
Abstract
Mitochondrial dysfunction is implicated in a variety of neurodegenerative diseases of the nervous system. Peroxisome proliferator–activated receptor-γ coactivator-1α (PGC-1α) is a regulator of mitochondrial function in multiple cell types. In sensory neurons, AMP-activated protein kinase (AMPK) augments PGC-1α activity and this pathway is depressed in diabetes leading to mitochondrial dysfunction and neurodegeneration. Antimuscarinic drugs targeting the muscarinic acetylcholine type 1 receptor (M1R) prevent/reverse neurodegeneration by inducing nerve regeneration in rodent models of diabetes and chemotherapy-induced peripheral neuropathy (CIPN). Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) is an upstream regulator of AMPK activity. We hypothesized that antimuscarinic drugs modulate CaMKKβ to enhance activity of AMPK, and PGC-1α, increase mitochondrial function and thus protect from neurodegeneration. We used the specific M1R antagonist muscarinic toxin 7 (MT7) to manipulate muscarinic signaling in the dorsal root ganglia (DRG) neurons of normal rats or rats with streptozotocin-induced diabetes. DRG neurons treated with MT7 (100 nM) or a selective muscarinic antagonist, pirenzepine (1 μM), for 24 h showed increased neurite outgrowth that was blocked by the CaMKK inhibitor STO-609 (1 μM) or short hairpin RNA to CaMKKβ. MT7 enhanced AMPK phosphorylation which was blocked by STO-609 (1 μM). PGC-1α reporter activity was augmented up to 2-fold (p < 0.05) by MT7 and blocked by STO-609. Mitochondrial maximal respiration and spare respiratory capacity were elevated after 3 h of exposure to MT7 (p < 0.05). Diabetes and CIPN induced a significant (p < 0.05) decrease in corneal nerve density which was corrected by topical delivery of MT7. We reveal a novel M1R-modulated, CaMKKβ-dependent pathway in neurons that represents a therapeutic target to enhance nerve repair in two of the most common forms of peripheral neuropathy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
The field of innervation of intraepidermal nerve fibers (IENFs) within the skin is plastic and maintained through a combination of collateral sprouting and regeneration that is regulated partly by neurotrophic factors [1, 2]. Distal dying-back of nerve fibers is observed in many peripheral neuropathies including those associated with diabetes, chemotherapy-induced peripheral neuropathy (CIPN), Friedreich’s ataxia, Charcot-Marie-Tooth disease type 2, and human immunodeficiency virus (HIV). There are no therapies for any of these neuropathies. Intriguingly, all of these diseases display some degree of mitochondrial dysfunction [3, 4].
The true prevalence of diabetic sensory neuropathy is not known and reports vary in their estimates from 10 to 90% in diabetic patients, depending on the criteria and methods used to define neuropathy [5]. The human and economic burden of diabetic neuropathy and its consequences in the form of painful neuropathy, foot ulceration, and amputation are considerable for both patients and healthcare systems [6]. The most common form of diabetic neuropathy in type 1 and type 2 patients, symmetrical sensorimotor polyneuropathy, exhibits pathological changes in both large and small fibers of peripheral nerves. Some of the earliest pathological features include the appearance of swellings in unmyelinated fibers that project into the epidermis, and there is dying-back of these distal axons leading to reduced IENF density in skin [5, 7,8,9,10]. Further, defective axon sprouting and regeneration impedes tissue re-innervation [11]. These indices of nerve pathology are also observed in rodent models of both type 1 and type 2 diabetes [12, 13], allowing for investigation of both putative pathogenic mechanisms and potential therapies.
Depending on the specific drug and dose regimen, CIPN can afflict between 20 and 85% of cancer patients receiving these drugs at standard doses and nearly 100% of patients at higher doses. Many patients with CIPN experience neuropathic pain, presenting as allodynia, hyperalgesia, and spontaneous pain. CIPN may resolve with cessation of chemotherapy, but in 30–70% of cases, it persists for months to years after treatment has concluded, or even increases in severity [14]. Patients with CIPN develop distal dying back of nerve fibers leading to loss of IENF, and this pathology is also seen in rodent models of CIPN [3].
Retraction or degeneration of IENF has been suggested to be the result of impaired mitochondrial function and local energy production. Growth cone motility is required to maintain fields of innervation and consumes 50% of ATP supplies in neurons due to high rates of local actin treadmilling [15]. In particular, unmyelinated axons are more energetically demanding than myelinated axons, consuming 2.5–10-fold more energy per action potential [16]. Mitochondria are known to concentrate in regions of high metabolic demand [17, 18], and sensory terminal boutons are packed with mitochondria [19, 20]. Our work in rodent models of type 1 and 2 diabetes has demonstrated that hyperglycemia triggers nutrient excess in neurons that, in turn, mediates a phenotypic change in mitochondria through alteration of the AMP-activated protein kinase (AMPK)/peroxisome proliferator–activated receptor γ coactivator-1α (PGC-1α) signaling axis [21]. A well-characterized upstream activator of AMPK is Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) which directly phosphorylates AMPK at its activation domain [22, 23] and forms a complex with AMPK [24]. This vital energy-sensing metabolic pathway modulates mitochondrial function, biogenesis, and regeneration [25,26,27]. The bioenergetic phenotype of mitochondria in diabetic neurons is characterized by inner membrane depolarization, reduced expression of respiratory chain components [28, 29], and suboptimal spare respiratory capacity [29,30,31], without remarkable ultrastructural alterations [32, 33].
Studies in peripheral nervous system (PNS) neurons demonstrate that acetylcholine (ACh) signaling through muscarinic receptors acts as a regulator of electrical activity and axonal growth cone motility during development. In sympathetic neurons, ACh activation of muscarinic acetylcholine type 1 receptor (M1R) mobilizes internal Ca2+ stores leading to closure of M-type K+ channels (Kv7 subtypes) and an enhancement of slow depolarization and discharge [34]. In embryonic neurons, ACh can modulate axonal growth in a positive or negative manner based upon context [35,36,37]. Furthermore, both adult sensory ganglia and the target of sensory neurons, keratinocytes within the epidermis, synthesize and secrete ACh [38, 39]. Adult rat sensory neurons of the dorsal root ganglia (DRG) express a peripheral form of choline acetyl transferase (pChAT), exhibit ChAT activity, have low AChE activity, and express multiple muscarinic receptors including M1R [38, 40, 41]. It is therefore becoming increasingly recognized that adult sensory neurons are cholinergic and amenable to manipulation by drugs that modulate cholinergic systems.
We have recently shown that selective or specific antagonists of M1R, including pirenzepine and muscarinic toxin 7 (MT7), induce a dose-dependent elevation of neurite outgrowth in adult sensory neurons [42,43,44]. Importantly, these drugs were also able to afford protection against several different forms of peripheral neuropathy. The exact mechanism of M1R antagonist–driven neurite outgrowth and nerve repair is, as yet, poorly described. To advance understanding of the downstream consequences of M1R antagonism, we tested the hypothesis that MT7, the only specific M1R antagonist (or allosteric modulator) [45], enhances mitochondrial function to drive neurite outgrowth via activation of the CaMKKβ/AMPK signal transduction pathway. Our attention was drawn to this possibility by the recent report that polymorphisms in the CaMKKβ gene are associated with susceptibility to HIV neuropathy [46]. To test our hypothesis, we performed mechanistic studies in vitro using adult sensory neuron cultures and assessed the capacity of MT7 to prevent neurodegeneration and drive nerve fiber repair in animal models of type 1 diabetes and CIPN.
Methods
Adult Rat DRG Sensory Neuron Culture
Animal protocols carefully followed the Canadian Committee on Animal Care (CCAC) guidelines. Adult male Sprague Dawley rats, approximately 5–8 weeks old and weighing 275–325 g, were fasted overnight then made diabetic with a single i.p. injection of 85 mg/kg streptozotocin (STZ) (Sigma-Aldrich, Oakville, ON, Canada) which causes degeneration of beta cells within the islets of Langerhans of the pancreas and induces experimental type 1 diabetes mellitus. Within weeks of induction, STZ-diabetic rats develop deficits in nerve conduction velocity and changes in pain perception threshold that reflect the clinical condition [47] and neuropathy in this model is not a consequence of direct STZ-induced neurotoxicity [48]. We confirmed the onset of sensory neuropathy using paw withdrawal time to a radiant heat stimulus [49], and only STZ-induced diabetic rats with blood glucose levels > 19 mM were selected for dissection and cell culture. Sensory neurons were isolated from the DRG and dissociated using previously described methods [42]. DRG neurons isolated from ganglia from all levels from these rats were plated onto poly-d-l-ornithine hydrobromide and laminin-coated multi-well plates for neuronal survival and axon outgrowth studies and 25 mm glass cover slips for immunocytochemistry. Neurons were grown in defined Hams F-12 medium (Life Technologies, Burlington, ON, Canada) with modified Bottenstein and Sato’s N2 medium (with no insulin) containing 0.1 mg/ml transferrin, 20 nM progesterone, 100 μM putrescine, 30 nM sodium selenite, and 1 mg/ml BSA, and supplemented with the following neurotrophic factors (NTFs): 0.1 ng/ml nerve growth factor (NGF), 0.1 ng/ml neurotrophin-3 (NT-3), and 1 ng/ml glial cell line–derived neurotrophic factor (GDNF) (all obtained from Sigma-Aldrich, Oakville, ON, Canada). Neurons derived from control animals were exposed to 10 mM glucose and 0.1 nM insulin. Neurons from STZ-induced diabetic rats were cultured in the presence of 25 mM glucose without adding insulin to mimic physiological conditions of diabetes in rats. Cultures were treated with MT7 (Alomone Labs, Jerusalem, Israel), STO-609 (Tocris, Minneapolis, MN, USA), or pirenzepine (Sigma-Aldrich, Oakville, ON, Canada).
Assessment of Total Neurite Outgrowth
Rat neurons grown on glass cover slips were imaged directly for GFP fluorescence or fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4) for 15 min at room temperature and permeabilized with 0.3% Triton X-100 in PBS for 5 min. Cells were then incubated in blocking buffer (Roche, Indianapolis, IN, USA) diluted with FBS and 1.0 mM PBS (1:1:3) for 1 h then rinsed three times with PBS. The primary antibody used was against β-tubulin isotype III, which is neuron-specific (1:1000; Sigma-Aldrich, Oakville, ON, Canada). For CaMKKβ immunocytochemistry, we used antibody at a dilution of 1:1000 (Santa Cruz Biotechnology, Dallas, TX, USA). The antibody was added to all wells, and plates were incubated at 4 °C overnight. The following day, the coverslips were incubated with CY3-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1 h at room temperature and then mounted and imaged using a Carl Zeiss Axioscope-2 fluorescence microscope equipped with an AxioCam camera. Images were captured using AxioVision4.8 software. For GFP imaging, we used a Carl Zeiss LSM510 confocal microscope. Quantification of total neurite outgrowth was performed by measuring the mean pixel area of captured images using ImageJ software (adjusted for the cell body signal). All values were adjusted for neuronal number post acquisition [42]. In this culture system, the level of total neurite outgrowth has been validated to be directly related to an arborizing form of axonal plasticity and homologous to collateral sprouting in vivo [50].
Luciferase Reporter Constructs for PGC-1α, Mutant Construct for CaMKKβ, and Cell Transfection of Adult Sensory Neurons
Reporter plasmid with the wild-type PGC-1α promoter upstream from luciferase was kindly donated by Dr. Michael Czubryt (University of Manitoba). DRG neurons (30 × 103) were transfected in triplicate with 1.8 μg of PGC-1α promoter/luciferase plasmid DNA and 0.2 μg of pCMV-Renilla (Promega, Madison, WI, USA) using the Amaxa electroporation kit for low numbers of cells according to the manufacturer’s instructions (ESBE Scientific, Toronto, ON, Canada). Neurons were lysed using passive lysis buffer provided with the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). The luciferase activity was measured using a luminometer (model LMAXII; Molecular Devices, Sunnyvale, CA, USA). Twenty microliters of each sample was loaded in a 96-well plate and was mixed with 100 μl of Luciferase Assay Reagent II, and firefly luciferase activity was first recorded. Then, 100 μl of Stop-and-Glo Reagent was added, and Renilla luciferase activity was measured. All values are normalized to Renilla luciferase activity. Plasmid over-expressing a wild type or mutant of CaMKKβ was also transfected into adult neurons using the Amaxa machine as described above. The mutant construct, comprised of multisite mutations at S129D, S133D, and S137D, and a wild-type construct were kindly donated by Dr. Anthony Means (Duke University Medical Center) as described [51]. The mutant construct exhibited mutations at S129, S133, and S137 leading to reduced phosphorylation of the enzyme that inhibited the autonomous activity of the enzyme.
Short Hairpin RNA–Based Blockade of CaMKKβ Signaling in DRG Neurons
For short hairpin RNA (shRNA)–based gene silencing, a GFP-expressing clone specific for CaMKKβ (clone ID: V3LMM_453709) was obtained from Open Biosystems (Lafayette, CO, USA). The pGIPz lentiviral system for mice and humans from the Open Biosystems database is held at the University of Manitoba, Winnipeg, Canada. A control scrambled shRNA unrelated to the CaMKKβ sequence was used as a negative control for lentiviral transduction [52]. For shRNA-based gene silencing studies, DRG neurons were transduced at 20× infectious unit in the presence of polybrene (8 μg/ml) for 2 h at 37 °C, complete medium was added, and neurons were cultured for additional 48 h. Fluorescence images derived from GFP-expressing virally transduced neurons were acquired by using a LSM510 confocal microscope (Carl Zeiss) with × 20 air objective. For dual transfection, neurons were initially transfected with plasmid for PGC-1α and, then after 48 h, were transduced with lentivirus carrying shRNA to CaMKKβ.
SiRNA-Based Silencing of CaMKKβ in Neurons
The lipid nanoparticle (LNP)-based small interfering RNA (siRNA)-mediated knockdown of CaMKKβ was achieved as described previously [53]. Briefly, the cells were transfected with a cocktail of 3 siRNAs designed against a variety of sites within the gene (see [53] for siRNA sequence data). The siRNAs were encapsulated in LNPs as described previously [54]. The siRNA-LNPs were prepared using a microfluidic micromixer by Precision NanoSystems Inc., Vancouver, BC, Canada. For the encapsulation, the desired amount of siRNAs (0.056 μg siRNA/μmol of lipid) was dissolved in the formulation buffer (25 mmol/l sodium acetate, pH 4.0). The siRNA/LNP preparation was added to the cultured cells where transfection reached 80–90% efficiency with no toxicity, and knockdown was achieved within 2 days in adult sensory neurons.
Western Blotting for CaMKKβ, P-AMPK, and Other Proteins
Rat DRG were harvested after treatments in ice-cold stabilization buffer containing 0.1 M PIPES (pH 6.9), 5 mM MgCl2, 5 mM EGTA, 0.5% Triton X-100, 20% glycerol, 10 mM NaF, 1 mM PMSF, and protease inhibitor cocktail. Mouse trigeminal ganglia were rapidly dissected, snap-frozen in liquid nitrogen, and stored at − 70 °C until assay. Protein assay was performed using the DC protein assay (Bio-Rad, Hercules, CA, USA), and Western blot analysis was conducted. The samples (5 μg total protein/lane) were resolved on a 10% SDS-PAGE gel and electroblotted (100 V, 1 h) onto a nitrocellulose membrane. Blots were then blocked in 5% non-fat milk (containing 0.05% Tween) overnight at 4 °C, rinsed in TBS-T, and then incubated with the primary antibodies to the following proteins: CaMKKβ (1:1000, Santa Cruz Biotechnology), CaMKKα (1:1000, Santa Cruz Biotechnology), phosphorylated CaMK1 (Thr-177, P-CaMK1; 1:1000; LifeSpan BioSciences, Seattle, WA, USA), phosphorylated CaMKIV (Thr-196 and Thr-200, P-CaMKIV; 1:750; Abcam, Cambridge, MA, USA), phosphorylated AMPK (P-AMPK on Thr-172; 1:500; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA; Cell Signaling Technology, Boston, MA, USA), total AMPK (T-AMPK; 1:500, Santa Cruz Biotechnology), PGC-1α (1:500, Santa Cruz Biotechnology), and phosphorylated acetyl coenzyme A carboxylase (P-ACC; 1:2000; Abcam, Cambridge, MA, USA). Total extracellular–regulated protein kinase (T-ERK; 1:2000, Santa Cruz Biotechnology) and/or actin were probed as loading controls. Previous studies have shown that the expression of T-ERK does not change in intact DRG or cultures from diabetic rats [55]. Secondary antibody was applied for 1 h at room temperature after 5–6 washes of 10 min in PBS-T. The blots were rinsed, incubated in Western Blotting Luminol Reagent (Santa Cruz Biotechnologies, CA, USA), and imaged using a Bio-Rad ChemiDoc image analyzer.
Measurement of Mitochondrial Respiration in Cultured DRG Neurons
An XF24 Analyzer (Seahorse Biosciences, Billerica, MA, USA) was used to measure the neuronal bioenergetic function of rat cultures. The XF24 creates a transient 7-μl chamber in specialized 24-well microplates that allows for the oxygen consumption rate (OCR) to be monitored in real time. Culture medium was changed 1 h before the assay to unbuffered Dulbecco’s modified Eagle’s medium (DMEM, pH 7.4) supplemented with 1 mM pyruvate and 10 mM d-glucose. Neuron density in the range of 2000–4000 cells per well gave a linear OCR. Oligomycin (1 μM), carbonylcyanide-p-trifluoromethoxyphenyl hydrazone (FCCP; 1 μM), and rotenone (1 μM) + antimycin A (1 μM) were injected sequentially through ports in the Seahorse FluxPak cartridges. Each loop was started by mixing for 3 min and then delayed for 2 min, and OCR was measured for 3 min. This allowed determination of the basal level of oxygen consumption, the amount of oxygen consumption linked to ATP production, the level of non-ATP–linked oxygen consumption (proton leak), the maximal respiratory capacity, and the non-mitochondrial oxygen consumption. Oligomycin inhibits the ATP synthase leading to a build-up of the proton gradient that inhibits electron flux and determines the coupling efficiency. Injecting FCCP to the culture determines the maximal capacity to reduce oxygen. Finally, rotenone (complex I inhibitor) + antimycin A (complex III inhibitor) was injected to inhibit the flux of electrons and stop the oxygen consumption at cytochrome c oxidase. The remaining OCR revealed after this intervention is primarily non-mitochondrial. Data are expressed as OCR in pmol/min/mg protein.
Corneal Confocal Microscopy
Type 1 diabetes was induced in female Swiss Webster mice (Envigo, USA) by injection of STZ (Sigma-Aldrich, St Louis, MO, USA) at 6–8 weeks of age in 0.9% saline after overnight fast at 90–100 mg/kg i.p. on two consecutive days, with hyperglycemia (blood glucose > 15 mmol/l confirmed 3 days later in blood obtained by tail prick using a strip-operated reflectance meter) (OneTouch Ultra, LifeScan). CIPN was induced in female Swiss Webster mice by four injections of 5 mg/kg i.p. oxaliplatin given on alternate days for a cumulative dose of 20 mg/kg and confirmed by calculating the 50% paw withdrawal threshold to von Frey filaments, as described in detail elsewhere [56]. Mice were treated with MT7 or vehicle after 6 weeks of diabetes or 4 weeks after the cessation of oxaliplatin treatment. MT7 was dissolved in 0.1 M sodium phosphate buffer to a concentration of 25 ng/ml, and 30 μl of this solution was delivered to the surface of one eye daily, Monday–Friday, for 2 weeks while the contralateral eye received a similar volume of vehicle. Corneal nerves of the sub-basal nerve plexus were imaged in anesthetized mice using a Heidelberg Retina Tomograph 3 with Rostock Cornea Module (Heidelberg Engineering, Heidelberg Germany) as described elsewhere [57]. Corneal nerve density was measured in 5 consecutive images (2-μm intervals) of the sub-basal nerve plexus (SBNP) between the corneal epithelium and stroma and quantified either as occupancy using an 8 × 8 grid layered onto each image [56] or as pixel density following tracing of all visible nerves using ImageJ software [58].
Statistical Analysis
For each neuronal culture experiment, one rat (age-matched normal or diabetic) was used with cultures performed in replicate and such experiments were independently repeated at least 2–3 times. Data are expressed as mean ± SEM and were subjected to one-way ANOVA with post hoc comparisons using Dunnett’s test for dose-response experiments and for comparison of treatment groups to a single control group. Tukey’s post hoc test was used to determine group means that were significantly different between multiple treatment groups. For in vivo studies, paired or unpaired Student’s t tests were performed as defined by the a priori experimental design. GraphPad Prism software was used to perform the statistical analysis.
Results
Expression Profile of CaMKs and CaMKKs in Nervous Tissue
Initially, expression of CaMKKβ in the DRG of adult normal age-matched rats was confirmed. Figure 1a shows that DRG expressed CaMKKβ along with known phosphorylation targets of CaMKKβ, namely CaMK1 and CaMKIV. Interestingly, the relative levels of expression in the DRG and sciatic nerve of CaMKKβ were higher than those seen in the brain. This corresponded with higher levels of phosphorylated CaMK1 and CaMKIV. The CaMKKα isoform was also expressed, although at low levels compared with other tissues.

CaMKKβ was expressed by adult sensory neurons and mediated neurite outgrowth. a Tissues from an adult normal age-matched rat were analyzed for expression of different forms of CaMK and CaMKKs. sc = nerve sciatic nerve, cord = spinal cord, DRG = lumbar DRG. Each tissue was analyzed in triplicate. b Cultured adult sensory neurons were transduced with lentivirus over-expressing scrambled sequence (scr) or shRNA to CaMKKβ for 48 h. Cells were fixed and then immunostained with antibody to CaMKKβ. Bar = 100 μm. c In a parallel study, culture lysates were analyzed for CaMKKβ expression using Western blotting and are expressed relative to T-ERK. Values are means ± SEM, n = 3 replicate cultures. d Effect of shRNA to CaMKKβ on levels of total neurite outgrowth (adjusted for cell number). Constructs co-expressed GFP, and so the GFP fluorescence signal was used for quantification of neurite outgrowth. Values are means ± SEM, n = 3 replicate cultures; *p < 0.05 vs shRNA to CaMKKβ by Student’s t test. e Neurons were transfected for 48 h with GFP-expressing plasmids to wild-type (WT) CaMKKβ or multisite mutant CaMKKβ-S129D, S133D, S137D. Neurite outgrowth is expressed as means ± SEM, n = 3 replicate cultures; *p < 0.05 vs mutant by Student’s t test
CaMKKβ Silencing Restricts Neurite Outgrowth of Adult Sensory Neurons
Dissociated adult sensory neurons derived from a normal adult rat were transduced with lentivirus over-expressing a scrambled sequence or shRNA to CaMKKβ. After 48 h, cultures were fixed and immunostained for CaMKKβ. Figure 1b shows loss of CaMKKβ expression in the presence of shRNA. In a parallel experiment, cell lysates were collected and CaMKKβ protein levels detected by Western blotting. Figure 1c shows an approximately 50% decrease of CaMKKβ in the presence of shRNA. In a supporting experiment, neurons were live-imaged for GFP fluorescence after 48 h of lentiviral transduction and neurite outgrowth was quantified. Figure 1d shows that over-expression of shRNA to CaMKKβ reduced neurite outgrowth in the presence of a cocktail of neurotrophic factors by at least 60% compared with scrambled sequence. A similar inhibition of neurite outgrowth occurred following over-expression of the kinase-dead mutant CaMKKβ-S129D, S133D, S137D (Fig. 1e).
Pharmacological Blockade of CaMKKβ Inhibits Neurite Outgrowth Driven by M1R Antagonists Pirenzepine and MT7
In a previous study, we demonstrated that selective or specific M1R antagonists increased neurite outgrowth in adult sensory neurons [42]. In Fig. 2a, treatment for 24 h with the selective M1R antagonist, pirenzepine at 1.0 μM, increased neurite outgrowth of normal adult sensory neurons by approximately 50% and this effect was dose-dependently blocked by the CaMKK inhibitor, STO-609. In a follow-up study, 1 μM pirenzepine elevated neurite outgrowth by approximately 50–60% and the CaMKKβ inhibitor, STO-609, completely blocked neurite outgrowth at 1 μM (Fig. 2b) whereas STO-609 alone had no effect on background neurite outgrowth supported by the cocktail of neurotrophic factors. To confirm this neuritogenic pathway was relevant in the setting of diabetic neuropathy, sensory neuron cultures were prepared from a 3–5-month-old STZ-diabetic rat and exposed to the specific M1R antagonist MT7 at 100 nM, with/without 1 μM STO-609 for 24 h. Figure 2c presents representative images of cultures immunostained for neuron-specific β-tubulin III. MT7 induced an approximately 40% elevation in neurite outgrowth that was completely blocked by STO-609 (Fig. 2d), replicating the pattern seen in the DRG from normal rats (Fig. 2b).

Neurite outgrowth was raised by pirenzepine and MT7 and blocked by the CaMKKβ inhibitor, STO-609. a Cultured sensory neurons from a normal rat were exposed to 1 μM pirenzepine and a range of STO-609 concentrations for 24 h (pretreated with STO-609 for 1 h). Cells were fixed and immunostained for β-tubulin III, and neurite outgrowth was assessed. Values are means ± SEM, n = 8–10 replicate cultures; *p < 0.05 vs 1.0 μM and 3.0 μM STO-609 by one-way ANOVA with Tukey’s post hoc test. b Neurons from a normal rat were cultured for 24 h with 1 μM pirenzepine (PZ) or 100 nM MT7 in the presence of 1 μM STO-609, and neurite outgrowth was analyzed. Values are means ± SEM, n = 5–6 replicate cultures; *p < 0.05 vs control, 1.0 μM STO alone, or PZ+STO-609 by one-way ANOVA with Tukey’s post hoc test. c, d Sensory neurons derived from a 3–5-month STZ-diabetic rat were cultured for 24 h in the presence of 100 nM MT7 with/without 1 μM STO-609. Fluorescent images were taken from cells immunostained for β-tubulin III; bar = 100 μm. In d, bar charts of data where values are means ± SEM, n = 6 replicate cultures; *p < 0.05 vs other groups by one-way ANOVA with Tukey’s post hoc test
MT7 Activated AMPK in a Dose and Time–Dependent Manner and Was Mediated Via CaMKKβ
CaMKKβ is an endogenous activator of AMPK [22, 23], so we determined if MT7 could activate AMPK in cultures derived from STZ-induced diabetic rats. In Fig. 3a, Western blots reveal a marked dose-dependent elevation in phosphorylation of AMPK following 1 h of MT7 treatment. Quantification of P-AMPK relative to T-AMPK or T-ERK revealed an at least 5-fold elevation at 100 nM MT7 (Fig. 3b). The endogenous target of activated AMPK, P-ACC, was also significantly enhanced (Fig. 3c). A time course for the effect of 100 nM MT7 was performed and revealed a remarkable elevation in P-AMPK levels following 6 h of treatment (Fig. 3d, e). At 1 h, 100 nM MT7 caused a 2-fold elevation in P-AMPK, although this was not statistically significant and not as robust as compared with the data revealed in Fig. 3a. To confirm the MT7-dependent phosphorylation was mediated via CaMKKβ, neuronal cultures were treated with 100 nM MT7 for 3 h in the presence/absence of 1 μM STO-609. The inhibitor completely blocked the MT7 induction of P-AMPK levels (Fig. 3f). To confirm that CaMKKβ was regulating AMPK phosphorylation status, we used lipid nanoparticles to deliver a cocktail of siRNAs to CaMKKβ to normal DRG cultures. This approach gives a superior level of knockdown for molecular endpoints, such as gene expression analysis, compared with the shRNA technique, since 80–90% of neurons were affected. Following 48 h of treatment with the siRNAs, the level of CaMKKβ protein was clearly depleted (Fig. 4a). This was a specific knockdown since the related CaMKKα protein was not affected (Fig. 4c). Using standard Western blotting, the knockdown of CaMKKβ also caused a reduction of the P-AMPK signal (Fig. 4b).

MT7 elevated phosphorylation of AMPK in a dose and time–dependent manner. a Sensory neurons derived from a 3–5-month STZ-diabetic rat were cultured overnight and then treated with varying doses of MT7 for 1 h. Western blots are shown for P-AMPK, T-AMPK, and T-ERK. b Levels of expression of these proteins presented relative to T-ERK or T-AMPK. c Samples from the experiment in b were probed for P-ACC. d Neurons from a diabetic rat were cultured overnight and then exposed to 100 nM MT7 for varying times. e Levels of P-AMPK presented relative to T-ERK and T-AMPK. For b, c, and e, values are means ± SEM, n = 3 replicate cultures; *p < 0.05 vs control by one-way ANOVA with Dunnett’s post hoc test. f Pharmacological blockade of CaMKKβ prevents MT7-dependent phosphorylation of AMPK. Neurons from a control rat were cultured overnight and then pretreated with 1 μM STO-609 for 2 h and then treated with 100 nM MT7 for 3 h. Levels of P-AMPK presented relative to T-ERK and T-AMPK. Values are means ± SEM, n = 3 replicate cultures; *p < 0.05 vs other groups by one-way ANOVA with Tukey’s post hoc test

Knockdown of CaMKKβ reduces AMPK phosphorylation. a–c Immunoblots showing the relative expression of a CaMKKβ, b pAMPK (on Thr-172), and c CaMKKα in control (scrambled siRNA) and CaMKKβ siRNA–transfected cultured adult rat primary DRG neurons (GAPDH used as a loading control in a and c from normal rats). Adult rat DRGs were freshly dissected, dissociated, and transfected with siRNAs using Amaxa nucleofection reagent and cultured in defined media for 48 h. The total proteins were separated by SDS-PAGE and immunoblotted
MT7 Elevated Transcriptional Activity of the AMPK target, PGC-1α, Via CaMKKβ
The pathway downstream from activation of AMPK that mediates mitochondrial biogenesis and refurbishment includes activation of PGC-1α leading to enhancement of its cotranscriptional activities [25, 26]. Cultures derived from STZ-diabetic rats were exposed to varying doses of MT7 for 30 min and cell lysates analyzed using a dual-luciferase assay for PGC-1α transcriptional activity. Figure 5a shows that 30–100 nM MT7 significantly elevated PGC-1α reporter activity. Figure 5b demonstrates that 100 nM MT7 significantly raised PGC-1α transcriptional activity as early as 15 min and this was sustained for up to 3 h. We used two approaches to causally link MT7 activation of PGC-1α transcriptional activity to upstream signaling via CaMKKβ. Neuronal cultures from STZ-diabetic rats were cotreated with either 100 nM MT7 and shRNA to CaMKKβ (Fig. 5c) or 1 μM STO-609 (Fig. 5d). Under both conditions of CaMKKβ inhibition, the ability of MT7 to enhance PGC-1α transcriptional activity was abolished. Finally, in support of the dual-luciferase reporter approach, we used Western blotting to determine levels of PGC-1α protein in lysates from neuronal cultures derived from STZ-diabetic rats. Treatment with 100 nM MT7 for 6 h significantly increased PGC-1α protein levels (Fig. 5e).

MT7 enhanced transcriptional activity of PGC-1α through a CaMKKβ-dependent pathway. a Neurons from a diabetic rat were transfected with reporter plasmid for PGC-1α and, then after 48 h, were treated with varying doses of MT7 for 1 h. Cell lysates underwent dual-luciferase reporter assay for PGC-1α and data presented relative to PGL3 internal control plasmid (indicated by black bar). Values are means ± SEM, n = 3 replicate cultures; *p < 0.05 vs no treatment by one-way ANOVA with Tukey’s post hoc test. b Time course for the effect of 100 nM MT7 on PGC-1α transcriptional activity. Values are means ± SEM, n = 3 replicate cultures; *p < 0.05 vs all time points by one-way ANOVA with Tukey’s post hoc test. c Neurons from a diabetic rat were transfected with the PGC-1α reporter plasmid and, then after 48 h, were transduced with lentivirus over-expressing shRNA to CaMKKβ. After 48 h, cultures were treated with 100 nM MT7 for 1 h and the reporter activity was determined. Values are means ± SEM, n = 3 replicate cultures; **p < 0.01 vs other groups by one-way ANOVA with Tukey’s post hoc test. d Neurons from a diabetic rat were transfected with the PGC-1α plasmid for 48 h. After this period, neurons were then pretreated with 1 μM STO-609 for 2 h and then treated with 100 nM MT7 for 1 h and PGC-1α reporter activity was determined. Values are means ± SEM, n = 3 replicate cultures; **p < 0.01 vs other groups by one-way ANOVA with Tukey’s post hoc test. e Diabetic neurons were cultured overnight and then exposed to 100 nM MT7 for varying periods of time. A crude nuclear sample was collected and subjected to Western blotting for PGC-1α. Values presented relative to T-ERK and are means ± SEM, n = 3 replicate cultures; *p < 0.05 vs control by one-way ANOVA with Dunnett’s post hoc test
MT7 Augmented Mitochondrial Function Through a CaMKKβ-Dependent Pathway
Neuronal cultures derived from STZ-diabetic rats were treated for 3 h with 100 nM MT7 ± STO-609 followed by assessment of bioenergetic profile using the Seahorse XF24 Analyzer. Maximal respiration and spare respiratory capacity were significantly increased by MT7, and this effect was blocked by STO-609 (Fig. 6; basal respiration and the coupling efficiency were unaffected by MT7 treatment).

MT7 improves mitochondrial function in sensory neurons cultured from STZ-induced diabetic rats. a Oxygen consumption rate (OCR) was measured in the DRG from a 6-week-old diabetic rat cultured overnight and in the presence or absence of 100 nM MT7 for 1 h and in the presence or absence of a 3-h STO-609 (3 μM) pretreatment. OCR in pmol/min was normalized to mg of protein. b Maximal respiration and c spare respiratory capacity were determined after subtracting the non-mitochondrial OCR. Values are mean ± SEM, n = 6–7 replicate cultures, and adjusted to total protein levels. *p < 0.05 control vs MT-7 (one-way ANOVA with Tukey’s post hoc test)
Topical MT7 Reverses Corneal Fiber Loss in STZ-Diabetic and CIPN Mice
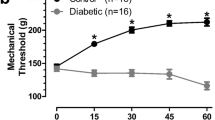
Nerve density in the corneal sub-basal plexus was measured by corneal confocal microscopy before and 4 weeks after induction of diabetes. Four weeks of diabetes caused a significant (p < 0.05, paired Student’s t test) reduction in corneal nerve density (measured as occupancy) that also persisted at week 6 of diabetes (Fig. 7a). Topical delivery of MT-7 for 10 days significantly (p < 0.05, paired Student’s t test) increased corneal nerve density to values similar to those before the onset of diabetes. In a separate study, corneal nerve density (measured as pixels per unit area) was quantified in mice that had been treated with vehicle (control) or oxaliplatin 3 weeks earlier. Oxaliplatin-injected mice exhibited CIPN, as illustrated by the presence of increased sensitivity to the touch of von Frey filaments (50% paw withdrawal threshold for vehicle group = 1.50 ± 0.1 vs CIPN group = 0.76 ± 0.14 g: p < 0.01 by unpaired Student’s t test, N = 8–9/group) and tended towards lower corneal nerve density than control mice (Fig. 7b). At this time, both control and CIPN mice were treated topically with MT7 to one eye for 2 weeks. MT7 treatment significantly increased corneal nerve density of CIPN mice compared to pre-MT7 treatment values (p < 0.01 by paired Student’s t test: Fig. 7b) but was without significant effect on nerve fiber density in control mice. At the end of the study, the ipsilateral and contralateral trigeminal ganglia were removed and subjected to Western blotting to quantify P-AMPK levels. Figure 8 shows that, in oxaliplatin-treated mice (CIPN), MT7 caused a significant elevation of P-AMPK levels in the ipsilateral trigeminal ganglion when compared to the contralateral trigeminal ganglion (corresponding to the vehicle-treated eye), when adjusted for T-AMPK (similar results were obtained when adjusted to T-ERK or total protein; data not shown). Also, note that P-AMPK levels were reduced in the trigeminal ganglia of the untreated side in CIPN mice vs control mice (although this effect did not reach statistical significance). MT7 treatment to one eye of normal mice did not result in significant differences of P-AMPK levels between the ipsilateral and contralateral trigeminal ganglia.

Topical MT7 promotes increased corneal nerve density in diabetic and CIPN mice. a Corneal nerve density measured by confocal microscopy in female Swiss Webster mice before induction of diabetes (day 0) and 28 days later. The same mice underwent further corneal nerve measurements after 42 days of diabetes before receiving daily delivery of MT7 to one eye (30 μl of 25 ng/ml solution), Monday–Friday, for 2 weeks. Sub-basal nerve plexus (SBNP) occupancy was calculated by tracing nerves and using a grid overlay system [56]. b Corneal nerve density (SBNP) was measured by confocal microscopy in control or oxaliplatin-treated (CIPN) female Swiss Webster mice before (pre) and 2 weeks after (+MT7) daily delivery of MT7 to one eye (30 μl of 25 ng/ml solution), Monday–Friday, for 2 weeks. In this study, the SBNP was measured by tracing all nerves and recording total pixels/image area [58]. Data are group mean + SEM of N = 7–9/group. Within-group statistical comparisons before and after treatment with STZ or MT7 by paired Student’s t test; *p < 0.05, **p < 0.01

Topical MT7 to the eye restored AMPK phosphorylation in the ipsilateral trigeminal ganglia (TG) in CIPN mice (oxaliplatin-treated; CIPN). Control or CIPN mice were treated topically to the left eye (ipsilateral) with MT7 using a stock of 25 ng/ml, and 30 μl of this solution was delivered to the surface of one eye daily, Monday–Friday, for 2 weeks. The right (contralateral) eye received vehicle. At the end of the study, the ipsilateral and contralateral TG from control (Ctrl) or CIPN mice were isolated and subjected to a Western blotting. b Western blot band intensities of phosphorylated AMPK (P-AMPK) and total AMPK (T-AMPK) were normalized to T-AMPK or T-ERK, respectively. The +MT7 is the treated (ipsilateral) eye, and without MT7 reflects the untreated (contralateral) eye. Data are mean ± SEM of N = 4 (ganglia were combined from two mice; 8 mice dissected to give n = 4); **p < 0.01 by paired Student’s t test
Discussion
Our previous work identified the AMPK/PGC-1α signaling cassette as an important target for the ability of M1R antagonists to enhance mitochondrial activity and promote neurite outgrowth from adult sensory neurons and to protect animal models against peripheral neuropathy [42]. The present study demonstrates for the first time that blockade of the M1R by the specific antagonist MT7 drives axonal outgrowth via CaMKKβ and subsequent activation of the AMPK/PGC-1α axis to augment mitochondrial function. Our in vitro assay for neurite outgrowth of DRG neurons is a useful model of axonal plasticity in vivo since it replicates the process of collateral sprouting [50]. Thus, this assay can be a predictor of efficacy with respect to drug effects on IENF density (see [42]) and fiber levels in the cornea. In this regard, topical MT7 also reversed loss of sensory nerves in the cornea of mice with neuropathy caused by diabetes or oxaliplatin-induced CIPN. Interestingly, the topical application of MT7 to one eye of CIPN mice enhanced AMPK activation only in the ipsilateral trigeminal ganglion, suggesting the importance of the interaction of MT7 with distal peripheral regions of sensory nerve fibers.
This is the first demonstration of a role for CaMKKβ in directing neurite outgrowth in adult peripheral sensory neurons. Previous studies in embryonic CNS neurons have shown that CaMKKβ is implicated in a variety of pathways driving neurite outgrowth and axonal plasticity. The phosphorylation state of CaMKKβ also impacts the formation of neurites and polarity during differentiation [51]. Knockout or pharmacological blockade of CaMKKβ in embryonic cerebellar granule cells [51], hippocampal neurons [59], and neuroblastoma cells [60] resulted in inhibition of neurite outgrowth, with CaMKKβ localized to neurites and growth cones [51, 60]. Activation of CaMKI, a primary downstream target of CaMKKβ, has also been mechanistically linked to the development of axonal projections in both embryonic cortical and hippocampal neurons [61]. Our studies extend this work to the PNS and place CaMKKβ downstream of the M1R and upstream of AMPK and PGC-1α in a signaling cascade that drives neurite outgrowth from adult sensory neurons.
Other than driving neurite growth, activation of the CaMKKβ/AMPK signaling pathway has also been implicated in protection from neurological impairments such as ischemic stroke and hypoxic-ischemic encephalopathy [62, 63]. A potentially key finding in understanding this neuroprotective property is that CaMKKβ lies upstream from AMPK to augment mitochondrial function. Previous studies have highlighted that activation of AMPK can elevate neurite outgrowth. For example, activation of AMPK by resveratrol elevates neurite outgrowth in embryonic sensory neurons [64]. Interestingly, this resveratrol-triggered pathway to AMPK activation in sensory neurons was independent of CaMKK [64], although in cortical neurons, CaMKK was activated upstream from AMPK, demonstrating the importance of cellular context when considering these signaling cassettes. Our previous work in adult sensory neurons showed that resveratrol-driven neurite outgrowth could be blocked with adenovirus-mediated delivery of dominant negative mutants of AMPK [29]. Furthermore, in STZ-induced diabetic rats, resveratrol therapy activated AMPK and mitochondrial function in the DRG and protected from sensory neuropathy [29]. In adult sensory neurons, M1R blockade also augments mitochondrial function via AMPK and protects from sensory neuropathy [42]. In the present study, MT7 elevated AMPK phosphorylation by at least 2–3-fold within 1 h and, at longer time points, caused a very robust elevation of P-AMPK that exceeded levels previously seen with the classical AMPK activator, resveratrol. In contrast, acute treatment with MT7 for up to 1 h caused no significant activation of AMPK, suggesting that there are several intermediate steps upstream of AMPK, presumably proximal to CaMKKβ, that are mediated by antagonism of the M1R (discussed below).
In this study, the majority of the work was performed with DRG cultures derived from diabetic rats. The rationale for this approach was that we have found responses to antimuscarinic drugs to be far more robust in diabetic cultures, particularly with respect to activation of AMPK and augmentation of mitochondrial function. Previous in vitro and in vivo studies with DRG neurons have revealed that the type 1 or type 2 diabetic state triggers downregulation of AMPK [29, 65]. This hyperglycemia-induced downregulation of AMPK and PGC-1α is also observed in the skeletal muscle, liver, kidney, and cardiac tissue [29, 66,67,68,69,70,71,72]. Therefore, we surmise that this suppression of the key energy sensor in the neuron thus engenders greater sensitivity to putative stimulators of AMPK and mitochondrial function.
PGC-1α is a key mediator of AMPK activation to enhance mitochondrial function, and activation of PGC-1α is neuroprotective in animal models of Huntington and Alzheimer’s diseases [73,74,75]. Further, impaired PGC-1α signaling has recently been implicated in Parkinson’s disease [75, 76] and causes CNS lesions linked to impaired neurite outgrowth and hyperactivity [77, 78]. PGC-1α was activated within 1 h by MT7 treatment in a CaMKKβ-dependent fashion. There was significant activation of PGC-1α transcriptional activity by 15 min which did precede evidence of any AMPK activation. Thus, it is feasible that other MT7 triggered pathways, independent of CaMKKβ, may be contributing to PGC-1α activation.
A potential therapeutic application of the consequences of M1R antagonism by MT7 is illustrated by our findings that MT7 delivered topically to the eye was effective in reversing reduced corneal nerve density caused by diabetes or CIPN. This, along with our previous finding that topical MT7 also prevented and reversed corneal nerve loss induced by the neurotoxic HIV-associated protein gp120 [42], supports the concept that M1R antagonism promotes peripheral nerve regenerative growth irrespective of the primary pathogenesis of the underlying neuropathy. Reduced corneal nerve density also occurs in patients with diabetic neuropathy [79] or cancer patients treated with oxaliplatin [80] and is emerging as an early biomarker for a diverse collection of neurodegenerative diseases in the PNS and CNS [81]. The promotion of corneal nerve regrowth in disease models by topical application of antimuscarinics may therefore serve as a predictor of their broader efficacy in peripheral nerves. Indeed, in the case of diabetic neuropathy, systemic delivery of the M1R-selective antagonist pirenzepine both prevents and reverses multiple functional and structural indices of peripheral neuropathy in diabetic rodents [42]. It is pertinent to note that MT7 was also very effective in activating AMPK in the trigeminal ganglia of CIPN mice, but only on the ipsilateral side. Access of MT7 to the cell bodies of the trigeminal ganglion is unlikely to be via a systemic route, otherwise the contralateral trigeminal would also have shown elevated AMPK activity. This supports the idea that specific M1R blockade, and subsequent activation of AMPK within the cell body, is mediated via an initial signal derived from the nerve ending in the cornea. This has important implications in terms of a future therapeutic approach in humans with peripheral neuropathy as topical delivery of M1R antagonists to sensory nerve endings may prove sufficient to activate AMPK in neuronal cell bodies and promote neuroprotection and nerve regrowth without requiring concurrent systemic exposure at concentrations likely to block M1R in other organs.
The mechanism by which M1R antagonism leads to activation of CaMKKβ and subsequent mobilization of the AMPK/PGC-1α signaling axis can only be speculative at this juncture. Key requirements for CaMKKβ activation were a rise in intracellular Ca2+ and corresponding availability of calmodulin [27]. It is unlikely this source of Ca2+ is intracellular, for example from the endoplasmic reticulum (ER), since M1R antagonism blocks ACh-induced ER Ca2+ release driven by IP3, which itself is generated via G protein/PLC-dependent PIP2 breakdown. Many Ca2+ channels interact with PIP2 at the membrane, including diverse TRP channels [82] many of which are non-selective cation channels [83]. Interestingly, TRPM3 is one of the few TRP-type cation channels that is closed under low PIP2 levels—as observed in the presence of ACh-induced IP3 generation [84, 85]. Thus, antagonism of the M1R could lead to opening of TRPM3 and a local rise in [Ca2+]. TRPM3 is expressed in human and rodent DRG sensory neurons with a function linked to thermal sensory perception [83, 86, 87]. TRPM3 also has an intracellular binding site for Ca2+ and calmodulin that could act as a scaffold for CaMKKβ activation [88, 89]. Whether TRPM3 serves as a Ca2+ modulator following M1R antagonism by MT7 is currently under investigation. Another possible mediator of cytoplasmic Ca2+ following M1R blockade is activation of STIM2, a Ca2+ sensor that can trigger CaMKKβ and AMPK [90].
Conclusions
We have demonstrated that CaMKKβ is a key signaling molecule that mediates neurite outgrowth and drives axonal repair in neurodegenerative diseases of the peripheral nervous system. This enzyme lies upstream from the critical energy sensing pathway comprising AMPK/PGC-1α which regulates mitochondrial function to enhance axonal growth and protect from neurodegeneration. The ability of a topically applied antimuscarinic drug to activate this pathway in vivo suggests novel approaches to promote nerve regeneration in diseases of the PNS.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Albers KM, Davis BM (2007) The skin as a neurotrophic organ. Neuroscientist 13(4):371–382. https://doi.org/10.1177/10738584070130040901
Taylor AM, Ribeiro-da-Silva A (2011) GDNF levels in the lower lip skin in a rat model of trigeminal neuropathic pain: implications for nonpeptidergic fiber reinnervation and parasympathetic sprouting. Pain 152(7):1502–1510. https://doi.org/10.1016/j.pain.2011.02.035
Bennett GJ, Doyle T, Salvemini D (2014) Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol 10(6):326–336. https://doi.org/10.1038/nrneurol.2014.77
Cashman CR, Hoke A (2015) Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci Lett 596:33–50. https://doi.org/10.1016/j.neulet.2015.01.048
Vinik AI, Casellini C, Nevoret ML (2016) Alternative quantitative tools in the assessment of diabetic peripheral and autonomic neuropathy. Int Rev Neurobiol 127:235–285. https://doi.org/10.1016/bs.irn.2016.03.010
Malik RA (2016) Wherefore art thou, O treatment for diabetic neuropathy? Int Rev Neurobiol 127:287–317. https://doi.org/10.1016/bs.irn.2016.03.008
Lauria G, Morbin M, Lombardi R, Borgna M, Mazzoleni G, Sghirlanzoni A, Pareyson D (2003) Axonal swellings predict the degeneration of epidermal nerve fibers in painful neuropathies. Neurology 61(5):631–636
Schmidt RE (2002) Neuropathology and pathogenesis of diabetic autonomic neuropathy. Int Rev Neurobiol 50:257–292
Zherebitskaya E, Akude E, Smith DR, Fernyhough P (2009) Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes 58(6):1356–1364
Zochodne DW (2016) Sensory neurodegeneration in diabetes: beyond glucotoxicity. Int Rev Neurobiol 127:151–180. https://doi.org/10.1016/bs.irn.2016.03.007
Zochodne DW (2015) Diabetes and the plasticity of sensory neurons. Neurosci Lett 596:60–65. https://doi.org/10.1016/j.neulet.2014.11.017
Christianson JA, Ryals JM, Johnson MS, Dobrowsky RT, Wright DE (2007) Neurotrophic modulation of myelinated cutaneous innervation and mechanical sensory loss in diabetic mice. Neuroscience 145(1):303–313. https://doi.org/10.1016/j.neuroscience.2006.11.064
Jolivalt CG, Lee CA, Ramos KM, Calcutt NA (2008) Allodynia and hyperalgesia in diabetic rats are mediated by GABA and depletion of spinal potassium-chloride co-transporters. Pain 140(1):48–57. https://doi.org/10.1016/j.pain.2008.07.005
Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, Colvin LA, Fallon M (2014) Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain 155(12):2461–2470. https://doi.org/10.1016/j.pain.2014.09.020
Bernstein BW, Bamburg JR (2003) Actin-ATP hydrolysis is a major energy drain for neurons. J Neurosci 23(1):1–6
Wang SS, Shultz JR, Burish MJ, Harrison KH, Hof PR, Towns LC, Wagers MW, Wyatt KD (2008) Functional trade-offs in white matter axonal scaling. J Neurosci 28(15):4047–4056. https://doi.org/10.1523/JNEUROSCI.5559-05.2008
Chen H, Chan DC (2006) Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol 18(4):453–459. https://doi.org/10.1016/j.ceb.2006.06.004
Mironov SL (2007) ADP regulates movements of mitochondria in neurons. Biophys J 92(8):2944–2952. https://doi.org/10.1529/biophysj.106.092981
Kruger L, Perl ER, Sedivec MJ (1981) Fine structure of myelinated mechanical nociceptor endings in cat hairy skin. J Comp Neurol 198(1):137–154. https://doi.org/10.1002/cne.901980112
Ribeiro-da-Silva A, Kenigsberg RL, Cuello AC (1991) Light and electron microscopic distribution of nerve growth factor receptor-like immunoreactivity in the skin of the rat lower lip. Neuroscience 43(2–3):631–646
Fernyhough P (2015) Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Curr Diab Rep 15(11):89. https://doi.org/10.1007/s11892-015-0671-9
Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG (2005) Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2(1):9–19. https://doi.org/10.1016/j.cmet.2005.05.009
Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D (2005) Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2(1):21–33. https://doi.org/10.1016/j.cmet.2005.06.005
Green MF, Anderson KA, Means AR (2011) Characterization of the CaMKKbeta-AMPK signaling complex. Cell Signal 23(12):2005–2012. https://doi.org/10.1016/j.cellsig.2011.07.014
Feige JN, Auwerx J (2007) Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol 17(6):292–301. https://doi.org/10.1016/j.tcb.2007.04.001
Hardie DG (2008) AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 32 Suppl 4:S7–12. doi:https://doi.org/10.1038/ijo.2008.116
Marcelo KL, Means AR, York B (2016) The Ca2+/calmodulin/CaMKK2 axis: nature’s metabolic CaMshaft. Trends Endocrinol Metab 27(10):706–718. https://doi.org/10.1016/j.tem.2016.06.001
Akude E, Zherebitskaya E, Chowdhury SK, Smith DR, Dobrowsky RT, Fernyhough P (2011) Diminished superoxide generation is associated with respiratory chain dysfunction and changes in the mitochondrial proteome of sensory neurons from diabetic rats. Diabetes 60(1):288–297. https://doi.org/10.2337/db10-0818
Roy Chowdhury SK, Smith DR, Saleh A, Schapansky J, Marquez A, Gomes S, Akude E, Morrow D et al (2012) Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 135(Pt 6):1751–1766. https://doi.org/10.1093/brain/aws097
Ma J, Farmer KL, Pan P, Urban MJ, Zhao H, Blagg BS, Dobrowsky RT (2014) Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J Pharmacol Exp Ther 348(2):281–292. https://doi.org/10.1124/jpet.113.210435
Urban MJ, Pan P, Farmer KL, Zhao H, Blagg BS, Dobrowsky RT (2012) Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy. Exp Neurol 235(1):388–396. https://doi.org/10.1016/j.expneurol.2012.03.005
Kalichman MW, Powell HC, Mizisin AP (1998) Reactive, degenerative, and proliferative Schwann cell responses in experimental galactose and human diabetic neuropathy. Acta Neuropathol 95(1):47–56
Kamiya H, Zhang W, Sima AA (2006) Degeneration of the Golgi and neuronal loss in dorsal root ganglia in diabetic BioBreeding/Worcester rats. Diabetologia 49(11):2763–2774. https://doi.org/10.1007/s00125-006-0379-0
Brown DA, Passmore GM (2009) Neural KCNQ (Kv7) channels. Br J Pharmacol 156(8):1185–1195. https://doi.org/10.1111/j.1476-5381.2009.00111.x
Tata AM, Cursi S, Biagioni S, Augusti-Tocco G (2003) Cholinergic modulation of neurofilament expression and neurite outgrowth in chick sensory neurons. J Neurosci Res 73(2):227–234. https://doi.org/10.1002/jnr.10650
Yang H, Kunes S (2004) Nonvesicular release of acetylcholine is required for axon targeting in the Drosophila visual system. Proc Natl Acad Sci U S A 101(42):15213–15218. https://doi.org/10.1073/pnas.0308141101
Erskine L, McCaig CD (1995) Growth cone neurotransmitter receptor activation modulates electric field-guided nerve growth. Dev Biol 171(2):330–339. https://doi.org/10.1006/dbio.1995.1285
Bernardini N, Levey AI, Augusti-Tocco G (1999) Rat dorsal root ganglia express m1-m4 muscarinic receptor proteins. J Peripher Nerv Syst 4(3–4):222–232
Grando SA, Pittelkow MR, Schallreuter KU (2006) Adrenergic and cholinergic control in the biology of epidermis: physiological and clinical significance. J Invest Dermatol 126(9):1948–1965. https://doi.org/10.1038/sj.jid.5700151
Bellier JP, Kimura H (2007) Acetylcholine synthesis by choline acetyltransferase of a peripheral type as demonstrated in adult rat dorsal root ganglion. J Neurochem 101(6):1607–1618. https://doi.org/10.1111/j.1471-4159.2007.04458.x
Hanada K, Kishimoto S, Bellier JP, Kimura H (2013) Peripheral choline acetyltransferase in rat skin demonstrated by immunohistochemistry. Cell Tissue Res 351(3):497–510. https://doi.org/10.1007/s00441-012-1536-z
Calcutt NA, Smith DR, Frizzi K, Sabbir MG, Chowdhury SK, Mixcoatl-Zecuatl T, Saleh A, Muttalib N et al (2017) Selective antagonism of muscarinic receptors is neuroprotective in peripheral neuropathy. J Clin Invest 127(2):608–622. https://doi.org/10.1172/JCI88321
Sabbir MG, Calcutt NA, Fernyhough P (2018) Muscarinic acetylcholine type 1 receptor activity constrains neurite outgrowth by inhibiting microtubule polymerization and mitochondrial trafficking in adult sensory neurons. Front Neurosci 12:402. https://doi.org/10.3389/fnins.2018.00402
Sabbir MG, Fernyhough P (2018) Muscarinic receptor antagonists activate ERK-CREB signaling to augment neurite outgrowth of adult sensory neurons. Neuropharmacology 143:268–281. https://doi.org/10.1016/j.neuropharm.2018.09.020
Max SI, Liang JS, Potter LT (1993) Stable allosteric binding of m1-toxin to m1 muscarinic receptors. Mol Pharmacol 44(6):1171–1175
Gaff J, Octaviana F, Ariyanto I, Cherry C, Laws SM, Price P (2019) Polymorphisms in CAMKK2 associate with susceptibility to sensory neuropathy in HIV patients treated without stavudine. J Neuro-Oncol 25:814–824. https://doi.org/10.1007/s13365-019-00771-w
Calcutt NA, Jorge MC, Yaksh TL, Chaplan SR (1996) Tactile allodynia and formalin hyperalgesia in streptozotocin-diabetic rats: effects of insulin, aldose reductase inhibition and lidocaine. Pain 68(2–3):293–299
Marshall AG, Lee-Kubli C, Azmi S, Zhang M, Ferdousi M, Mixcoatl-Zecuatl T, Petropoulos IN, Ponirakis G et al (2017) Spinal disinhibition in experimental and clinical painful diabetic neuropathy. Diabetes 66(5):1380–1390. https://doi.org/10.2337/db16-1181
Galbraith JA, Mrosko BJ, Myers RR (1993) A system to measure thermal nociception. J Neurosci Methods 49(1–2):63–68. https://doi.org/10.1016/0165-0270(93)90109-5
Smith DS, Skene JH (1997) A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci 17(2):646–658
Green MF, Scott JW, Steel R, Oakhill JS, Kemp BE, Means AR (2011) Ca2+/calmodulin-dependent protein kinase kinase beta is regulated by multisite phosphorylation. J Biol Chem 286(32):28066–28079. https://doi.org/10.1074/jbc.M111.251504
Saleh A, Shan L, Halayko AJ, Kung S, Gounni AS (2009) Critical role for STAT3 in IL-17A-mediated CCL11 expression in human airway smooth muscle cells. J Immunol 182(6):3357–3365
Sabbir MG (2018) Loss of Ca2+/calmodulin dependent protein kinase kinase 2 leads to aberrant transferrin phosphorylation and trafficking: a potential biomarker for Alzheimer’s disease. Front Mol Biosci 5:99. https://doi.org/10.3389/fmolb.2018.00099
Rungta RL, Choi HB, Lin PJ, Ko RW, Ashby D, Nair J, Manoharan M, Cullis PR et al (2013) Lipid nanoparticle delivery of siRNA to silence neuronal gene expression in the brain. Mol Ther Nucleic Acids 2:e136. https://doi.org/10.1038/mtna.2013.65
Chowdhury SK, Zherebitskaya E, Smith DR, Akude E, Chattopadhyay S, Jolivalt CG, Calcutt NA, Fernyhough P (2010) Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 59(4):1082–1091. https://doi.org/10.2337/db09-1299
Jolivalt CG, Frizzi KE, Guernsey L, Marquez A, Ochoa J, Rodriguez M, Calcutt NA (2016) Peripheral neuropathy in mouse models of diabetes. Curr Protoc Mouse Biol 6(3):223–255. https://doi.org/10.1002/cpmo.11
Chen DK, Frizzi KE, Guernsey LS, Ladt K, Mizisin AP, Calcutt NA (2013) Repeated monitoring of corneal nerves by confocal microscopy as an index of peripheral neuropathy in type-1 diabetic rodents and the effects of topical insulin. J Peripher Nerv Syst 18(4):306–315. https://doi.org/10.1111/jns5.12044
Jolivalt CG, Marquez A, Quach D, Navarro Diaz MC, Anaya C, Kifle B, Muttalib N, Sanchez G et al (2019) Amelioration of both central and peripheral neuropathy in mouse models of type 1 and type 2 diabetes by the neurogenic molecule NSI-189. Diabetes 68(11):2143–2154. https://doi.org/10.2337/db19-0271
Nakamuta S, Funahashi Y, Namba T, Arimura N, Picciotto MR, Tokumitsu H, Soderling TR, Sakakibara A et al (2011) Local application of neurotrophins specifies axons through inositol 1,4,5-trisphosphate, calcium, and Ca2+/calmodulin-dependent protein kinases. Sci Signal 4(199):ra76. https://doi.org/10.1126/scisignal.2002011
Cao W, Sohail M, Liu G, Koumbadinga GA, Lobo VG, Xie J (2011) Differential effects of PKA-controlled CaMKK2 variants on neuronal differentiation. RNA Biol 8(6):1061–1072. https://doi.org/10.4161/rna.8.6.16691
Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G et al (2004) Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci 24(15):3786–3794. https://doi.org/10.1523/JNEUROSCI.3294-03.2004
Duan J, Cui J, Yang Z, Guo C, Cao J, Xi M, Weng Y, Yin Y et al (2019) Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3beta/Nrf2 signaling. J Neuroinflammation 16(1):24. https://doi.org/10.1186/s12974-019-1406-7
Zhang Y, Xu N, Ding Y, Doycheva DM, Zhang Y, Li Q, Flores J, Haghighiabyaneh M et al (2019) Chemerin reverses neurological impairments and ameliorates neuronal apoptosis through ChemR23/CAMKK2/AMPK pathway in neonatal hypoxic-ischemic encephalopathy. Cell Death Dis 10(2):97. https://doi.org/10.1038/s41419-019-1374-y
Dasgupta B, Milbrandt J (2007) Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A 104(17):7217–7222. https://doi.org/10.1073/pnas.0610068104
Schartner E, Sabbir MG, Saleh A, Silva RV, Roy Chowdhury S, Smith DR, Fernyhough P (2018) High glucose concentration suppresses a SIRT2 regulated pathway that enhances neurite outgrowth in cultured adult sensory neurons. Exp Neurol 309:134–147. https://doi.org/10.1016/j.expneurol.2018.08.001
Bugger H, Abel ED (2010) Mitochondria in the diabetic heart. Cardiovasc Res 88(2):229–240. https://doi.org/10.1093/cvr/cvq239
Gao AW, Canto C, Houtkooper RH (2014) Mitochondrial response to nutrient availability and its role in metabolic disease. EMBO Mol Med 6(5):580–589. https://doi.org/10.1002/emmm.201303782
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E et al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34(3):267–273. https://doi.org/10.1038/ng1180ng1180
Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I et al (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100(14):8466–8471. https://doi.org/10.1073/pnas.10329131001032913100
Richardson DK, Kashyap S, Bajaj M, Cusi K, Mandarino SJ, Finlayson J, DeFronzo RA, Jenkinson CP et al (2005) Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem 280(11):10290–10297. https://doi.org/10.1074/jbc.M408985200
Szendroedi J, Phielix E, Roden M (2012) The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 8(2):92–103. https://doi.org/10.1038/nrendo.2011.138
Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, Andreyev A, Quach T et al (2013) AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 123(11):4888–4899. https://doi.org/10.1172/JCI66218
Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA et al (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 26(13):3169–3179. https://doi.org/10.1038/sj.emboj.7601758
St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K et al (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127(2):397–408. https://doi.org/10.1016/j.cell.2006.09.024
Tsunemi T, La Spada AR (2012) PGC-1alpha at the intersection of bioenergetics regulation and neuron function: from Huntington’s disease to Parkinson’s disease and beyond. Prog Neurobiol 97(2):142–151. https://doi.org/10.1016/j.pneurobio.2011.10.004
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL et al (2011) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 144(5):689–702. https://doi.org/10.1016/j.cell.2011.02.010
Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF et al (2005) PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3(4):e101. https://doi.org/10.1371/journal.pbio.0030101
Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S et al (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119(1):121–135. https://doi.org/10.1016/j.cell.2004.09.013
Quattrini C, Tavakoli M, Jeziorska M, Kallinikos P, Tesfaye S, Finnigan J, Marshall A, Boulton AJ et al (2007) Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes 56(8):2148–2154. https://doi.org/10.2337/db07-0285
Ferdousi M, Azmi S, Petropoulos IN, Fadavi H, Ponirakis G, Marshall A, Tavakoli M, Malik I et al (2015) Corneal confocal microscopy detects small fibre neuropathy in patients with upper gastrointestinal cancer and nerve regeneration in chemotherapy induced peripheral neuropathy. PLoS One 10(10):e0139394. https://doi.org/10.1371/journal.pone.0139394
Petropoulos IN, Ponirakis G, Khan A, Gad H, Almuhannadi H, Brines M, Cerami A, Malik RA (2019) Corneal confocal microscopy: ready for prime time. Clin Exp Optom. https://doi.org/10.1111/cxo.12887
Rohacs T (2014) Phosphoinositide regulation of TRP channels. Handb Exp Pharmacol 223:1143–1176. https://doi.org/10.1007/978-3-319-05161-1_18
Nilius B, Owsianik G (2011) The transient receptor potential family of ion channels. Genome Biol 12(3):218. https://doi.org/10.1186/gb-2011-12-3-218
Toth BI, Konrad M, Ghosh D, Mohr F, Halaszovich CR, Leitner MG, Vriens J, Oberwinkler J et al (2015) Regulation of the transient receptor potential channel TRPM3 by phosphoinositides. J Gen Physiol 146(1):51–63. https://doi.org/10.1085/jgp.201411339
Badheka D, Rohacs T (2015) TRPM3 joins the ranks of PI(4,5)P2 sensitive ion channels. Channels (Austin) 9(5):233–234. https://doi.org/10.1080/19336950.2015.1089072
Straub I, Krugel U, Mohr F, Teichert J, Rizun O, Konrad M, Oberwinkler J, Schaefer M (2013) Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol Pharmacol 84(5):736–750. https://doi.org/10.1124/mol.113.086843
Vriens J, Owsianik G, Hofmann T, Philipp SE, Stab J, Chen X, Benoit M, Xue F et al (2011) TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 70(3):482–494. https://doi.org/10.1016/j.neuron.2011.02.051
Holendova B, Grycova L, Jirku M, Teisinger J (2012) PtdIns(4,5)P2 interacts with CaM binding domains on TRPM3 N-terminus. Channels (Austin) 6(6):479–482. https://doi.org/10.4161/chan.22177
Holakovska B, Grycova L, Jirku M, Sulc M, Bumba L, Teisinger J (2012) Calmodulin and S100A1 protein interact with N terminus of TRPM3 channel. J Biol Chem 287(20):16645–16655. https://doi.org/10.1074/jbc.M112.350686
Chauhan AS, Liu X, Jing J, Lee H, Yadav RK, Liu J, Zhou Y, Gan B (2019) STIM2 interacts with AMPK and regulates calcium-induced AMPK activation. FASEB J 33(2):2957–2970. https://doi.org/10.1096/fj.201801225R
Acknowledgments
We thank Dr. Michael Czubryt (University of Manitoba) and Dr. Anthony Means (Duke University Medical Center) for kindly donating plasmids. We are grateful to St Boniface Hospital Research for the support.
Funding
This work was supported by grant no. MOP-130282 from the Canadian Institutes of Health Research to P.F. and grant no. NS081082 from the National Institutes of Health to N.A.C.
Author information
Authors and Affiliations
Contributions
A.S. supervised and performed the lentivirus transduction and plasmid transfection, neurite outgrowth, and PGC-1α reporter studies. M.G.S. performed the nanoparticle transfection and Western blotting. M.-R.A. performed the Western blotting and data analysis on the trigeminal ganglia samples from in vivo experiments. D.S. supervised the maintenance of diabetic rats, performed the cell culture experiments (for example, Seahorse work), and aided in the data analysis and statistics. S.R. performed the Seahorse analysis. L.T. performed the Western blot work on P-AMPK, PGC-1α, and several neurite outgrowth experiments. J.B. performed the Western blotting for CaMK and CaMKKβ isoforms in different tissues. E.G., M.Z.K, and K.F. maintained and treated the diabetic and CIPN mice, collected and analyzed the corneal nerve images, and quantified, analyzed, and interpreted the resulting data. N.A.C. supervised the corneal confocal work and aided in the writing of the manuscript. P.F. supervised all the cell culture work and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
All animal protocols carefully followed Canadian Committee on Animal Care (CCAC) or Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) guidelines.
Conflict of Interest
The authors, P.F. and N.A.C., declare they are directors and shareholders in WinSanTor Inc., which has licensed IP from the University of Manitoba and UCSD. This IP includes some of the data presented in the current manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saleh, A., Sabbir, M.G., Aghanoori, MR. et al. Muscarinic Toxin 7 Signals Via Ca2+/Calmodulin-Dependent Protein Kinase Kinase β to Augment Mitochondrial Function and Prevent Neurodegeneration. Mol Neurobiol 57, 2521–2538 (2020). https://doi.org/10.1007/s12035-020-01900-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01900-x