoxicity of this environment. Cunha [172] reported the direct relationship of A2AR in neurodegeneration, since this damage positively regulates this receptor and increases the release of ATP, triggering an increase in receptor activity and synaptotoxicity.
Situations such as neuroinflammation, reactive astrogliosis, synaptic dysfunction, and increased susceptibility can be caused by the direct or indirect activation of these receptors. Thus, the positive regulation of A2AR can still cause loss of function of astrocytes and microglia, since this activity causes the interruption of metabolic support, triggering the progression of brain damage. Therefore, due to their involvement in neurotransmission, inflammation, and immune responses, the study of these transmitters and receptors can assist in the development and application of a therapy for MS [173].
Experiments in rats indicated an increase in ATP hydrolysis after brain injuries during EAE. Thus, the modulatory action of ATP, ADP, and AMP is related to a decrease in ATP levels and an increase in serum AMP. The physiological and pathological relationships of these adenine nucleotides demonstrate a sensitive communication network between the brain and the vascular system, directly interfering in the development of human neuropathologies [131].
From this perspective, studies suggest the involvement of ecto-5′-nucleotidase in the neuroinflammatory process linked to MS [174], as well as its activities altered in the demyelination process. This process is also repeated with NTPDase in both synaptosomes and in platelets, these data were obtained in research with mice demyelinated by ethidium bromide and treated with INF-B [135]. Thus, important thrombo regulatory enzymes are visualized associated with pathological conditions of the CNS, especially after demyelination of the CNS [175], added to treatment with a drug capable of regulating the hydrolysis of the adenine nucleotide in platelets and synaptosomes.
Changes such as these illustrate the involvement of these enzymes in axonal damage and in the demyelination process in MS [175], given that evidence also indicates a reduction in E-NPP activity and an increase in AMP hydrolysis in the platelets of rats and that after The modulation of this hydrolysis occurred as well as enzymatic modification of E-NPP and ecto-5′-nucleotide [176]. On the other hand, studies include an increased production of adenosine to an anti-inflammatory response in rats with EAE [174], as well as the role of IL-10 in its recovery, since the increased levels of this interleukin in the CNS, being able to infiltrate immune cells and stimulating the expression of CXCL12 in astrocytes, act to restrict inflammation and promote myelin repair [177].
Regarding purinergic receptors, P2X7R and P2Y1R stood out for their activity in oligodendrocyte progenitor cells [178], implying in the processes of myelination and remyelination. Thus, it was demonstrated that overexpression of P2X7R, due to sensitivity to ATP, acts directly in the destruction of oligodendrocytes and in the cell death process, being circumvented by treatment with P2X7 antagonists in the pathology of chronic EAE significantly reducing demyelination [179]. Grygorowicz et al. [180] sharing this therapy also found a reduction in astrogliosis and an improvement in the neurological symptoms of EAE, results possibly from the reduction in the release of potentially neurotoxic substances such as ATP and glutamate, justifying how astroglia can influence the pathogenesis of both EAE as well as MS by purinergic and glutamatergic mechanisms [135].
ADP signaling during EAE is responsible for the negative gene and protein regulation of NTPDases2, P2Y1, and P2Y12, being responsible for increasing the severity of symptoms [181]. However, although complex and contradictory, the effects of A2AR on neuroinflammation and neuronal damage seem to be a possible therapeutic target for MS. Liu et al. [182] reported an attenuation of EAE by activating this receptor, which is also associated with an increase in intracellular Ca + 2 in lymphocytes, regulating the inflammatory process by reducing the infiltration of inflammatory cells and demyelination in the spinal cord. Due to its complexity, a dual role is perceived in autoimmune neuroinflammation, since the protective role of the receptor occurs only in the initial stages of EAE; as the disease progresses, its activation triggers the permanence of tissue damage in the inflamed CNS [183].
Thus, caffeine, a non-selective antagonist for adenosine receptors, has been highlighted in the literature as a chronic treatment for both EAE and MS in animal models. The protective effect is associated with positive regulation of A1R in the brain and an increase in the level of extracellular adenosine [184], unrelated to A2AR [185]. Thus, coffee consumption becomes one of the important topics in the pathophysiology of MS in order to reduce brain damage [186], supporting the view that extracellular nucleotides and purine nucleosides play fundamental roles in the immune system and in the inflammatory response of MS, as well as EAE, and can assist in understanding the pathophysiological complex of this disorder and the development of effective therapies.
Cholinergic Anti-inflammatory Pathway as MS Therapy
Aiming at a modulating therapy of the inflammatory process, recent clinical studies have introduced the intrinsic cholinergic anti-inflammatory pathway in different pathologies. Several studies have linked the inactivating enzymatic activity of ACh with markers of pathological conditions due to the contribution in pathways that modulate inflammatory and immunological responses [135]. The anti-inflammatory cholinergic pathway is modulated by stimulation of the acetylcholine alpha 7 nicotinic receptor (α7 nAChR) by nicotine or ACh, resulting in neural inhibition of inflammation [187]. In addition, Fujii et al. [142] suggested cholinergic manipulation as a regulator of immune function, using specific agonists and antagonists for immune cells. However, the action of α7 nAChR must be taken into account, as well as the possible resistance to cholinesterase 3 inhibitors (RIC-3) expressed in the immune system, which directly implies in MS therapy (Table 1).
The ACh molecule and the activation of α7nAChRs are able to suppress T cell differentiation [142], reduce the levels of TNF-α and IL-1 in macrophages [188], plus a reduction in pro-inflammatory cytokines [143] and a blocking of NFkB activity in monocytes and macrophages [155]. In T cells, the actions of ACh and nicotinic receptors are able to reduce the levels of proinflammatory cytokines [145] and the activity of Th1 and Th17, while in B cells, they act to decrease the production of antibodies [143]. Being able to reduce the intensity of inflammation by modulating the infiltration of monocytes and macrophages, ACh also reduces the autoimmune responses in NK cells [136] (Fig. 3) (Table 1).

Relationship of nicotinic receptors in immune and neuronal cells. Cholinergic activity in immune and neuronal cells, highlighted in the text, can be pro-inflammatory and anti-inflammatory. Thus, in macrophages, the activation of nAChRs by the ACh molecule is observed, providing a blockade of the secretion of pro-inflammatory cytokines such as TNF-a and IL-1, as well as a blockade of NFkB and lymphocyte differentiation. In T cells, there is the expression of both receptors and ACh modulates anti-inflammatory actions by nAChRs, blocking the release of pro-inflammatory cytokines and the activation of Th1 and Th17. However, the activation of mAChRs provides an increase in the concentration of c-fos, NOS, and IL-2. In B cells, nAChRs act by blocking pro-inflammatory cytokines and the production of antibodies, reducing inflammation, and in oligodendrocytes, mAChRs have neuroplastic and control action on cell proliferation and differentiation
In neuronal cells, such as oligodendrocytes, ACh and mAChRs act to control cell proliferation and differentiation [189], in addition to the neuroplastic action of this neurotransmitter on axons [144]. The muscarinic receptor also acts in association with ACh in T cells; however, it provides an increase in inflammatory cytokines [143, 145] and an increase in levels of c-fos, IL-2, nitrogen oxide species (NOS) [188] (Fig. 3). Thus, it is suggested that the increased plasma and tissue concentration of AChE and BuChE can be read as a marker of low-grade systemic inflammation, both local and systemic, given the inactivation of ACh [139]. In addition, produced by the liver, BuChE has still been closely associated with a prognosis of diseases that cause neurodegeneration in addition to inflammation [190] (Table 1).
Understanding the mechanisms of action and the regulators of the relationship between the immune system and the cholinergic may be responsible for the application of new immunomodulatory agents or extend the application of cholinergic agents [191]. In addition, it is also necessary to analyze the responses generated by the system, since the activation of mAChR, in contrast to the nicotinic receptor, can increase the proliferation of T cells and the production of pro-inflammatory mediators. Thus, both pro-effects and anti-inflammatory actions can be generated, being dependent on the activated receptor [143] (Table 1).
Purinergic Modulation as Therapy in MS
In the purinergic system, the activity of ATP and its receptors is observed in immune cells, such as macrophages and monocytes. In this way, when activating P2X7, it is able to increase the release of IL-1α; IL-1β; IL-6; IL-18; TNF-α and enable the activation of NFκB [192]. Activating the P2X4 receptor in macrophages is responsible for the release of prostaglandin E2 (PGE2) [193], myelin phagocytosis, and brain-derived neurotrophic factor (BDNF), favoring remyelination [194], in addition to providing lymphocyte apoptosis with P2X7 activation [193] (Fig. 4) (Table 2).

Modulation of purinergic receptors in immune and neuronal cells. Purinergic receptors are present in a large majority of cells, including immune and neuronal cells. Thus, with inflammation and cell lysis mediated by viral infection, extracellular ATP is released and overactivation of P2 receptors, in an analogous manner, with the metabolization of this nucleotide, the adenosine nucleoside (Ado) can be formed that acts on the P1R. Thus, in myeloid-derived suppressor cells, P2X7R increases the release of ROS, IL-1β, TGF-β, arginase-1. B cells express P2X which, activated by Ado, provide the blocking of NFkB, IKK, cAMP, and PKA. Expressing P2X7, its activation in T cells induces apoptosis, A2AR acts in a pro-inflammatory and cytotoxic way, while A2BR blocks the secretion of IL-2. In macrophages, with the expression of P1R and P2R, while P2X4R acts on PGE2 and BDNF, P2X7R acts by increasing the release of pro-inflammatory cytokines and the activation of NFkB. In addition, with anti-inflammatory actions, A2AR blocks the secretion of TNF-a, and the release of nitrogen and oxygen intermediates, while A2BR acts on angiogenesis and on increasing VEGF. In oligodendrocytes, P2X7R provides greater permeability of calcium, differentiation of myelin and excitotoxicity. A2AR causes immunosuppression and A2BR causes axonal dysfunction and an increase in neurotrophic factors
In myeloid-derived suppressor cells, P2X7 triggers the release of reactive oxygen species (ROS); IL-1β; arginase-1; TGF-β [193] and the activation of the inflammasome [194], already in neuronal cells such as oligodendrocyte, the activity of P2X7 by the nucleotide stimulates the differentiation and formation of myelin, in addition to increasing the permeability to calcium, causing excitotoxicity [189] and contributing to excitotoxicity the death of primary oligodendrocytes [195]. On the other hand, adenosine upon activating A2BR results in an increase in cells involved in the immunosuppression process [193]. In addition to the increase in intracellular calcium, the proliferation of myelin by oligodendrocytes, the reduction of oligodendrocyte precursor cells (OPCs) [125], as well as the increase in neurotrophic factors and axonal dysfunction by activating A2AR [128, 130] (Fig. 4) (Table 2).
The activity of the nucleoside in monocytes and macrophages is capable of reducing the levels of TNF-α and producing active nitrogen and oxygen intermediates [196]. In the case of A2AR activation in macrophages, vascular endothelial growth factor (VEGF) secretion and angiogenesis are also increased [193]. In T cells, A2AR activation reduces cytokine activation, proliferation, and release, as well as the resulting cytotoxic and A2BR decreases levels of IL-2 production [196]. Adenosine activity in B cells, on the other hand, triggers NFkB blockade and increases adenosine 3′, cyclic 5′-monophosphate (cAMP) and cAMP-dependent protein kinase A (PKA), inhibiting IkB kinase (IKK) phosphorylation [192, 196] [183, 187] (Fig. 4) (Table 2).
Future Therapeutic Proposals
Compiling the acquired knowledge, it is possible to score both ACh and purine actions in different types of cells. Thus, the positive or negative modulation of receptors or changes in the levels of extracellular molecules can result in functions that result in a better prognosis in the pathogenesis of MS. Thus, future studies may apply therapies that use P2X7 receptor antagonists, as is the case of study [197], which demonstrated beneficial anti-inflammatory effects in neurodegeneration in animal models, since the activation of P2X7 is closely related to the worsening of neurodegeneration [189, 192,193,194,195] (Table 2). In addition, the use of nicotinic or muscarinic receptor agonists is associated with a reduction in the inflammatory condition and a modulation of immune cells [142, 145, 155, 164, 188, 189] (Table 1). Finally, A2B receptor agonists, as well as an extracellular increase in adenosine, can result in imusupression and reduced release of pro-inflammatory cytokines, as well as release of anti-inflammatory substances [125, 193] (Table 2). Thus, more clinical studies are needed for the application of this therapy in patients with MS, justified by its urgency in a COVID-19 pandemic scenario, given its relationship with the severity of the disease.
Conclusion
Therefore, considering the pro-inflammatory context resulting from SARS-CoV-2 infection, the persistence of this condition can spread myelin degeneration and lead to worsening of MS or even trigger the manifestation of the disease. In mild cases of COVID-19, the exacerbation of MS can be temporary; however, the risk of disease recurrence or progression can be significant. Still, treatment with DMD, immunosuppressants, and corticosteroids for long periods can influence the immune response and can lead to complicated cases of COVID-19. Thus, the modulation of purinergic receptors and the activation of the cholinergic anti-inflammatory pathway can act as possible therapies to reduce neuronal inflammation. From this perspective, the use of P2X7 antagonists, ACh agonists, and P1 receptor agonists or greater adenosine bioavailability stands out.
References
Chen G, Wu D, Guo W et al (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 130:2620–2629. https://doi.org/10.1172/JCI137244
Thakolwiboon S, Zhao-Fleming H, Pan J et al (2020) Disease-modifying therapies during the COVID-19 outbreak: a narrative review of international and national recommendations. Int J MS Care 22:151–157. https://doi.org/10.7224/1537-2073.2020-037
Willis MD, Robertson NP (2020) Multiple sclerosis and the risk of infection: considerations in the threat of the novel coronavirus, COVID-19/SARS-CoV-2. J Neurol 267:1567–1569. https://doi.org/10.1007/s00415-020-09822-3
Zila I, Mokra D, Kopincova J et al (2017) Vagal-immune interactions involved in cholinergic anti-inflammatory pathway. Physiol Res 66:S139–S145
Yamada M, Ichinose M (2018) The cholinergic pathways in inflammation: a potential pharmacotherapeutic target for COPD. Front Pharmacol 9
World Health Organization. Modes of transmission of virus causing COVID‐19: implications for IPC precaution recommendations. In: https://www.who.int/news-room/commentaries/detail/modes-of-transmission-of-virus-causing-covid-19-implications-for-ipc-precaution-recommendations. Accessed 17 May 2021
Steyn E, Mohamed Z, Husselman C (2013) Non-invasive vagus nerve stimulation for the treatment of acute asthma exacerbations - results from an initial case series Int J Emerg Med 6 https://doi.org/10.1186/1865-1380-6-7
Trapp BD, Peterson J, Ransohoff RM et al (1998) Axonal Transection in the Lesions of Multiple Sclerosis. N Engl J Med 338:278–285. https://doi.org/10.1056/nejm199801293380502
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG (2000) Multiple sclerosis. N Engl J Med 343:938–952. https://doi.org/10.1056/NEJM200009283431307
McFarland HF, Martin R (2007) Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol 8:913–919
Lublin FD, Reingold SC (1996) Defining the clinical course of multiple sclerosis: results of an international survey. Neurology 46:907–911
Mulakayala N, Rao P, Iqbal J et al (2013) Synthesis of novel therapeutic agents for the treatment of multiple sclerosis: a brief overview. Eur J Med Chem 60:170–186
Hemmer B, Selter, (2013) Update on immunopathogenesis and immunotherapy in multiple sclerosis. ImmunoTargets Ther 2:21. https://doi.org/10.2147/itt.s31813
Gran B, Hemmer B, Martin R (1999) Molecular mimicry and multiple sclerosis - a possible role for degenerate t cell recognition in the induction of autoimmune responses. J Neural Transm Suppl 19–31 https://doi.org/10.1007/978-3-7091-6369-6_3
Wucherpfennig KW, Strominger JL (1995) Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80:695–705. https://doi.org/10.1016/0092-8674(95)90348-8
Hemmer B, Archelos JJ, Hartung HP (2002) New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 3:291–301. https://doi.org/10.1038/nrn784
O’Connor KC, Bar-Or A, Hafler DA (2001) The neuroimmunology of multiple sclerosis: possible roles of T and B lymphocytes in immunopathogenesis. J Clin Immunol 21:81–92. https://doi.org/10.1023/A:1011064007686
Serafini B, Rosicarelli B, Magliozzi R et al (2004) Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 14:164–174. https://doi.org/10.1111/j.1750-3639.2004.tb00049.x
Amadio S, Montilli C, Magliozzi R et al (2010) P2Y12 receptor protein in cortical gray matter lesions in multiple sclerosis. Cereb Cortex 20:1263–1273. https://doi.org/10.1093/cercor/bhp193
Lucchinetti C, Brock W, Parisi J et al (2000) Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination Ann Neurol 47 https://doi.org/10.1002/1531-8249
Kinzel S, Weber MS (2016) B cell-directed therapeutics in multiple sclerosis: rationale and clinical evidence. CNS Drugs 30:1137–1148
Wang D, Hu B, Hu C et al (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA - J Am Med Assoc 323:1061–1069. https://doi.org/10.1001/jama.2020.1585
Mao L, Jin H, Wang M et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 77:683–690. https://doi.org/10.1001/jamaneurol.2020.1127
Giacomelli A, Pezzati L, Conti F et al (2020) Self-reported olfactory and taste disorders in patients with severe acute respiratory coronavirus 2 infection: a cross-sectional study. Clin Infect Dis 71:889–890
Filatov A, Sharma P, Hindi F, Espinosa PS (2020) Neurological complications of coronavirus disease (COVID-19): encephalopathy. Cureus 12 https://doi.org/10.7759/cureus.7352
Pleasure SJ, Green AJ, Josephson SA (2020) The spectrum of neurologic disease in the severe acute respiratory syndrome coronavirus 2 pandemic infection: neurologists move to the frontlines. JAMA Neurol 77:679–680
Saleki K, Banazadeh M, Saghazadeh A, Rezaei N (2020) The involvement of the central nervous system in patients with COVID-19. Rev Neurosci 31:453–456. https://doi.org/10.1515/revneuro-2020-0026
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11:995–998
Paniz-Mondolfi A, Bryce C, Grimes Z et al (2020) Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol 92:699–702
Netland J, Meyerholz DK, Moore S et al (2008) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol 82:7264–7275. https://doi.org/10.1128/jvi.00737-08
Zubair AS, McAlpine LS, Gardin T et al (2020) Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a review. JAMA Neurol 77:1018–1027
Montalvan V, Lee J, Bueso T, et al (2020) Neurological manifestations of COVID-19 and other coronavirus infections: a systematic review. Clin. Neurol. Neurosurg. 194
Sankowski R, Mader S, Valdés-Ferrer SI (2015) Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosci 9. https://doi.org/10.3389/fncel.2015.00028
Briguglio M, Bona A, Porta M et al (2020) Disentangling the hypothesis of host dysosmia and SARS-CoV-2: the bait symptom that hides neglected neurophysiological routes. Front Physiol 11 https://doi.org/10.3389/fphys.2020.00671
Alberti P, Beretta S, Piatti M et al (2020) Guillain-Barré syndrome related to COVID-19 infection. Neurol Neuroimmunol NeuroInflammation 7. https://doi.org/10.1212/NXI.0000000000000741
Zanin L, Saraceno G, Panciani PP et al (2020) SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir (Wien) 162:1491–1494. https://doi.org/10.1007/s00701-020-04374-x
Moriguchi T, Harii N, Goto J et al (2020) A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis 94:55–58. https://doi.org/10.1016/j.ijid.2020.03.062
Palao M, Fernández-Díaz E, Gracia-Gil J et al (2020) Multiple sclerosis following SARS-CoV-2 infection Mult Scler Relat Disord 45 https://doi.org/10.1016/j.msard.2020.102377
Donati D (2020) Viral infections and multiple sclerosis. Drug Discov Today Dis Model 32:27–33
Andersen O, Lygner PE, Bergström T et al (1993) Viral infections trigger multiple sclerosis relapses: a prospective seroepidemiological study. J Neurol 240:417–422. https://doi.org/10.1007/BF00867354
McKay KA, Jahanfar S, Duggan T et al (2017) Factors associated with onset, relapses or progression in multiple sclerosis: a systematic review. Neurotoxicology 61:189–212. https://doi.org/10.1016/j.neuro.2016.03.020
Marrodan M, Alessandro L, Farez MF, Correale J (2019) The role of infections in multiple sclerosis. Mult Scler J 25:891–901
Dendrou CA, Fugger L, Friese MA (2015) Immunopathology of multiple sclerosis. Nat Rev Immunol 15:545–558
Barres BA, Hart IK, Coles HSR et al (1992) Cell death and control of cell survival in the oligodendrocyte lineage. Cell 70:31–46. https://doi.org/10.1016/0092-8674(92)90531-G
Compston A, Coles A (2008) Multiple sclerosis. Lancet 372:1502–1517
Frohman EM, Racke MK, Raine CS (2006) Multiple sclerosis — the plaque and its pathogenesis. N Engl J Med 354:942–955. https://doi.org/10.1056/nejmra052130
Fischer MT, Wimmer I, Höftberger R et al (2013) Disease-specific molecular events in cortical multiple sclerosis lesions. Brain 136:1799–1815. https://doi.org/10.1093/brain/awt110
Lassmann H, Van Horssen J, Mahad D (2012) Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 8:647–656
Link H (1998) The cytokine storm in multiple sclerosis. Mult Scler 4:12–15
Imitola J, Chitnis T, Khoury SJ (2005) Cytokines in multiple sclerosis: from bench to bedside. Pharmacol Ther 106:163–177
Goverman J (2009) Autoimmune T cell responses in the central nervous system. Nat Rev Immunol 9:393–407
Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA (2004) Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 199:971–979. https://doi.org/10.1084/jem.20031579
Minagar A, Alexander JS (2003) Blood-brain barrier disruption in multiple sclerosis. Mult Scler 9:540–549
Pérez-Cerdá F, Sánchez-Gómez MV, Matute C (2016) The link of inflammation and neurodegeneration in progressive multiple sclerosis. Mult Scler Demyelinating Disord 1:1–8. https://doi.org/10.1186/s40893-016-0012-0
Milo R, Korczyn AD, Manouchehri N, Stüve O (2020) The temporal and causal relationship between inflammation and neurodegeneration in multiple sclerosis. Mult Scler J 26:876–886
Magliozzi R, Howell O, Vora A et al (2007) Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130:1089–1104. https://doi.org/10.1093/brain/awm038
Zorec R, Županc TA, Verkhratsky A (2019) Astrogliopathology in the infectious insults of the brain. Neurosci Lett 689:56–62
Pekny M, Pekna M, Messing A et al (2016) Astrocytes: a central element in neurological diseases. Acta Neuropathol 131:323–345
Rosciszewski G, Cadena V, Murta V et al (2018) Toll-like receptor 4 (TLR4) and triggering receptor expressed on myeloid cells-2 (TREM-2) activation balance astrocyte polarization into a proinflammatory phenotype. Mol Neurobiol 55:3875–3888. https://doi.org/10.1007/s12035-017-0618-z
Wheeler DL, Sariol A, Meyerholz DK, Perlman S (2018) Microglia are required for protection against lethal coronavirus encephalitis in mice. J Clin Invest 128:931–943. https://doi.org/10.1172/JCI97229
Zhou Z, Ren L, Zhang L et al (2020) Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27 883 890.e2 https://doi.org/10.1016/j.chom.2020.04.017
Moore JB, June CH (2020) Cytokine release syndrome in severe COVID-19. Science (80-) 368:473–474
Azkur AK, Akdis M, Azkur D et al (2020) Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy Eur J Allergy Clin Immunol 75:1564–1581
Coperchini F, Chiovato L, Croce L et al (2020) The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev 53:25–32
Diao B, Wang C, Tan Y et al (2020) Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol 11. https://doi.org/10.3389/fimmu.2020.00827
Giamarellos-Bourboulis EJ, Netea MG, Rovina N et al (2020) Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 27:992-1000.e3. https://doi.org/10.1016/j.chom.2020.04.009
Dosch SF, Mahajan SD, Collins AR (2009) SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-κB pathway in human monocyte macrophages in vitro. Virus Res 142:19–27. https://doi.org/10.1016/j.virusres.2009.01.005
Berger JR (2020) COVID-19 and the nervous system. J Neurovirol 26:143–148
Zindler E, Zipp F (2010) Neuronal injury in chronic CNS inflammation. Best Pract Res Clin Anaesthesiol 24:551–562
Yadav SK, Mindur JE, Ito K, Dhib-Jalbut S (2015) Advances in the immunopathogenesis of multiple sclerosis. Curr Opin Neurol 28:206–219
Raony Í, Figueiredo CS, Pandolfo de P et al (2020) Psycho-neuroendocrine-immune interactions in COVID-19: potential impacts on mental health. Front Immunol 11 https://doi.org/10.3389/fimmu.2020.01170
Wu Y, Xu X, Chen Z et al (2020) Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun 87:18–22
Calsolaro V, Edison P (2016) Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimer’s Dement 12:719–732
Heneka MT, Golenbock D, Latz E et al (2020) Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimer’s Res Ther 12. https://doi.org/10.1186/s13195-020-00640-3
Boziki MK, Mentis AFA, Shumilina M et al (2020) COVID-19 immunopathology and the central nervous system: implication for multiple sclerosis and other autoimmune diseases with associated demyelination Brain Sci 10 https://doi.org/10.3390/brainsci10060345
Kurtzke JF (1993) Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev 6:382–427
Serrano-Castro PJ, Estivill-Torrús G, Cabezudo-García P et al (2020) Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: a delayed pandemic? Neurologia 35:245–251
Jung YJ, Tweedie D, Scerba MT, Greig NH (2019) Neuroinflammation as a factor of neurodegenerative disease: thalidomide analogs as treatments. Front Cell Dev Biol 7
Wang K, Song F, Fernandez-Escobar A et al (2018) The properties of cytokines in multiple sclerosis: pros and cons. Am J Med Sci 356:552–560
Shindler KS, Kenyon LC, Dutt M et al (2008) Experimental optic neuritis induced by a demyelinating strain of mouse hepatitis virus. J Virol 82:8882–8886. https://doi.org/10.1128/jvi.00920-08
Bender SJ, Weiss SR (2010) Pathogenesis of murine coronavirus in the central nervous system. J Neuroimmune Pharmacol 5:336–354
Kishore A, Kanaujia A, Nag S et al (2013) Different mechanisms of inflammation induced in virus and autoimmune-mediated models of multiple sclerosis in C57BL6 mice Biomed Res Int 2013 https://doi.org/10.1155/2013/589048
Duffy L, O’Reilly SC (2016) Toll-like receptors in the pathogenesis of autoimmune diseases: Recent and emerging translational developments. ImmunoTargets Ther 5:69–80
Libbey JE, Cusick MF, Fujinami RS (2014) Role of pathogens in multiple sclerosis. Int Rev Immunol 33:266–283
Houtman JJ, Hinze HC, Fleming JO (1995) Demyelination induced by murine coronavirus JHM infection of congenitally immunodeficient mice Advances in Experimental Medicine and Biology Springer New York LLC 159 163
Boucher A, Desforges M, Duquette P, Talbot PJ (2007) Long-term human coronavirus-myelin cross-reactive T-cell clones derived from multiple sclerosis patients. Clin Immunol 123:258–267. https://doi.org/10.1016/j.clim.2007.02.002
Xia H, Lazartigues E (2008) Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem 107:1482–1494
Iroegbu JD, Ifenatuoha CW, Ijomone OM (2020) Potential neurological impact of coronaviruses: implications for the novel SARS-CoV-2. Neurol Sci 41:1329–1337. https://doi.org/10.1007/s10072-020-04469-4
Mohammadi S, Moosaie F, Aarabi MH (2020) Understanding the immunologic characteristics of neurologic manifestations of SARS-CoV-2 and potential immunological mechanisms. Mol Neurobiol 57:5263–5275
Mares J, Hartung HP (2020) Multiple sclerosis and COVID-19. Biomed Pap 164:217–225
Zheng C, Kar I, Chen CK et al (2020) Multiple sclerosis disease-modifying therapy and the COVID-19 pandemic: implications on the risk of infection and future vaccination. CNS Drugs 34:879–896. https://doi.org/10.1007/s40263-020-00756-y
Kieseier BC (2011) The mechanism of action of interferon-β in relapsing multiple sclerosis. CNS Drugs 25:491–502
Hung IFN, Lung KC, Tso EYK et al (2020) Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet 395:1695–1704. https://doi.org/10.1016/S0140-6736(20)31042-4
Riederer F (2017) Ocrelizumab versus placebo in primary progressive multiple sclerosis. J fur Neurol Neurochir und Psychiatr 18:30–31
Cross AH, Naismith RT (2014) Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med 275:350–363
Fox RJ, Miller DH, Phillips JT et al (2012) Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 367:1087–1097. https://doi.org/10.1056/nejmoa1206328
Gilli F, Li L, Royce DB et al (2017) Treatment of Theiler’s virus-induced demyelinating disease with teriflunomide. J Neurovirol 23:825–838. https://doi.org/10.1007/s13365-017-0570-8
Bowen JD, Brink J, Brown TR et al (2020) COVID-19 in MS: Initial observations from the Pacific Northwest. Neurol Neuroimmunol neuroinflammation 7 https://doi.org/10.1212/NXI.0000000000000783
Luca B, Tommaso G, Bavaro DF, et al (2020) Seroconversion and indolent course of COVID-19 in patients with multiple sclerosis treated with fingolimod and teriflunomide. J Neurol Sci 416
Maghzi AH, Houtchens MK, Preziosa P et al (2020) COVID-19 in teriflunomide-treated patients with multiple sclerosis. J Neurol 267:2790–2796. https://doi.org/10.1007/s00415-020-09944-8
Möhn N, Saker F, Bonda V et al (2020) Mild COVID-19 symptoms despite treatment with teriflunomide and high-dose methylprednisolone due to multiple sclerosis relapse. J Neurol 267:2803–2805
Nesbitt C, Rath L, Yeh WZ, et al (2020) MSCOVID19: Using social media to achieve rapid dissemination of health information. Mult Scler Relat Disord 45
Naismith RT, Wundes A, Ziemssen T et al (2020) Diroximel fumarate demonstrates an improved gastrointestinal tolerability profile compared with dimethyl fumarate in patients with relapsing–remitting multiple sclerosis: results from the randomized, double-blind, phase III EVOLVE-MS-2 study. CNS Drugs 34:185–196. https://doi.org/10.1007/s40263-020-00700-0
Diebold M, Altersberger V, Décard BF et al (2019) A case of progressive multifocal leukoencephalopathy under dimethyl fumarate treatment without severe lymphopenia or immunosenescence. Mult Scler J 25:1682–1685. https://doi.org/10.1177/1352458519852100
Mantero V, Abate L, Basilico P, et al (2020) COVID‐19 in dimethyl fumarate‐treated patients with multiple sclerosis. J Neurol
Cohen JA, Barkhof F, Comi G et al (2010) Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 362:402–415. https://doi.org/10.1056/nejmoa0907839
Paolicelli D, Manni A, Iaffaldano A, Trojano M (2020) Efficacy and safety of oral therapies for relapsing-remitting multiple sclerosis. CNS Drugs 34:65–92
National Multiple Sclerosis Society No Title. https://www.nationalmssociety.org/what-you-need-to-know-about-coronavirus-(COVID-19)/DMT-guidelines-for-coronavirus-(COVID-19)-and. Accessed 10 Apr 2021
Gomez-Mayordomo V, Montero-Escribano P, Matías-Guiu JA et al (2021) Clinical exacerbation of SARS-CoV2 infection after fingolimod withdrawal. J Med Virol 93:546–549. https://doi.org/10.1002/jmv.26279
Valencia-Sanchez C, Wingerchuk DM (2020) A fine balance: immunosuppression and immunotherapy in a patient with multiple sclerosis and COVID-19. Mult Scler Relat Disord 42. https://doi.org/10.1016/j.msard.2020.102182
Sigrist CJ, Bridge A, Mercier Le P (2020) A potential role for integrins in host cell entry by SARS-CoV-2 Antiviral Res 177 https://doi.org/10.1016/j.antiviral.2020.104759
Giovannoni G, Hawkes C, Lechner-Scott J, et al (2020) The COVID-19 pandemic and the use of MS disease-modifying therapies. Mult Scler Relat Disord 39
Simões JLB, Bagatini MD (2021) Purinergic signaling of ATP in COVID-19 associated Guillain-Barré Syndrome. J Neuroimmune Pharmacol 16:48–58
Hauser SL, Bar-Or A, Comi G et al (2017) Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 376:221–234. https://doi.org/10.1056/nejmoa1601277
Derfuss Tobias Serum immunoglobulin levels and risk of serious infections in the pivotal phase III trials of ocrelizumab in multiple sclerosis and their open-label extensions. Paper presented at the 2019 European Committee for Treatment and Research in Multiple Sclerosi. https://onlinelibrary.ectrims-congress.eu/ectrims/2019/stockholm/279399/tobias.derfuss.serum.immunoglobulin.levels.and.risk.of.serious.infecti. Accessed 27 May 2021
Chen C, Zhang XR, Ju ZY, He WF (2020) Advances in the research of mechanism and related immunotherapy on the cytokine storm induced by coronavirus disease 2019. Zhonghua Shao Shang Za Zhi 36:471–475. https://doi.org/10.3760/cma.j.cn501120-20200224-00088
Safavi F, Nourbakhsh B, Azimi AR (2020) B-cell depleting therapies may affect susceptibility to acute respiratory illness among patients with multiple sclerosis during the early COVID-19 epidemic in Iran. Mult Scler Relat Disord 43 https://doi.org/10.1016/j.msard.2020.102195
Prompetchara E, Ketloy C, Palaga T (2020) Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pacific J Allergy Immunol 38:1–9
Meca-Lallana V, Aguirre C, Río B et al (2020) COVID-19 in 7 multiple sclerosis patients in treatment with ANTI-CD20 therapies. Mult Scler Relat Disord 44. https://doi.org/10.1016/j.msard.2020.102306
Quinti I, Lougaris V, Milito C et al (2020) A possible role for B cells in COVID-19? Lesson from patients with agammaglobulinemia. J Allergy Clin Immunol 146:211-213.e4. https://doi.org/10.1016/j.jaci.2020.04.013
Gross CC, Ahmetspahic D, Ruck T et al (2016) Alemtuzumab treatment alters circulating innate immune cells in multiple sclerosis. Neurol Neuroimmunol NeuroInflammation 3. https://doi.org/10.1212/NXI.0000000000000289
Guevara C, Villa E, Cifuentes M, et al (2020) Mild COVID-19 infection in a patient with multiple sclerosis and severe depletion of T-lymphocyte subsets due to alemtuzumab. Mult Scler Relat Disord 44
Carandini T, Pietroboni AM, Sacchi L et al (2020) Alemtuzumab in multiple sclerosis during the COVID-19 pandemic: a mild uncomplicated infection despite intense immunosuppression. Mult Scler J 26:1268–1269
Dalla Costa G, Leocani L, Montalban X et al (2020) Real-time assessment of COVID-19 prevalence among multiple sclerosis patients: a multicenter European study. Neurol Sci 41:1647–1650. https://doi.org/10.1007/s10072-020-04519-x
Amadio S, Apolloni S, D’Ambrosi N, Volonté C (2011) Purinergic signalling at the plasma membrane: a multipurpose and multidirectional mode to deal with amyotrophic lateral sclerosis and multiple sclerosis. J Neurochem 116:796–805
Burnstock G (2017) Purinergic signalling and neurological diseases: an update. CNS Neurol Disord - Drug Targets 16:257–265. https://doi.org/10.2174/1871527315666160922104848
Al-Rashida M, Iqbal J (2014) Therapeutic potentials of ecto-nucleoside triphosphate diphosphohydrolase, ecto-nucleotide pyrophosphatase/phosphodiesterase, ecto-5′-nucleotidase, and alkaline phosphatase inhibitors. Med Res Rev 34:703–743. https://doi.org/10.1002/med.21302
Rodrigues RJ, Almeida T, Díaz-Hernández M et al (2016) Presynaptic P2X1-3 and α3-containing nicotinic receptors assemble into functionally interacting ion channels in the rat hippocampus. Neuropharmacology 105:241–257. https://doi.org/10.1016/j.neuropharm.2016.01.022
Bono MR, Fernández D, Flores-Santibáñez F et al (2015) CD73 and CD39 ectonucleotidases in T cell differentiation: beyond immunosuppression. FEBS Lett 589:3454–3460
Ribeiro JA, Sebastião AM (2010) Modulation and metamodulation of synapses by adenosine. Acta Physiol 199:161–169
Laketa D, Savić J, Bjelobaba I et al (2015) Brain injury alters ectonucleotidase activities and adenine nucleotide levels in rat serum. J Med Biochem 34:215–222. https://doi.org/10.2478/jomb-2014-0025
Cieślak M, Komoszyński M (2011) The role of ecto-purines in inflammation leading to demyelination - new means for therapies against multiple sclerosis. Neurol Neurochir Pol 45:489–499. https://doi.org/10.1016/s0028-3843(14)60318-2
Reardon C (2016) Neuro-immune interactions in the cholinergic anti-inflammatory reflex. Immunol Lett 178:92–96
Chu C, Artis D, Chiu IM (2020) Neuro-immune interactions in the tissues. Immunity 52:464–474
Polachini CRN, Spanevello RM, Schetinger MRC, Morsch VM (2018) Cholinergic and purinergic systems: a key to multiple sclerosis? J Neurol Sci 392:8–21
Jiang W, Li D, Han R et al (2017) Acetylcholine-producing NK cells attenuate CNS inflammation via modulation of infiltrating monocytes/macrophages. Proc Natl Acad Sci U S A 114:E6202–E6211. https://doi.org/10.1073/pnas.1705491114
Antonelli T, Tomasini MC, Castellazzi M et al (2013) Biological markers in cerebrospinal fluid for axonal impairment in multiple sclerosis: acetylcholinesterase activity cannot be considered a useful biomarker. Neurol Sci 34:769–771. https://doi.org/10.1007/s10072-012-1265-9
Anglister L, Etlin A, Finkel E et al (2008) Cholinesterases in development and disease. Chem Biol Interact 175:92–100. https://doi.org/10.1016/j.cbi.2008.04.046
Das UN (2012) Acetylcholinesterase and butyrylcholinesterase as markers of low-grade systemic inflammation. Ann Hepatol 11:409–411
Renard PY, Jean L (2017) Probing the cholinergic system to understand neurodegenerative diseases. Future Med Chem 9:131–133
Dineley KT, Pandya AA, Yakel JL (2015) Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci 36:96–108
Fujii T, Mashimo M, Moriwaki Y et al (2017) Physiological functions of the cholinergic system in immune cells. J Pharmacol Sci 134:1–21
Nizri E, Brenner T (2013) Modulation of inflammatory pathways by the immune cholinergic system. Amino Acids 45:73–85
Picciotto MR, Higley MJ, Mineur YS (2012) Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76:116–129
Kawashima K, Fujii T (2003) The lymphocytic cholinergic system and its biological function. Life Sci 72:2101–2109. https://doi.org/10.1016/S0024-3205(03)00068-7
Richardson PJ, Brown SJ (1987) ATP Release from affinity-purified rat cholinergic nerve terminals. J Neurochem 48:622–630. https://doi.org/10.1111/j.1471-4159.1987.tb04138.x
Cunha RA, Ribeiro JA, Sebastião AM (1994) Purinergic modulation of the evoked release of [3H]acetylcholine from the hippocampus and cerebral cortex of the rat: role of the ectonucleotidases. Eur J Neurosci 6:33–42. https://doi.org/10.1111/j.1460-9568.1994.tb00245.x
Salgado AI, Cunha RA, Ribeiro JA (2000) Facilitation by P2 receptor activation of acetylcholine release from rat motor nerve terminals: interaction with presynaptic nicotinic receptors. Brain Res 877:245–250. https://doi.org/10.1016/S0006-8993(00)02679-2
Cunha RA, Milusheva E, Vizi ES et al (1994) Excitatory and inhibitory effects of A1 and A2A adenosine receptor activation on the electrically evoked [3H]acetylcholine release from different areas of the rat hippocampus. J Neurochem 63:207–214. https://doi.org/10.1046/j.1471-4159.1994.63010207.x
Correia-de-Sá P, Sebastião AM, Ribeiro JA (1991) Inhibitory and excitatory effects of adenosine receptor agonists on evoked transmitter release from phrenic nerve endings of the rat. Br J Pharmacol 103:1614–1620. https://doi.org/10.1111/j.1476-5381.1991.tb09836.x
Barajas-López C, Espinosa-Luna R, Zhu Y (1998) Functional interactions between nicotinic and P2X channels in short-term cultures of guinea-pig submucosal neurons. J Physiol 513:671–683. https://doi.org/10.1111/j.1469-7793.1998.671ba.x
Rodrigues RJ, Tomé AR, Cunha RA (2015) ATP as a multi-target danger signal in the brain. Front Neurosci 9
Garção P, Szabõ EC, Wopereis S et al (2013) Functional interaction between pre-synaptic α6β2-containing nicotinic and adenosine A2A receptors in the control of dopamine release in the rat striatum. Br J Pharmacol 169:1600–1611. https://doi.org/10.1111/bph.12234
Oliveira L, Timóteo MA, Correia-de-Sá P (2002) Modulation by adenosine of both muscarinic M1-facilitation and M2-inhibition of [3H]-acetylcholine release from the rat motor nerve terminals. Eur J Neurosci 15:1728–1736. https://doi.org/10.1046/j.1460-9568.2002.02020.x
Van Westerloo DJ (2010) The vagal immune reflex: a blessing from above. Wien Med Wochenschr 160:112–117
Huang G, Wang Y, Vogel P, Chi H (2015) Control of IL-17 receptor signaling and tissue inflammation by the p38α-MKP-1 signaling axis in a mouse model of multiple sclerosis. Sci Signal 8:ra24. https://doi.org/10.1126/scisignal.aaa2147
Bari M, Di Reale M, Di Nicola M et al (2016) Dysregulated homeostasis of acetylcholine levels in immune cells of RR-multiple sclerosis patients. Int J Mol Sci 17 https://doi.org/10.3390/ijms17122009
Darvesh S, LeBlanc AM, Macdonald IR et al (2010) Butyrylcholinesterase activity in multiple sclerosis neuropathology. Chem Biol Interact 187:425–431. https://doi.org/10.1016/j.cbi.2010.01.037
Kooi EJ, Prins M, Bajic N et al (2011) Cholinergic imbalance in the multiple sclerosis hippocampus. Acta Neuropathol 122:313–322. https://doi.org/10.1007/s00401-011-0849-4
Hasselmo ME (2006) The role of acetylcholine in learning and memory. Curr Opin Neurobiol 16:710–715
Lindblom RPF, Aeinehband S, Ström M et al (2016) Complement receptor 2 is increased in cerebrospinal fluid of multiple sclerosis patients and regulates C3 function. Clin Immunol 166–167:89–95. https://doi.org/10.1016/j.clim.2016.04.003
Aeinehband S, Lindblom RPF, Al NF et al (2015) Complement component C3 and butyrylcholinesterase activity are associated with neurodegeneration and clinical disability in multiple sclerosis. PLoS ONE 10 https://doi.org/10.1371/journal.pone.0122048
Li Y, Hai S, Zhou Y, Dong BR (2015) Cholinesterase inhibitors for rarer dementias associated with neurological conditions. Cochrane Database Syst Rev 2015
Nizri E, Hamra-Amitay Y, Sicsic C et al (2006) Anti-inflammatory properties of cholinergic up-regulation: a new role for acetylcholinesterase inhibitors. Neuropharmacology 50:540–547. https://doi.org/10.1016/j.neuropharm.2005.10.013
Zabrodskii PF (2011) Effect of acetylcholine on mortality of mice from sepsis and proinflammatory cytokine production. Bull Exp Biol Med 150:340–342. https://doi.org/10.1007/s10517-011-1137-y
Tyagi E, Agrawal R, Nath C, Shukla R (2010) Inhibitory role of cholinergic system mediated via α7 nicotinic acetylcholine receptor in LPS-induced neuro-inflammation. Innate Immun 16:3–13. https://doi.org/10.1177/1753425909104680
Mazzanti CM, Spanevello RM, Pereira LB et al (2006) Acetylcholinesterase activity in rats experimentally demyelinated with ethidium bromide and treated with interferon beta. Neurochem Res 31:1027–1034. https://doi.org/10.1007/s11064-006-9112-0
Mazzanti CM, Spanevello R, Ahmed M et al (2007) Cyclosporine A inhibits acetylcholinesterase activity in rats experimentally demyelinated with ethidium bromide. Int J Dev Neurosci 25:259–264. https://doi.org/10.1016/j.ijdevneu.2007.02.005
Fields RD, Dutta DJ, Belgrad J, Robnett M (2017) Cholinergic signaling in myelination. Glia 65:687–698
Burnstock G (2015) Purinergic signalling in neuroregeneration. Neural Regen Res 10:1919. https://doi.org/10.4103/1673-5374.165300
Grygorowicz T, Sulejczak D, Struzyńska L (2011) Expression of purinergic P2X7 receptor in rat brain during the symptomatic phase of experimental autoimmune encephalomyelitis and after recovery of neurological deficits. Acta Neurobiol Exp (Wars) 71:65–73
Cunha RA (2016) How does adenosine control neuronal dysfunction and neurodegeneration? J Neurochem 139:1019–1055
Schetinger MRC, Morsch VM, Bonan CD, Wyse ATS (2007) NTPDase and 5′-nucleotidase activities in physiological and disease conditions: new perspectives for human health. BioFactors 31:77–98
Lavrnja I, Laketa D, Savic D et al (2015) Expression of a second ecto-5′-nucleotidase variant besides the usual protein in symptomatic phase of experimental autoimmune encephalomyelitis. J Mol Neurosci 55:898–911. https://doi.org/10.1007/s12031-014-0445-x
Spanevello RM, Mazzanti CM, Maldonado PA et al (2007) Activities of enzymes that hydrolyze adenine nucleotides in platelets from rats experimentally demyelinated with ethidium bromide and treated with interferon-β. Life Sci 80:1109–1114. https://doi.org/10.1016/j.lfs.2006.11.049
Leal CAM, Leal DBR, Adefegha SA et al (2016) Effects of chlorogenic acid on adenine nucleotides hydrolyzing enzyme activities and expression in platelets of rats experimentally demyelinated with ethidium bromide. Biomed Pharmacother 81:363–370. https://doi.org/10.1016/j.biopha.2016.04.003
Blaževski J, Petković F, Momčilović M et al (2013) High interleukin-10 expression within the central nervous system may be important for initiation of recovery of Dark Agouti rats from experimental autoimmune encephalomyelitis. Immunobiology 218:1192–1199. https://doi.org/10.1016/j.imbio.2013.04.004
Agresti C, Meomartini ME, Amadio S et al (2005) Metabotropic P2 receptor activation regulates oligodendrocyte progenitor migration and development. Glia 50:132–144. https://doi.org/10.1002/glia.20160
Matute C, Torre I, Pérez-Cerdá F et al (2007) P2X7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci 27:9525–9533. https://doi.org/10.1523/JNEUROSCI.0579-07.2007
Grygorowicz T, Wełniak-Kamińska M, Struzyńska L (2016) Early P2X7R-related astrogliosis in autoimmune encephalomyelitis. Mol Cell Neurosci 74:1–9. https://doi.org/10.1016/j.mcn.2016.02.003
Zhang Z, Yang D, Zhan M et al (2017) Deletion of type 3 adenylyl cyclase perturbs the postnatal maturation of olfactory sensory neurons and olfactory cilium ultrastructure in mice. Front Cell Neurosci 11 https://doi.org/10.3389/fncel.2017.00001
Liu Y, Zou H, Zhao P et al (2016) Activation of the adenosine A2A receptor attenuates experimental autoimmune encephalomyelitis and is associated with increased intracellular calcium levels. Neuroscience 330:150–161. https://doi.org/10.1016/j.neuroscience.2016.05.028
Kim DG, Krenz A, Toussaint LE et al (2016) Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J Neuroinflammation 13 https://doi.org/10.1186/s12974-015-0467-5
Rivera-Oliver M, Díaz-Ríos M (2014) Using caffeine and other adenosine receptor antagonists and agonists as therapeutic tools against neurodegenerative diseases: a review. Life Sci 101:1–9
Chen GQ, Chen YY, Wang XS et al (2010) Chronic caffeine treatment attenuates experimental autoimmune encephalomyelitis induced by guinea pig spinal cord homogenates in Wistar rats. Brain Res 1309:116–125. https://doi.org/10.1016/j.brainres.2009.10.054
Tsutsui S, Schnermann J, Noorbakhsh F et al (2004) A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J Neurosci 24:1521–1529. https://doi.org/10.1523/JNEUROSCI.4271-03.2004
Nicolussi EM, Huck S, Lassmann H, Bradl M (2009) The cholinergic anti-inflammatory system limits T cell infiltration into the neurodegenerative CNS, but cannot counteract complex CNS inflammation. Neurobiol Dis 35:24–31. https://doi.org/10.1016/j.nbd.2009.03.010
Reale M, de Angelis F, di Nicola M et al (2012) Relation between pro-inflammatory cytokines and acetylcholine levels in relapsing-remitting multiple sclerosis patients. Int J Mol Sci 13:12656–12664. https://doi.org/10.3390/ijms131012656
Marinelli C, Bertalot T, Zusso M, et al (2016) Systematic review of pharmacological properties of the oligodendrocyte lineage. Front. Cell. Neurosci. 10
Santarpia L, Grandone I, Contaldo F, Pasanisi F (2013) Butyrylcholinesterase as a prognostic marker: a review of the literature. J Cachexia Sarcopenia Muscle 4:31–39
Millet T, Roger PL, Eran N et al (2016) Role of the α7 nicotinic acetylcholine receptor and RIC-3 in the cholinergic anti-inflammatory pathway Cent Nerv Syst Agents Med Chem 17 https://doi.org/10.2174/1871524916666160829114533
Bours MJL, Swennen ELR, Di Virgilio F et al (2006) Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112:358–404
Di Virgilio F, Vuerich M (2015) Purinergic signaling in the immune system. Auton Neurosci Basic Clin 191:117–123
Domercq M, Matute C (2019) Targeting P2X4 and P2X7 receptors in multiple sclerosis. Curr Opin Pharmacol 47:119–125
Domercq M, Zabala A, Matute C (2019) Purinergic receptors in multiple sclerosis pathogenesis. Brain Res Bull 151:38–45
Safarzadeh E, Jadidi-Niaragh F, Motallebnezhad M, Yousefi M (2016) The role of adenosine and adenosine receptors in the immunopathogenesis of multiple sclerosis. Inflamm Res 65:511–520
Oliveira-Giacomelli ágatha, Naaldijk Y, Sardá-Arroyo L, et al (2018) Purinergic receptors in neurological diseases with motor symptoms: Targets for therapy. Front Pharmacol 9:325
Acknowledgements
JLBS is grateful to the Federal University of Fronteira Sul for the research grant that promotes the production of this and other publications.
Author information
Authors and Affiliations
Contributions
JLBS had the idea for the article. JLBS, JBA, and MDB performed the literature search and data analysis and drafted and critically revised the work.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Informed consent and consent to publish were obtained from each of the participants.
Research Involving Human Participants and/or Animals
Not applicable.
Informed Consent
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Simões, J.L.B., de Araújo, J.B. & Bagatini, M.D. Anti-inflammatory Therapy by Cholinergic and Purinergic Modulation in Multiple Sclerosis Associated with SARS-CoV-2 Infection. Mol Neurobiol 58, 5090–5111 (2021). https://doi.org/10.1007/s12035-021-02464-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02464-0