
Abstract
Brain-derived neurotrophic factor (BDNF) is one of the most studied neurotrophins in the healthy and diseased brain. As a result, there is a large body of evidence that associates BDNF with neuronal maintenance, neuronal survival, plasticity, and neurotransmitter regulation. Patients with psychiatric and neurodegenerative disorders often have reduced BDNF concentrations in their blood and brain. A current hypothesis suggests that these abnormal BDNF levels might be due to the chronic inflammatory state of the brain in certain disorders, as neuroinflammation is known to affect several BDNF-related signaling pathways. Activation of glia cells can induce an increase in the levels of pro- and antiinflammatory cytokines and reactive oxygen species, which can lead to the modulation of neuronal function and neurotoxicity observed in several brain pathologies. Understanding how neuroinflammation is involved in disorders of the brain, especially in the disease onset and progression, can be crucial for the development of new strategies of treatment. Despite the increasing evidence for the involvement of BDNF and neuroinflammation in brain disorders, there is scarce evidence that addresses the interaction between the neurotrophin and neuroinflammation in psychiatric and neurodegenerative diseases. This review focuses on the effect of acute and chronic inflammation on BDNF levels in the most common psychiatric and neurodegenerative disorders and aims to shed some light on the possible biological mechanisms that may influence this effect. In addition, this review will address the effect of behavior and pharmacological interventions on BDNF levels in these disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brain disorders are among the major causes of disability and morbidity worldwide. According to recent projections, the incidence of such diseases will increase in the next decades [1]. The lack of adequate treatment turns these diseases into a significant problem worldwide, and the absence of effective treatment can partly be ascribed to our incomplete knowledge of the etiology of most brain disorders. Many mental diseases, however, are tightly associated with environmental stimuli, such as stress [2]. Challenging events can pose a significant burden on individuals, especially those more sensitive to its effects. Although acute stress can have benefits (e.g., enhanced attention, memory), it can also become life threatening when stressful events become a routine part of the life of individuals. A number of studies have already shown that stress is associated with metabolic changes, cardiovascular risk, endocrine abnormalities, mood changes, and impairment of cognitive functions (i.e., mild cognitive impairment), leading to an increased risk of developing psychiatric and neurologic disorders [2, 3]. Chronic stress can lead to the activation of pro-inflammatory microglia, releasing cytokines and pro-inflammatory substances, and recruitment of peripheral immune cells to the brain, thus creating the inflammatory environment that is characteristic for many brain pathologies.
In order to cope with stressful events, brain cells release several substances that can promote neuronal survival, such as antiinflammatory cytokines, growth factors and neurotrophic factors. One of the best-studied neurotrophins is the brain-derived neurotrophic factor (BDNF). Brain pathologies are usually associated with a downregulation of BDNF release, resulting in reduced BDNF levels in the brain and in blood. BDNF has been suggested as a candidate biomarker of pathological conditions, and therapy efficacy, as most of current treatments are accompanied by a significant change in blood BDNF levels. However, there is still a gap in our understanding of the physiological mechanisms that lead to changes in BDNF levels under pathological conditions.
This review will summarize our current knowledge of BDNF in the pathophysiology of the most common brain disorders. Since neuroinflammation has been considered an important mediator for the onset and progression of many brain pathologies, this review will also attempt to explore the interaction between neuroinflammation and BDNF expression in the brain.
BDNF Expression and Function
BDNF is a member of the neurotrophin family, which also includes neural growth factor (NGF) and neurotrophins 3 and 4. The Bdnf gene is comprised of a common 3′-exon that encodes the pro-BDNF region of the protein, and several species-dependent 5′-noncoding, promoter-regulated regions, terminating in a coding 5′-exon that encompasses the gene expression [4, 5]. Bdnf gene expression is strongly regulated by a wide array of endogenous and exogenous stimuli (e.g., stress, physical activity, brain injury, diet). BDNF is translated as a pro-neurotrophin (pro-BDNF) that can be cleaved into mature BDNF in the cytoplasm by endoproteases or in the extracellular matrix by plasmin or matrix metalloproteinases (MMP). Both mature BDNF and pro-BDNF can be secreted and bind to the low affinity p75 neurotrophin receptor (p75NTR), which causes activation of the apoptosis cascade [6, 7]. On the other hand, cleaved, mature BDNF binds to its high-affinity receptor tyrosine kinase B (TrkB), activating several signaling cascades, including the Ras-mitogen-activated protein kinase (MAPK), the phosphatidylinositol-3-kinase (PI3K), and the phospholipase Cγ (PLC-γ) pathway. These signaling cascades induce an increase in Ca2+ intake, phosphorylation of transcription factors, and de novo expression of the Bdnf gene (Fig. 1) [8]. Although pro-BDNF can act as a signaling factor for the apoptotic cascade, it is not yet clear if pro-BDNF is secreted by neurons under normal conditions as its concentration in presynaptic terminals is relatively low when compared to mature BDNF. Indeed, in animal models, the concentration of mature BDNF can be ten times higher than the concentration of pro-BDNF [9, 10], which poses a question regarding the efficacy of pro-BDNF as a proper signaling factor.

BDNF induces survival-related signaling mechanisms: BDNF induces survival-related signaling mechanisms: In physiological conditions, binding of BDNF to TrkB receptor in either paracrine or autocrine signaling elicits three distinct downstream pathways. BDNF-dependent phospholipase C-gamma (PLC-γ) can induce short-term signaling by increasing Ca2+ neuronal response and inhibit inflammatory-dependent apoptosis cascade (dashed lines) by inhibition of glycogen synthase kinase 3-beta (GSK-3β). Induction of phosphatidylinositol 3-Phosphate (PI3K) induces transcription of BDNF mRNA by activating mTOR-dependent translation of BDNF. Additionally, BDNF can modulate gene regulation by activating NF-κB and CREB transcription factors by inducing Akt and Erk downstream pathways, respectively. Gene modulation induces neuronal survival, growth, long-term potentiation (LTP), and de novo expression of BDNF. In addition, BDNF-independent transactivation of TrkB can also play an important role in the neurotrophic pathway regulation by factors, such as adenosine, zinc, epidermal growth factor (EGF), glucocorticoids, and pituitary adenylate-cyclase-activating polypeptide (PACAP), further enhancing TrkB signaling in the synapse
BDNF has a wide array of functions within the brain and is highly abundant in several brain structures. In the brain, BDNF is involved in plasticity, neuronal survival, formation of new synapses, dendritic branching, and modulation of excitatory and inhibitory neurotransmitter profiles [11, 12]. BDNF is active at all stages of development and aging [13]. Knockout mice lacking BDNF rarely reach adulthood and, when they do, there is a development of several sensory impairments [14, 15]. BDNF is also found in peripheral organs, such as the heart, gut, thymus, and spleen [16, 17]. Around 90% of the BDNF in blood is stored within platelets [18]. Many brain pathologies cause reduction of BDNF protein levels both in the brain and serum of patients [19,20,21,22]. Unfortunately, it is still unclear whether BDNF protein levels measured in serum samples reflect BDNF levels in the brain, as studies in animal models gave contradictory results so far [23,24,25].
BDNF in Neuroinflammation
After inflammatory signaling (e.g., stress, pro-inflammatory signals), several signaling cascades are changed within the cell. This signaling generates a sequence of events that may eventually lead to neuronal malfunction and apoptosis. Microglia also participate actively in the development of pathological neuroinflammatory process by releasing pro-inflammatory cytokines, which contribute to the neurotoxicity. This cycle is repeated as long as the stressor is present, which can develop into serious consequences (e.g., cognitive impairment, behavioral dysfunction, neurological and psychiatric disorders). One of the main factors of inflammatory activation is the nuclear factor-kappa B (NF-κB), a transcription factor that induces the expression of several pro- and antiapoptotic genes, including Bdnf [26]. Interestingly, binding of BDNF to the TrkB receptor can also induce the expression of NF-κB, although the pathways for this modulation are yet unclear (Fig. 2). NF-κB is closely involved in the innate and adaptive immune response in several psychiatric and neurodegenerative diseases [27]. NF-κB is a regulator of, e.g., apoptosis, neuronal survival and proliferation, and migration and maturation of immune cells [28]. BDNF-induced NF-κB expression stimulates PLC-γ/PKC signaling through the activation of the kinases IKKα and IKKβ. These kinases phosphorylate the NF-κB inhibitory unit IκBα, resulting in the binding of ubiquitin and subsequently degradation of IκBα by proteasomes [29]. IκBα degradation induces the release of the NF-κB and formation of the p50/p65 dimer, which binds to the DNA and induces the expression of genes related to neuronal proliferation, survival, and inflammatory response [29, 30]. Furthermore, it is known that BDNF can also bind to the p75NTR. Even though the affinity of BDNF for the p75NTR receptor is several times lower than for TrkB [31], a p75NTR-mediated effect on NF-κB expression can be observed. Studies have shown that activation of p75NTR increases apoptotic and inflammatory signaling in neurons and glial cells by activation of c-Jun N-terminal kinases (JNK) and NF-κB expression, respectively [32, 33]. However, the effect the p75NTR has on neurotrophic signaling is still under debate, as there is no clear evidence on how large the role of the p75NTR is in mediating such processes.

BDNF response after inflammatory brain pathogenicity. In chronically stressful situations, such as brain pathologies, there is an induction of NF-κB-dependent pro-inflammatory activation of microglia after induction of the pattern recognition receptor (PRR) by the challenge (e.g., stress or pathology). Pro-inflammatory cytokines, especially IL-1, can directly bind microglial cells, which result in induction of the expression and release of several mediators, most of which are neurotoxic, including reactive oxygen (ROS) and nitrogen species (RNS), pro-inflammatory cytokines (such as tumor necrosis factor), and chemokines (such as CC-chemokine ligand 2, CCL2; also known as MCP1). Additionally, there is a decrease of BDNF signaling in the synaptic cleft, further reducing BDNF-dependent survival-related signaling (black dashed lines) and inhibition of apoptotic pathways, such as glycogen synthase kinase 3-beta (GSK-3: red dashed line). Such factors will lead to an increase of NF-κB complex binding to genes that express pro-inflammatory cytokines (e.g., interleukin 1-β, IL-6, IL-8, TNF). The effect of transactivation factors on the BDNF-independent maintenance of TrkB is not clear yet
Thus, the role of BDNF in neuroinflammation is strongly related to its ability to induce—and being induced by—NF-κB. However, the exact regulatory mechanisms are not yet clear.
BDNF and Aging
The aging process can lead to the impairment of several brain functions. Studies have reported a decrease in whole brain volume in elderly when compared with young adults, especially in brain regions related to cognition [34,35,36,37]. Upon aging, microglia increasingly adopt a pro-inflammatory state due to a decrease in the resting signaling by neurons and astrocytes [38, 39]. As a result, external stimuli (e.g., stress, trauma, infection) can submit the aged brain more easily into a state of mild chronic neuroinflammation, making the brain more prone to apoptotic signaling [40]. This can lead to volume loss and the associated cognitive impairment [41]. Animal studies in stress models, such as chronic stress, maternal separation, and social defeat, have confirmed that stress is associated with glial activation and that aging decreases cognitive function [42, 43]. The loss of volume is indicative of a reduction in the global neuronal network, and consequently a diminished brain plasticity that could support the brain in such events, thus reducing the cognitive function [44, 45].
It is known that BDNF plays a role in maintaining brain function by inducing survival signaling and neuroplasticity. Although the effect is more visible in younger population, elderly subjects also benefit from it, especially those cognitively, physically, and socially active, reducing the risk of age-related comorbidities [46]. Studies in animal models report an increase in brain BDNF levels when aged animals are submitted to protocols such as long-term environmental enrichment [47,48,49] or physical activity [50, 51]. Studies on aged mice demonstrated that animals heterozygous for BDNF showed decreased fear extinction learning [52] and conditioned fear learning [53] when compared with young heterozygous mice, but the mechanism responsible for the behavioral effects is not completely clear yet. Better understanding of the processes that are modulated by BDNF may facilitate the development of novel therapeutic methods that could help to prevent or counteract the aging effects on the human brain.
BDNF in Psychiatric Disorders
Whenever the brain is challenged by harmful events, its coping mechanisms are activated in order to revert the system to homeostasis. When these coping mechanisms fail, e.g., due to excessive damage, enhanced sensitivity, and chronic or recurrent exposure to stimuli, a disturbance of normal brain function may occur that can lead to the onset of neuropsychiatric disorders (Table 1). Although psychiatric diseases can display wide spectra of symptoms, common phenomena in these disorders are a disarray of excitatory/inhibitory neurotransmitter signaling and loss of neuronal function, leading to mood and behavioral disturbances, and cognitive impairment.
In humans, BDNF is known to be a useful biomarker for several psychiatric disorders [54]. Most chronic psychiatric diseases are accompanied by changes in BDNF levels, but it is still unclear if changes in BDNF levels are the cause or the result of the disturbances of normal brain function. Therefore, the effects of BDNF levels have been investigated in animal models. Heterozygous BDNF mice show increased weight gain, aggressiveness, anxiety, and contextual memory impairment and therefore have been suggested as an animal model for mood disorders [55]. These results suggest that BDNF could be a key player in the development of several symptoms associated with psychiatric disorders. This hypothesis is supported by the fact that effective treatment of these symptoms resulted in normalization of BDNF levels [56]. In this section, we will further discuss the role of BDNF in the main psychiatric disorders: major depressive disorder, bipolar disorder, and schizophrenia.
Major Depressive Disorder
Major depressive disorder (MDD) is a common psychiatric disease characterized by abnormal behavior, anhedonia, sleep and dietary problems, cognitive impairment, and, in more severe cases, suicidal tendencies. Biologically, depression is related to a decrease of neurotransmitter signaling in the brain, dysfunction of hypothalamus pituitary adrenal axis (HPA-axis), increase in inflammatory signaling, and reduction in hippocampal volume.
Both MDD patients [57,58,59] and animal models of depression [60, 61] show a remarkable reduction in serum BDNF levels. Karege and colleagues have shown that this decrease is not related to platelet-associated BDNF release in the bloodstream [62], suggesting that reduced BDNF levels in the brain rather than a reduction in the peripheral release of BDNF by platelets are the cause of altered protein levels in blood. The magnitude of the decrease in plasma BDNF levels is associated with disease duration [63], but it is not clear yet whether the severity of symptoms is related to BDNF levels. BDNF single nucleotide polymorphism (val66met), however, is associated with the severity of depression in patients [64]. Successful antidepressant treatment is usually associated with an increase in BDNF levels in serum and plasma [65, 66], whereas treatment failure is associated with a lack of response of plasma BDNF levels. Thus, BDNF seems an important player in the pathophysiology and might be a biomarker for monitoring treatment response in depression [65, 67]. Epidemiological studies show that a third of all MDD patients experience no changes in the symptoms when treated with the most commonly used antidepressants. Postmortem studies revealed that these treatment-resistant patients have significantly lower BDNF levels, especially in BDNF-rich brain structures, such as the hippocampus [68,69,70]. Treatment with the rapidly acting antidepressant ketamine was able to increase plasma BDNF levels to the level of healthy controls [71, 72]. Ketamine is an inhibitor of NMDA receptors, inducing rapid, glutamate-dependent Ca2+ signaling and activation of cAMP response element-binding protein (CREB). Clearly, there is a need to increase our knowledge on the role of BDNF in MDD.
Induction of a pro-inflammatory response by systemic application of lipopolysaccharide (LPS) causes depressive-like symptoms (i.e., sickness behavior) in rodents [73, 74], which may also affect BDNF levels. Increased pro-inflammatory signaling leads to a reduction in the mRNA expression of Bdnf and other neurotrophins in plasticity-related brain structures, especially cortical regions [75]. The effects of reduced BDNF expression levels in mice, treated with a systemic LPS injection, can be counteracted by induction of TrkB-mediated signaling with the agonist 7,8-dihydroxyflavone, which leads to a reduction of depressive-like behavior [76]. Gibney and colleagues have found increased expression of interleukins IL-1β, IL-6, and tumor necrosis factor-α (TNF-α) and reduced expression of Bdnf genes in depressive-like rats 6 h after an inflammatory challenge. In this study, expression of cytokines returned to baseline levels after 48 h, but Bdnf mRNA remained low in frontal cortex and hippocampus [77]. In humans, chronic stress is one of the main precursors of depressive symptoms [78, 79]. In laboratory stress-conditioning, depressed patients show a higher inflammatory response, characterized by increased IL-6 release and NF-κB DNA binding, than healthy controls [80]. Depressive symptoms can also be caused by treatment that stimulate the immune system, such as interferon-α (IFN-α). Patients treated with IFN-α were shown to have decreased serum BDNF levels in combination with increased protein levels of the cytokines IL-1 and IL-2 [81, 82]. Interestingly, individuals that had higher BDNF levels at baseline showed better resilience to IFN-α-induced MDD.
These preclinical and clinical findings show that long-term exposure to stress or inflammation leads to a decrease in BDNF levels, reducing the capacity of the neurons to cope with further challenges (i.e., neuronal plasticity), and ultimately leading to a decreased function and neuronal death. Interestingly, treatment with antidepressants can result in an antiinflammatory response throughout the brain, mitigating the inflammatory unbalance to homeostatic levels and normalizing BDNF concentrations [83, 84]. However, further research is needed to understand the mechanisms involved in the regulation of BDNF by neuroinflammation.
Bipolar Disorder
Bipolar disorder (BD) is characterized by fluctuations of mood throughout lifetime, oscillating between depressive, euthymic, and manic episodes. BD is associated with cognitive impairment and other comorbidities that affect the quality of life of the individual [85,86,87]. BD is characterized by alterations in dopaminergic and glutamatergic neurotransmitter systems, mitochondrial dysfunction, and increased oxidative stress, which in turn are related to neuroinflammation, neurotoxicity and eventually neuronal death [87]. Two recent meta-analyses have shown that serum and plasma levels of BDNF in BD patients during depressive and manic episodes are decreased, but no difference in BDNF levels between BD patients in an euthymic episode and healthy controls was found [88, 89]. It is known that treatment with mood stabilizers increases BDNF levels in prefrontal cortex and hippocampus of animals by inducing promoter IV-driven expression [90, 91]. Also in humans, treatment for the manic or depressive phases of BD is associated with an increase in serum BDNF levels [92, 93].
Recent studies have shown an association between in manic and depressive stages of BD and a pro-inflammatory profile of immune cells [94, 95]. Steiner and colleagues have found that suicidal mood disorder patients had a significant increase of microglial cell density in the dorsolateral prefrontal cortex, anterior cingulate gyrus, and mediodorsal thalamus compared to healthy controls and non-suicidal, mood disorder patients [96]. The presence of immune cells clusters in these brain regions suggests a strong inflammatory response, which could trigger the suicidal predisposition of these patients [96]. Although plenty of literature is available on BDNF or neuroinflammation in BD, there is a severe lack of studies regarding the association between both mechanisms on BD. Only two studies have analyzed both BDNF and cytokine levels in BD patients, with somewhat different conclusions. Patas and colleagues have shown an association between both serum BDNF and plasma IL-6 levels with a depressive episode associated with melancholic trait [97], while Wang and colleagues have found an association for serum BDNF levels, but not for IL-1β or IL-6 [98]. Clearly, more studies are needed to elucidate the interaction between neuroinflammation, BDNF, and disease symptoms in BD.
Interestingly, the most commonly used therapeutic drugs—lithium and valproate—were able to reverse the inflammatory state in mood disorders [99,100,101]. The most common assumption is that lithium and valproate can inhibit glycogen synthase kinase 3 (GSK-3) and sodium channel function, respectively [102]. Inhibition of GSK-3 activity by lithium increases cellular levels of BDNF. GSK-3 can inhibit mammalian target-of-rapamycin (mTOR)—an important modulator of BDNF-dependent neuronal plasticity and survival—and thus affect proper BDNF signaling and impair optimal cellular function. In the manic phase of BD, there is a remarkable increase in PKC-mediated signaling, which is associated with BDNF-dependent Ca2+ induction. PKC isozymes are involved in the pro-inflammatory response mediated by macrophages [103] and, more recently, microglia [104] through activation of the NF-κB inflammation pathway. Treatment of BD patients with lithium or valproate inhibits PKC upregulation, normalizing its level to that of euthymic subjects, and increases BDNF levels [105]. PKC inhibitor tamoxifen enhances the capacity of lithium to reduce symptoms of mania in BD [105,106,107]. PKC inhibition probably suppresses the expression of NF-κB and consequently resolves the NF-κB-mediated inhibition of Bdnf expression, resulting in an increase in peripheral BDNF levels in BD. However, the mechanisms underlying the increase in BDNF levels in response to treatment should still be further investigated. Yet, current evidence suggest that a decrease in BDNF levels can be considered as a biomarker for both depressive and manic stages of BD.
Schizophrenia
Schizophrenia is a disease characterized by disturbances in the proper perception of a person’s surroundings. Schizophrenia is associated with a high suicide rate and accounts for a large number of hospitalizations, causing a significant burden to healthcare systems worldwide. The symptoms of schizophrenia comprise positive (e.g., hallucinations, delusions, confused thoughts, concentration impaired) negative (e.g., depression, anhedonia, self-neglect) and cognitive effects (e.g., memory, attention, reason impairments). Schizophrenic patients exhibit a decreased activation of γ-aminobutyric acid (GABA) signaling [108], inducing impaired neuronal activation, especially in dopaminergic neurons [109]. The etiology of schizophrenia is not fully understood yet and symptoms can vary between individual patients, making the diagnosis of schizophrenia challenging.
A recent meta-analysis revealed that serum BDNF levels in both drug-naïve and medicated schizophrenic patients are reduced. Serum BDNF levels in schizophrenic patients decrease with age but were independent of the dosage of medication [110]. However, it remains unclear if and how BDNF levels in the brain are altered in schizophrenic patients. Some studies report increased BDNF levels in frontal and temporal structures [111, 112], while others report decreased levels in the same brain structures [108, 113, 114]. Besides clinical observations, in vitro studies using the phencyclidine (PCP) psychosis model also give ambiguous results. Adachi and colleagues reported that exposure of cortical cultures to the non-competitive NMDA agonist PCP initially resulted in an increase in BDNF levels, whereas TrkB, ERK1/2, and Akt signaling were decreased [115]. In contrast, two other studies reported decreased BDNF mRNA expression in cortical slices after exposure to a low dose of PCP [116]. Taken together, in vitro, in vivo and clinical results seem to indicate that plasma BDNF levels are decreased in schizophrenic patients, but data on brain BDNF levels are contradictory.
Schizophrenia is a multifactorial disease, in which both genetic and environmental factors play a role. It is well established that physical and mental distresses can trigger psychotic behavior in (genetically) vulnerable patients [117]. These triggers can induce inflammatory changes that are associated with reduced neurotransmitter signaling, increased oxidative stress, and reduced synaptic branching [118]. Mondelli and colleagues have shown in leukocytes of first-episode schizophrenic patients that childhood trauma and the number recent stressful life events were negatively correlated with BDNF mRNA levels. BDNF levels were also negatively correlated with IL-6 expression, suggesting an inflammation mediated decrease in BDNF expression, or vice versa. Moreover, BDNF, IL-6, and cortisol levels correlated inversely with hippocampal volume [119]. A postmortem study demonstrated that schizophrenic patients have an increased inflammatory profile in dorsolateral prefrontal cortex. The group of patients with high levels of neuroinflammation had lower expression of BDNF [120]. As psychotic episodes are related with increased neuroinflammation and activated microglia [120, 121], it can be hypothesized that pro-inflammatory cytokines may be modulating BDNF mRNA expression via interaction of NF-κB or CREB transcription factors. It is known that schizophrenia patients have upregulated genes for inflammatory cytokines [122, 123], and downregulated Bdnf gene transcription [124, 125]. Neuroinflammation could be the key factor for the decrease of Bdnf gene expression in schizophrenic patients, as pro-inflammatory cytokines increase methylation of Bdnf gene, leading to a decrease of CREB binding to the specific Bdnf site.
Remarkably, drug treatment that is effective in controlling disease progression in schizophrenic patients can have diverse effects on peripheral BDNF protein levels [126,127,128]. In addition, some studies suggest that baseline BDNF levels in schizophrenia patients might reflect the susceptibility towards available drug therapies [129, 130]. Clearly, the interaction between neuroinflammation, BDNF levels, and treatment response should be better understood, as this could lead to identification of new targets for improved therapies.
BDNF in Neurodegenerative Disorders
Despite research on neurodegenerative disorders has been increasing exponentially, there are still gaps in our knowledge on the etiology, onset and progression of most neurodegenerative diseases. Treatment is usually restricted to mitigation of the symptoms, rather than cure or delay of progression. Diagnosis of neurodegenerative diseases is usually based on subjective cognitive tests in combination with neuroimaging [131,132,133], but the current techniques are not able to successfully diagnose these diseases in their earlier stages, when the pathology is already present but does not cause symptoms yet. Attempts to discover new biomarkers for the diagnosis of early stages of the disease are ongoing [134,135,136]. Possibly, BDNF could qualify as such a biomarker.
In the following sections, we will discuss the role of BDNF in neurodegenerative disorders, focusing of Alzheimer’s disease, Parkinson’s disease, and epilepsy. We will also address the possible use of BDNF as a biomarker for diagnosis. In addition, we describe the role of neuroinflammation in the development of these diseases and explain how BDNF can help the brain to cope with inflammation.
Alzheimer’s Disease
AD is characterized by a progressive loss of neurons in the brain, leading to impairment in memory and general cognition. Hallmarks of AD pathology are deposition of amyloid-β plaques in the extracellular matrix, formation of tau-phosphorylated neurofibrillary tangles within the cell and neuritic plaques. Tangles and plaques disrupt the signaling activity of neurons, eventually leading to neuronal apoptosis. As the disease progress, the axonal transport is constantly reduced, and the general function of neurons is impaired. These changes decrease BDNF axonal transport, resulting in reduced availability of BDNF within the synaptic cleft and consequently diminished signaling through TrkB receptors [137, 138]. BDNF mRNA and protein levels are also reduced in cognition-related structures such as hippocampus and frontal cortex, which corroborates BDNF depletion to be involved in the cognitive deficit leading to AD dementia [139]. BDNF levels are also reduced in plasma of patients with mild cognitive impairment (MCI) [140] and AD [141]. AD patients with higher serum concentrations of BDNF showed less cognitive decline after 1 year; this effect was more pronounced in the more severe stages of the disease [142, 143]. These studies suggest that BDNF might be a predictor for the rate of disease progression in AD.
Neuroinflammation is a key factor in the development and progression of AD [144, 145]. Amyloid-β deposits induce pro-inflammatory activation of microglia which might be an effort of microglia to mitigate the antigen-related damage [146]. Some studies have reported inflammatory challenges as an associated risk factor for the development of dementia-related symptoms, as they show increased pro-inflammatory cytokines levels [147, 148]. On the other hand, treatment with antiinflammatory medication tends to mitigate cognitive impairment in animal models of amyloid-β injection [149, 150]. Interestingly, PET imaging studies have shown that subjects with high amounts of amyloid-β, but no dementia, had decreased microglia activation [151,152,153], indicating a fundamental participation of microglia in the development and progression of AD.
There is no clear evidence on how effective BDNF can be in controlling neuroinflammation-dependent progression of the disease. Prakash shows that rats injected with Aβ in the hippocampus have a remarkable decrease in BDNF and increased TNF-α, IL-6, and caspase-3 protein levels 3 weeks after intracerebroventricular injection. These changes were associated with inability lack of memory retention in Morris Water Maze test [154]. Another study confirmed the increase in pro-inflammatory cytokines and reduced antiinflammatory cytokines and BDNF protein levels and gene expression after Aβ1–42 injection [155]. In humans, two studies have shown an increase in pro-inflammatory cytokines and a decrease in BDNF levels in the serum of early- and late-onset AD, although there was no correlation with pro-inflammatory and neurotrophin results in both studies [141, 156].
It is known, however, that pro-inflammatory cytokines—especially IL-1β—can cause downregulation of BDNF expression in cognition-related brain structures, such as the hippocampus [157, 158]. As these cytokines are upregulated in AD, a decrease of BDNF levels is expected to occur in such brain structures, leading to a decrease in survival signaling and, consequently, neuronal death. Moreover, increase in hyper-phosphorylated tau impairs anterograde transport of BDNF to the axon, further decreasing BDNF signaling in the synaptic cleft. As AD is a disease that inflicts several different physiological effects in both the internal and external milieus of the brain, efficient analysis of the role of BDNF in these processes is challenging and the overall effect of BDNF on brain function may be variable.
Parkinson’s Disease
PD is characterized by movement impairment (bradykinesia, tremors, and rigidity), often combined with mild cognitive symptoms (decreased attention, executive function, memory) and mood disturbances (apathy, aggressiveness, anhedonia, depression) [159, 160]. The onset of PD is caused by the formation of aggregated α-synuclein plaques (i.e., Lewy bodies) in the substantia nigra pars compacta, leading to a progressive loss of dopaminergic neurons [161]. It is estimated that clinical symptoms start to appear when more than 50% of the neurons in the substantia nigra are already lost [162]. Movement symptoms are usually the first signs leading to the diagnosis of PD. As the disease progresses, more brain structures become affected by the neuronal loss and non-motor symptoms become evident [161, 163]. The severity of symptoms increases as the disease progresses and as a result, the patient loses gradually independence until patients become highly dependent on caregiver support in the late stages of the disease.
PD patients have lower concentrations of BDNF mRNA and protein in the substantia nigra pars compacta than healthy controls [164, 165]. Neurons with the lowest BDNF levels were suggested to be most prone to injury. Porritt and colleagues demonstrated that local inhibition of the production of BDNF with an antisense oligonucleotide leads to a significant loss of dopaminergic neurons in the substantia nigra pars compacta of rats, which suggests that BDNF has an important role in neuronal survival [166]. In contrast to the aforementioned studies, some reports describe that the BDNF levels in serum are increased in PD patients, especially in moderate to severe stages of the disease [167, 168]. This could mean that the CNS tries to cope with the loss of neurons by increasing BDNF production, resulting in enhanced serum levels of the protein. However, there is no direct evidence that supports this hypothesis.
The onset and progression of PD are also associated with neuroinflammation. Several studies in animal models of PD have reported increased microglial activation and pro-inflammatory cytokines [169, 170]. In humans, PD is associated with neuroinflammation in both postmortem [171] and in vivo analysis [172, 173]. Sawada and colleagues have found a remarkable increase of microglial cells in the hippocampus, amygdala, and entorhinal cortex of PD patients, which was associated with a decrease of BDNF mRNA expression and increased IL-6 in those regions [174]. Nagatsu has shown increased levels of IL-1β, IL-2, IL-6, and TNF-α in the striatum of PD patients, associated with a decreased BDNF protein levels in the same structure. Aggregated α-synuclein can induce an acute, local neuroinflammatory process in PD-associated brain structures, which suppresses BDNF expression and reduces BDNF protein levels. However, there is no evidence on how changes in BDNF levels in de brain affect the progression of PD and further analysis of the interaction between pro-inflammatory cytokines and BDNF is therefore necessary.
Epilepsy
Epilepsy patients are affected by seemingly unprovoked seizures but usually also suffer from significant mood and cognitive changes [175]. The symptoms of epilepsy are induced by a disarray of excitatory neuronal connections, generating deregulated firing, or lack of inhibition of excitatory neurons. Although there are several treatment strategies to reduce seizures, 30% of the patients show little to no response to common antiepileptic drugs.
Seizures have been associated with an increased expression of several neurotrophic-related genes, including transcription factors [176], neuropeptides [177], and growth factors [178]. Two recent reports have shown that BDNF gene expression is increased in the hippocampus and temporal cortex of temporal lobe epilepsy patients [179, 180]. Seizures also increased hippocampal and cortical BDNF protein levels in animal model of epilepsy [181,182,183]. BDNF seems to be involved in epileptogenesis by regulating several signaling pathways within excitatory neurons, increasing Ca2+ signaling and glutamate expression [13, 184]. Overexpression of BDNF may also contribute to the epilepsy-induced cortical network deregulation by increasing even further the plasticity and dendritic branching signaling and causing an overexcitement state of glutamatergic neurons, thus reducing seizure threshold [185]. This creates a positive feedback loop: as upregulated glutamate neurotransmitter increases BDNF signaling and expression, it further increases glutamate signaling. Other studies have shown that transient inhibition of the BDNF receptor TrkB after seizure induction prevents the development of temporal lobe epilepsy [186, 187]. These findings indicate that BDNF is intimately related to the pathogenesis of epilepsy, and new therapeutic methods should take BDNF into consideration. However, it is worth noting that BDNF may also play a part in protecting neurons against harmful stimuli; therefore, treatment aiming to reduce BDNF levels as a whole should be carefully considered.
In epilepsy, chronic seizures lead to excitotoxicity and neuronal apoptosis with associated gliosis [188]. Studies in animal models have reported that seizures are associated with increased activation of microglia, especially of the M1 subtype [189], and an increased release of pro-inflammatory mediators [190, 191]. Although studies that link BDNF with neuroinflammation in epilepsy are lacking, a hypothesis for such a link can be formulated based on existing knowledge. In healthy conditions, BDNF signaling induces the activation of transcription factor NF-κB, which in turn induces de novo expression of the Bdnf gene [26, 192]. It is possible that overexpression of BDNF leads to an inflammatory response mediated by NF-κB, leading to astrocyte activation, increased production of cytokines and neurotoxic reactive oxygen species, and local recruitment of activated microglia [193]. Activated pro-inflammatory microglia are known to elicit apoptotic response after chronic stimulation, which causes neurotoxicity and further neuronal death. As BDNF is overexpressed at the onset of seizures [194], there is also an increase in BDNF-mediated glutamate signaling, contributing to the systemic neuronal imbalance. Although there are several players involved in the development of seizures, BDNF might be an important factor in modulating the disease. BDNF inducing, or being induced, by NF-κB also highlights the importance of neuroinflammation in modulating BDNF-dependent regulation. However, there is a need for further researches to better understand the role of BDNF in epilepsy, especially regarding its role in modulation of inflammatory processes.
BDNF a Potential Therapy for Brain Pathologies
BDNF has been regarded as a possible biomarker for monitoring the onset, progression, and treatment of brain pathologies. However, recent studies suggest that BDNF may also be a potential target for new treatment strategies. Some promising results have been published from studies showing that therapeutic use of BDNF can, directly or indirectly, modulate changes within the brain [195]. An important finding is that peripheral BDNF is able to cross the blood-brain barrier [196], which is a prerequisite for BDNF-related therapy. However, the rate at which BDNF is taken up by the brain has not been quantified yet. Nonetheless, experimental therapy has been investigated in vivo and in vitro with promising results in animal models of AD [197], PD [198], and MDD [199] (a comprehensive review on BDNF drug delivery in brain pathologies and current stage of clinical trial can be found in [200]). For example, BDNF infusion into the hippocampus of adult rats was able to increase neurogenesis and regional neuronal activity [201]. Delivery of the BDNF mimetic 7-8-dihydroxiflavone is able to revert cognitive deficits in an AD animal model [202]. Also, gene transfection of BDNF into a 6-hydroxydopamine-induced unilateral lesion in the striatum was able to revert motor deficits in this animal model of PD [203].
The current need for improved treatments for brain disorders is pressing, and although large amounts of resources are spent on new therapies, few have been able to bring the desired results. The initial findings suggest that BDNF may be more than a biomarker for brain disorders; it may also become a possible target for the treatment of brain disorders. However, there is still a need for more information about the pharmacological features of BDNF-based substances in order to develop new treatments.
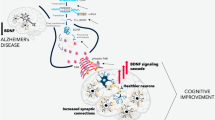
BDNF levels are activity-dependent, which means that the expression of BDNF changes under positive (e.g., physical activity, cognitive enhancement) and negative (e.g., obesity, sedentarism) behavioral and environmental stimuli. Several studies on both humans and animals show that physical activity, cognitive stimulation, and a balanced diet can stimulate BDNF expression [204,205,206,207]. Physical activity is the best studied positive factor for stimulation of BDNF expression. Thus, exercise can act as an inductor of neuronal plasticity, neurogenesis and neuronal survival [208, 209]. Several studies in both animals and humans have assessed the effects of physical activity on BDNF levels in psychiatric [210,211,212,213] and neurodegenerative diseases [214,215,216,217]. These studies indeed indicated that physical activity augments neuronal protection in brain disorders by stimulating BDNF expression. However, there are still some questions regarding the required time of the physical intervention, the optimal type of exercise (i.e., strength, endurance, aerobic exercises). Although less studied, diet can also provoke changes BDNF levels in physiological conditions. BDNF is known to affect the regulation of feeding and energy metabolism [218,219,220,221]. Decreased levels of BDNF were found in subjects consuming diets with high sugar and fat [222, 223]. On the other hand, dietary restriction (i.e., the maintenance of a balanced diet) or addition of supplementary substances (e.g., omega-3 fatty acids; resveratrol) can induce an increase in BDNF levels and reduce cognitive impairment in animal models [224,225,226]. It is not clear yet how large the effect of dietary management on BDNF protein levels in psychiatric or neurodegenerative disorders, but it is known that BDNF affects—and is affected by—dietary behavior. More research is needed in order to better understand the dietary influence on brain disorders.
Lifestyle changes that modulate BDNF levels in the brain may prove capable to control symptoms and delay progression in brain pathologies and thus might become cheap and easily accessible (adjuvant) treatment for these pathologies in the near future. However, a possible concern about such therapies is the compliance of the patients with the treatment. Most of behavioral therapies are based on long periods of treatment with a relatively slow improvement when compared with medications. This may reduce the motivation of patients to continue treatment, especially because most psychiatric and neurodegenerative disorders can be accompanied by mood symptoms (e.g., anhedonia, hopelessness, aggressiveness). Therefore, slow-acting lifestyle therapies could yield better results if they are combined with fast-acting pharmacological interventions.
Concluding Remarks
BDNF is involved in several processes that are essential for the optimal functioning of the brain. Several studies report altered brain and plasma BDNF levels in patients with various brain pathologies. However, the pathophysiological mechanisms that underlie these changes are still not fully understood yet. There is also no conclusive evidence that can discriminate whether changes in BDNF levels are a causative or the consequence of the disease onset. It is challenging to prove a causative role of BDNF in psychiatric and neurodegenerative disorders. Some circumstantial evidence suggest such a causative role for BDNF. Human populations with a genetic variant that decreases the BDNF concentration appear to be more susceptible to psychiatric disorders. However, it should be noted that BDNF is highly affected by environmental changes, which in turn may affect genetic findings. In animals, injection of BDNF in the hippocampus was shown to cause a decrease of depressive-like behavior. However, it remains unclear how these findings translate to the clinical situation, as current disease models are not able to properly reproduce complex human disorders, because they usually only mimic part of the symptoms (i.e., low predictive validity). Direct translation of the animal findings to humans would imply an intervention by intrathecal BDNF injection in the brain or spinal cord of patients. However, such an intervention would be highly invasive and the success would depend on the ability of BDNF to migrate to the target region. Alternatively, interventions that aim to stimulate the production of BDNF could be prospectively investigated in patients with low BDNF levels, either due to a genetic predisposition or environmental factors. BDNF levels should be monitored repetitively to determine if changes in BDNF levels precede alleviation of symptoms or vice versa. In a population with a genetic predisposition for low BDNF levels, preventive intervention with a BDNF stimulating treatment could also be considered, but this would require a large study population and a long follow-up time.
Evidence from preclinical studies and clinical trials suggests that treatment strategies aiming to increase brain BDNF levels could have a beneficial effect on many brain disorders. In this respect, epilepsy is an exception as it is associated with increased levels of BDNF. At present, pharmacological intervention by administration of exogenous BDNF is still challenging, but lifestyle changes could provoke the desired effect. Environmental and physiological stimuli, such as physical activity, social interactions, and sensory and cognitive stimuli, are powerful modifiers of neurotrophin levels, including BDNF levels, and have been shown to alleviate symptoms of brain pathologies in both humans and animal models. Plasma BDNF can be reliably assayed, and samples are easily collected and therefore BDNF has been proposed as a potential peripheral biomarker for the assessment of the status of the brain and the efficacy of treatment. However, discrepancies between brain and plasma BDNF alterations have been observed and need to be further elucidated before plasma BDNF can qualify as a suitable biomarker.
Neuroinflammation is regulated by factors that are also involved in modulation of BDNF expression. Both neuroinflammation and altered BDNF expression are common phenomena in many brain disorders. Remarkably, there are only few studies that have investigated the link between BDNF and neuroinflammation. Better understanding of the interaction between BDNF and neuroinflammation could open new ways for therapy management and could facilitate the development of new therapeutic strategies for brain diseases.
References
Whiteford HA, Degenhardt L, Rehm J et al (2013) Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet 382:1575–1586. https://doi.org/10.1016/S0140-6736(13)61611-6
Lupien SJ, McEwen BS, Gunnar MR, Heim C (2009) Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci 10:434–445. https://doi.org/10.1038/nrn2639
McEwen BS (2012) Brain on stress: how the social environment gets under the skin. Proc Natl Acad Sci U S A 109(Suppl):17180–17185. https://doi.org/10.1073/pnas.1121254109
Aid T, Kazantseva A, Piirsoo M et al (2007) Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res 85:525–535. https://doi.org/10.1002/jnr.21139
Pruunsild P, Kazantseva A, Aid T et al (2007) Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90:397–406. https://doi.org/10.1016/j.ygeno.2007.05.004
Barker PA (2009) Whither proBDNF? Nat Neurosci 12:105–106. https://doi.org/10.1038/nn0209-105
Lu B, Pang PT, Woo NH (2005) The yin and yang of neurotrophin action. Nat Rev Neurosci 6:603–614. https://doi.org/10.1038/nrn1726
Minichiello L (2009) TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10:850–860. https://doi.org/10.1038/nrn2738
Matsumoto T, Rauskolb S, Polack M et al (2008) Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci 11:131–133. https://doi.org/10.1038/nn2038
Dieni S, Matsumoto T, Dekkers M et al (2012) BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol 196:775–788. https://doi.org/10.1083/jcb.201201038
Edelmann E, Lessmann V, Brigadski T (2014) Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 76(Pt C):610–627. https://doi.org/10.1016/j.neuropharm.2013.05.043
Panja D, Bramham CR (2014) BDNF mechanisms in late LTP formation: a synthesis and breakdown. Neuropharmacology 76(Pt C):664–676. https://doi.org/10.1016/j.neuropharm.2013.06.024
Park H, Poo MM (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14:7–23. https://doi.org/10.1038/nrn3379
Ernfors P, Lee K-F, Jaenisch R (1994) Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368:147–150. https://doi.org/10.1038/368147a0
Jones KR, Fariñas I, Backus C, Reichardt LF (1994) Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76:989–999. https://doi.org/10.1016/0092-8674(94)90377-8
Lommatzsch M, Quarcoo D, Schulte-Herbrüggen O et al (2005) Neurotrophins in murine viscera: a dynamic pattern from birth to adulthood. Int J Dev Neurosci 23:495–500. https://doi.org/10.1016/j.ijdevneu.2005.05.009
Lommatzsch M, Braun A, Mannsfeldt A et al (1999) Abundant production of brain-derived neurotrophic factor by adult visceral epithelia. Implications for paracrine and target-derived neurotrophic functions. Am J Pathol 155:1183–1193. https://doi.org/10.1016/S0002-9440(10)65221-2
Fujimura H, Altar CA, Chen R et al (2002) Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb Haemost 87:728–734. https://doi.org/10.1016/j.bbi.2008.05.010
Jiang H, Chen S, Li C et al (2017) The serum protein levels of the tPA-BDNF pathway are implicated in depression and antidepressant treatment. Transl Psychiatry 7:e1079. https://doi.org/10.1038/tp.2017.43
Borba EM, Duarte JA, Bristot G et al (2016) Brain-derived neurotrophic factor serum levels and hippocampal volume in mild cognitive impairment and dementia due to Alzheimer disease. Dement Geriatr Cogn Dis Extra 91:559–567. https://doi.org/10.1159/000450601
Galvez-Contreras AY, Campos-Ordonez T, Lopez-Virgen V et al (2016) Growth factors as clinical biomarkers of prognosis and diagnosis in psychiatric disorders. Cytokine Growth Factor Rev. https://doi.org/10.1016/j.cytogfr.2016.08.004
Autry AE, Monteggia LM (2012) Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 64:238–258. https://doi.org/10.1124/pr.111.005108
Karege F, Schwald M, Cisse M (2002) Postnatal developmental profile of brain-derived neurotrophic factor in rat brain and platelets. Neurosci Lett 328:261–264. https://doi.org/10.1016/S0304-3940(02)00529-3
Sartorius A, Hellweg R, Litzke J et al (2009) Correlations and discrepancies between serum and brain tissue levels of neurotrophins after electroconvulsive treatment in rats. Pharmacopsychiatry 42:270–276. https://doi.org/10.1055/s-0029-1224162
Klein AB, Williamson R, Santini MA et al (2011) Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int J Neuropsychopharmacol 14:347–353. https://doi.org/10.1017/S1461145710000738
Marini AM, Jiang X, Wu X et al (2004) Role of brain-derived neurotrophic factor and NF-kappaB in neuronal plasticity and survival: from genes to phenotype. Restor Neurol Neurosci 22:121–130
Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD (2011) NF-κB in aging and disease. Aging Dis 2:449–465
Oeckinghaus A, Ghosh S (2009) The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1:1–14. https://doi.org/10.1101/cshperspect.a000034
Mattson MP, Meffert MK (2006) Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ 13:852–860. https://doi.org/10.1038/sj.cdd.4401837
Burstein E, Duckett CS (2003) Dying for NF-kappaB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr Opin Cell Biol 15:732–737. https://doi.org/10.1016/j.ceb.2003.10.005
Bernard-Gauthier V, Boudjemeline M, Rosa-Neto P et al (2013) Towards tropomyosin-related kinase B (TrkB) receptor ligands for brain imaging with PET: radiosynthesis and evaluation of 2-(4-[18F] fluorophenyl)-7,8-dihydroxy-4H-chromen-4-one and 2-(4-([N-methyl-11C]-dimethylamino)phenyl)-7,8-dihydroxy-4H-chromen-4-one. Bioorganic Med Chem 21:7816–7829. https://doi.org/10.1016/j.bmc.2013.10.012
Hempstead BL (2002) The many faces of p75NTR. Curr Opin Neurobiol 12:260–267. https://doi.org/10.1016/S0959-4388(02)00321-5
Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4:299–309. https://doi.org/10.1038/nrn1078
Giorgio A, Santelli L, Tomassini V et al (2010) Age-related changes in grey and white matter structure throughout adulthood. Neuroimage 51:943–951. https://doi.org/10.1016/j.neuroimage.2010.03.004
Kalpouzos G, Chételat G, Baron JC et al (2009) Voxel-based mapping of brain gray matter volume and glucose metabolism profiles in normal aging. Neurobiol Aging 30:112–124. https://doi.org/10.1016/j.neurobiolaging.2007.05.019
Tisserand DJ (2004) A voxel-based morphometric study to determine individual differences in gray matter density associated with age and cognitive change over time. Cereb Cortex 14:966–973. https://doi.org/10.1093/cercor/bhh057
Manard M, Bahri MA, Salmon E, Collette F (2016) Relationship between grey matter integrity and executive abilities in aging. Brain Res 1642:562–580. https://doi.org/10.1016/j.brainres.2016.04.045
Neumann H (2001) Control of glial immune function by neurons. Glia 36:191–199. https://doi.org/10.1002/glia.1108
Cardona AE, Pioro EP, Sasse ME et al (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924. https://doi.org/10.1038/nn1715
Niraula A, Sheridan JF, Godbout JP (2017) Microglia priming with aging and stress. Neuropsychopharmacology 42:318–333. https://doi.org/10.1038/npp.2016.185
von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J (2015) Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci 7:124
Wang J, Yuan J, Pang J et al (2016) Effects of chronic stress on cognition in male SAMP8 mice. Cell Physiol Biochem 39:1078–1086. https://doi.org/10.1159/000447816
Sousa VC, Vital J, Costenla AR et al (2014) Maternal separation impairs long term-potentiation in CA1-CA3 synapses and hippocampal-dependent memory in old rats. Neurobiol Aging 35:1680–1685. https://doi.org/10.1016/j.neurobiolaging.2014.01.024
Goh JO, Park DC (2009) Neuroplasticity and cognitive aging: the scaffolding theory of aging and cognition. Restor Neurol Neurosci 27:391–403
Calabrese F, Guidotti G, Racagni G, Riva MA (2013) Reduced neuroplasticity in aged rats: a role for the neurotrophin brain-derived neurotrophic factor. Neurobiol Aging 34:2768–2776. https://doi.org/10.1016/j.neurobiolaging.2013.06.014
Stern Y (2012) Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 11:1006–1012
Nithianantharajah J, Hannan AJ (2006) Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci 7:697–709. https://doi.org/10.1038/nrn1970
Cao W, Duan J, Wang X et al (2014) Early enriched environment induces an increased conversion of proBDNF to BDNF in the adult rat’s hippocampus. Behav Brain Res 265:76–83. https://doi.org/10.1016/j.bbr.2014.02.022
Jha S, Dong BE, Xue Y et al (2016) Antidepressive and BDNF effects of enriched environment treatment across ages in mice lacking BDNF expression through promoter IV. Transl Psychiatry 6:e896. https://doi.org/10.1038/tp.2016.160
Marlatt MW, Potter MC, Lucassen PJ, van Praag H (2012) Running throughout middle-age improves memory function, hippocampal neurogenesis, and BDNF levels in female C57BL/6J mice. Dev Neurobiol 72:943–952. https://doi.org/10.1002/dneu.22009
O’Callaghan RM, Griffin EW, Kelly AM (2009) Long-term treadmill exposure protects against age-related neurodegenerative change in the rat hippocampus. Hippocampus 19:1019–1029. https://doi.org/10.1002/hipo.20591
Psotta L, Lessmann V, Endres T (2013) Impaired fear extinction learning in adult heterozygous BDNF knock-out mice. Neurobiol Learn Mem 103:34–38. https://doi.org/10.1016/j.nlm.2013.03.003
Endres T, Lessmann V (2012) Age-dependent deficits in fear learning in heterozygous BDNF knock-out mice. Learn Mem 19:561–570. https://doi.org/10.1101/lm.028068.112
Chen S, Jiang H, Liu Y et al (2017) Combined serum levels of multiple proteins in tPA-BDNF pathway may aid the diagnosis of five mental disorders. Sci Rep 7:6871. https://doi.org/10.1038/s41598-017-06832-6
Lindholm JSO, Castrén E (2014) Mice with altered BDNF signaling as models for mood disorders and antidepressant effects. Front Behav Neurosci 8:143. https://doi.org/10.3389/fnbeh.2014.00143
Nuernberg GL, Aguiar B, Bristot G et al (2016) Brain-derived neurotrophic factor increase during treatment in severe mental illness inpatients. Transl Psychiatry 6:e985. https://doi.org/10.1038/tp.2016.227
Gonul AS, Akdeniz F, Taneli F et al (2005) Effect of treatment on serum brain-derived neurotrophic factor levels in depressed patients. Eur Arch Psychiatry Clin Neurosci 255:381–386. https://doi.org/10.1007/s00406-005-0578-6
Karege F, Perret G, Bondolfi G et al (2002) Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res 109:143–148. https://doi.org/10.1016/S0165-1781(02)00005-7
Shimizu E, Hashimoto K, Okamura N et al (2003) Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry 54:70–75. https://doi.org/10.1016/S0006-3223(03)00181-1
Chen Q, Luo Y, Kuang S et al (2017) Cyclooxygenase-2 signalling pathway in the cortex is involved in the pathophysiological mechanisms in the rat model of depression. Sci Rep 7:488. https://doi.org/10.1038/s41598-017-00609-7
Larsen MH, Mikkelsen JD, Hay-Schmidt A, Sandi C (2010) Regulation of brain-derived neurotrophic factor (BDNF) in the chronic unpredictable stress rat model and the effects of chronic antidepressant treatment. J Psychiatr Res 44:808–816. https://doi.org/10.1016/j.jpsychires.2010.01.005
Karege F, Bondolfi G, Gervasoni N et al (2005) Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol Psychiatry 57:1068–1072. https://doi.org/10.1016/j.biopsych.2005.01.008
Bus BAA, Molendijk ML, Tendolkar I et al (2015) Chronic depression is associated with a pronounced decrease in serum brain-derived neurotrophic factor over time. Mol Psychiatry 20:602–608. https://doi.org/10.1038/mp.2014.83
Hosang GM, Shiles C, Tansey KE et al (2014) Interaction between stress and the BDNFVal66Met polymorphism in depression: a systematic review and meta-analysis. BMC Med 12:7. https://doi.org/10.1186/1741-7015-12-7
Björkholm C, Monteggia LM (2016) BDNF—a key transducer of antidepressant effects. Neuropharmacology 102:72–79. https://doi.org/10.1016/j.neuropharm.2015.10.034
Molendijk ML, Bus BAA, Spinhoven P et al (2011) Serum levels of brain-derived neurotrophic factor in major depressive disorder: state–trait issues, clinical features and pharmacological treatment. Mol Psychiatry 16:1088–1095. https://doi.org/10.1038/mp.2010.98
Castrén E, Rantamäki T (2010) The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev. Neurobiol. 70:289–297
Brunoni AR, Machado-Vieira R, Zarate CA et al (2014) BDNF plasma levels after antidepressant treatment with sertraline and transcranial direct current stimulation: results from a factorial, randomized, sham-controlled trial. Eur Neuropsychopharmacol 24:1144–1151. https://doi.org/10.1016/j.euroneuro.2014.03.006
Piccinni A, Del Debbio A, Medda P et al (2009) Plasma brain-derived neurotrophic factor in treatment-resistant depressed patients receiving electroconvulsive therapy. Eur Neuropsychopharmacol 19:349–355. https://doi.org/10.1016/j.euroneuro.2009.01.002
Colla M, Kronenberg G, Deuschle M et al (2007) Hippocampal volume reduction and HPA-system activity in major depression. J Psychiatr Res 41:553–560. https://doi.org/10.1016/j.jpsychires.2006.06.011
Allen AP, Naughton M, Dowling J et al (2015) Serum BDNF as a peripheral biomarker of treatment-resistant depression and the rapid antidepressant response: a comparison of ketamine and ECT. J Affect Disord 186:306–311. https://doi.org/10.1016/j.jad.2015.06.033
Kim Y-K, Na K-S (2016) Role of glutamate receptors and glial cells in the pathophysiology of treatment-resistant depression. Prog Neuro-Psychopharmacol Biol Psychiatry 70:117–126. https://doi.org/10.1016/j.pnpbp.2016.03.009
Kent S, Bluthé RM, Kelley KW, Dantzer R (1992) Sickness behavior as a new target for drug development. Trends Pharmacol Sci 13:24–28. https://doi.org/10.1016/0165-6147(92)90012-U
Qin L, Wu X, Block ML et al (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55:453–462. https://doi.org/10.1002/glia.20467
Guan Z, Fang J (2006) Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav Immun 20:64–71. https://doi.org/10.1016/j.bbi.2005.04.005
Zhang JC, Wu J, Fujita Y et al (2014) Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int J Neuropsychopharmacol 18:1–12. https://doi.org/10.1093/ijnp/pyu077
Gibney SM, McGuinness B, Prendergast C et al (2013) Poly I: C-induced activation of the immune response is accompanied by depression and anxiety-like behaviours, kynurenine pathway activation and reduced BDNF expression. Brain Behav Immun 28:170–181. https://doi.org/10.1016/j.bbi.2012.11.010
Capuron L, Miller AH (2004) Cytokines and psychopathology: lessons from interferon-α. Biol Psychiatry 56:819–824. https://doi.org/10.1016/j.biopsych.2004.02.009
Raison CL, Miller AH (2011) Is depression an inflammatory disorder? Curr Psychiatry Rep 13:467–475. https://doi.org/10.1007/s11920-011-0232-0
Pace TWW, Mletzko TC, Alagbe O et al (2006) Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163:1630–1633. https://doi.org/10.1176/appi.ajp.163.9.1630
Lotrich FE, Albusaysi S, Ferrell RE (2013) Brain-derived Neurotrophic factor serum levels and genotype: association with depression during interferon-alpha treatment. Neuropsychopharmacology 38:985–995. https://doi.org/10.1038/npp.2012.263
Kenis G, Prickaerts J, van Os J et al (2011) Depressive symptoms following interferon-α therapy: mediated by immune-induced reductions in brain-derived neurotrophic factor? Int J Neuropsychopharmacol 14:247–253. https://doi.org/10.1017/S1461145710000830
Dahl J, Ormstad H, Aass HCD et al (2014) The plasma levels of various cytokines are increased during ongoing depression and are reduced to normal levels after recovery. Psychoneuroendocrinology 45:77–86. https://doi.org/10.1016/j.psyneuen.2014.03.019
Hannestad J, DellaGioia N, Bloch M (2011) The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology 36:2452–2459. https://doi.org/10.1038/npp.2011.132
Angst F, Stassen HH, Clayton PJ, Angst J (2002) Mortality of patients with mood disorders: follow-up over 34-38 years. J Affect Disord 68:167–181. https://doi.org/10.1016/S0165-0327(01)00377-9
Goldstein TR, Birmaher B, Axelson D et al (2009) Psychosocial functioning among bipolar youth. J Affect Disord 114:174–183. https://doi.org/10.1016/j.jad.2008.07.001
Berk M, Kapczinski F, Andreazza AC et al (2011) Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev 35:804–817
Fernandes BS, Molendijk ML, Köhler CA et al (2015) Peripheral brain-derived neurotrophic factor (BDNF) as a biomarker in bipolar disorder: a meta-analysis of 52 studies. BMC Med 13:289. https://doi.org/10.1186/s12916-015-0529-7
Munkholm K, Vinberg M, Kessing LV (2016) Peripheral blood brain-derived neurotrophic factor in bipolar disorder: a comprehensive systematic review and meta-analysis. Mol Psychiatry 21:216–228. https://doi.org/10.1038/mp.2015.54
Yasuda S, Liang M-H, Marinova Z et al (2009) The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry 14:51–59. https://doi.org/10.1038/sj.mp.4002099
Jornada LK, Moretti M, Valvassori SS et al (2010) Effects of mood stabilizers on hippocampus and amygdala BDNF levels in an animal model of mania induced by ouabain. J Psychiatr Res 44:506–510. https://doi.org/10.1016/j.jpsychires.2009.11.002
Fernandes BS, Gama CS, Maria Ceresér K et al (2011) Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis. J Psychiatr Res 45:995–1004. https://doi.org/10.1016/j.jpsychires.2011.03.002
Tunca Z, Ozerdem A, Ceylan D et al (2014) Alterations in BDNF (brain derived neurotrophic factor) and GDNF (glial cell line-derived neurotrophic factor) serum levels in bipolar disorder: the role of lithium. J Affect Disord 166:193–200. https://doi.org/10.1016/j.jad.2014.05.012
Muneer A (2016) Bipolar disorder: role of inflammation and the development of disease biomarkers. Psychiatry Investig 13:18–33. https://doi.org/10.4306/pi.2016.13.1.18
Munkholm K, Braüner JV, Kessing LV, Vinberg M (2013) Cytokines in bipolar disorder vs. healthy control subjects: a systematic review and meta-analysis. J Psychiatr Res 47:1119–1133. https://doi.org/10.1016/j.jpsychires.2013.05.018
Steiner J, Bielau H, Brisch R et al (2008) Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 42:151–157. https://doi.org/10.1016/j.jpsychires.2006.10.013
Patas K, Penninx BWJH, Bus BAA et al (2014) Association between serum brain-derived neurotrophic factor and plasma interleukin-6 in major depressive disorder with melancholic features. Brain Behav Immun 36:71–79. https://doi.org/10.1016/j.bbi.2013.10.007
Wang T-Y, Lee S-Y, Chen S-L et al (2016) Comparing clinical responses and the biomarkers of BDNF and cytokines between subthreshold bipolar disorder and bipolar II disorder. Sci Rep 6:27431. https://doi.org/10.1038/srep27431
Basselin M, Kim HW, Chen M et al (2010) Lithium modifies brain arachidonic and docosahexaenoic metabolism in rat lipopolysaccharide model of neuroinflammation. J Lipid Res 51:1049–1056. https://doi.org/10.1194/jlr.M002469
Boufidou F, Nikolaou C, Alevizos B et al (2004) Cytokine production in bipolar affective disorder patients under lithium treatment. J Affect Disord 82:309–313. https://doi.org/10.1016/j.jad.2004.01.007
Kim YK, Jung HG, Myint AM et al (2007) Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. J Affect Disord 104:91–95. https://doi.org/10.1016/j.jad.2007.02.018
Gould TD, Quiroz JA, Singh J et al (2004) Emerging experimental therapeutics for bipolar disorder: insights from the molecular and cellular actions of current mood stabilizers. Mol Psychiatry 9:734–755. https://doi.org/10.1038/sj.mp.4001518
Yadav M, Clark L, Schorey JS (2006) Macrophage’s proinflammatory response to a mycobacterial infection is dependent on sphingosine kinase-mediated activation of phosphatidylinositol phospholipase C, protein kinase C, ERK1/2, and phosphatidylinositol 3-kinase. J Immunol 176:5494–5503. https://doi.org/10.4049/jimmunol.176.9.5494
Song XM, Yu Q, Dong X et al (2017) Aldose reductase inhibitors attenuate β-amyloid-induced TNF-α production in microglia via ROS-PKC-mediated NF-κB and MAPK pathways. Int Immunopharmacol 50:30–37. https://doi.org/10.1016/j.intimp.2017.06.005
Zarate CA, Manji HK (2009) Protein kinase C inhibitors: rationale for use and potential in the treatment of bipolar disorder. CNS Drugs 23:569–582
Yildiz A, Guleryuz S, Ankerst DP et al (2008) Protein kinase C inhibition in the treatment of mania. Arch Gen Psychiatry 65:255–263. https://doi.org/10.1001/archgenpsychiatry.2007.43
Zarate CA, Singh JB, Carlson PJ et al (2007) Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord 9:561–570. https://doi.org/10.1111/j.1399-5618.2007.00530.x
Radhu N, Dominguez LG, Farzan F et al (2015) Evidence for inhibitory deficits in the prefrontal cortex in schizophrenia. Brain 138:483–497. https://doi.org/10.1093/brain/awu360
Grace AA (2016) Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci 17:524–532. https://doi.org/10.1038/nrn.2016.57
Green MJ, Matheson SL, Shepherd A et al (2011) Brain-derived neurotrophic factor levels in schizophrenia: a systematic review with meta-analysis. Mol Psychiatry 16:960–972. https://doi.org/10.1038/mp.2010.88
Iritani S, Niizato K, Nawa H et al (2003) Immunohistochemical study of brain-derived neurotrophic factor and its receptor, TrkB, in the hippocampal formation of schizophrenic brains. Prog Neuropsychopharmacol Biol Psychiatry 27:801–807. https://doi.org/10.1016/S0278-5846(03)00112-X
Takahashi M, Shirakawa O, Toyooka K et al (2000) Abnormal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients. Mol Psychiatry 5:293–300. https://doi.org/10.1038/sj.mp.4000718
Durany N, Michel T, Zöchling R et al (2001) Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr Res 52:79–86. https://doi.org/10.1016/S0920-9964(00)00084-0
Weickert CS, Hyde TM, Lipska BK et al (2003) Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry 8:592–610. https://doi.org/10.1038/sj.mp.4001308
Adachi N, Numakawa T, Kumamaru E et al (2013) Phencyclidine-induced decrease of synaptic connectivity via inhibition of BDNF secretion in cultured cortical neurons. Cereb Cortex 23:847–858. https://doi.org/10.1093/cercor/bhs074
Katanuma Y, Numakawa T, Adachi N et al (2014) Phencyclidine rapidly decreases neuronal mRNA of brain-derived neurotrophic factor. Synapse 68:257–265. https://doi.org/10.1002/syn.21735
Benros ME, Nielsen PR, Nordentoft M et al (2011) Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry 168:1303–1310. https://doi.org/10.1176/appi.ajp.2011.11030516
Khandaker GM, Cousins L, Deakin J et al (2015) Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry 2:258–270. https://doi.org/10.1016/S2215-0366(14)00122-9
Mondelli V, Cattaneo A, Murri MB et al (2011) Stress and inflammation reduce brain-derived neurotrophic factor expression in first-episode psychosis: a pathway to smaller hippocampal volume. J Clin Psychiatry 72:1677–1684. https://doi.org/10.4088/JCP.10m06745
Fillman SG, Cloonan N, Catts VS et al (2013) Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry 18:206–214. https://doi.org/10.1038/mp.2012.110
Doorduin J, de Vries EFJ, Willemsen ATM et al (2009) Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med 50:1801–1807. https://doi.org/10.2967/jnumed.109.066647
Sabunciyan S, Maher B, Bahn S et al (2015) Association of DNA methylation with acute mania and inflammatory markers. PLoS One 10:e0132001. https://doi.org/10.1371/journal.pone.0132001
Van Den Oord EJCG, Clark SL, Xie LY et al (2016) A whole methylome CpG-SNP association study of psychosis in blood and brain tissue. Schizophr Bull 42:1018–1026. https://doi.org/10.1093/schbul/sbv182
Mill J, Tang T, Kaminsky Z et al (2008) Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 82:696–711. https://doi.org/10.1016/j.ajhg.2008.01.008
Kordi-Tamandani DM, Sahranavard R, Torkamanzehi A (2012) DNA methylation and expression profiles of the brain-derived neurotrophic factor (BDNF) and dopamine transporter (DAT1) genes in patients with schizophrenia. Mol Biol Rep 39:1–5
Kudlek Mikulic S, Mihaljevic-Peles A, Sagud M, et al (2017) Brain-derived neurotrophic factor serum and plasma levels in the treatment of acute schizophrenia with olanzapine or risperidone: 6-week prospective study. Nord J psychiatry 1–8. https://doi.org/10.1080/08039488.2017.1340518
Li J, Ye F, Xiao W et al (2016) Increased serum brain-derived neurotrophic factor levels following electroconvulsive therapy or antipsychotic treatment in patients with schizophrenia. Eur Psychiatry 36:23–28. https://doi.org/10.1016/j.eurpsy.2016.03.005
Pırıldar Ş, Gönül AS, Taneli F, Akdeniz F (2004) Low serum levels of brain-derived neurotrophic factor in patients with schizophrenia do not elevate after antipsychotic treatment. Prog Neuro-Psychopharmacol Biol Psychiatry 28:709–713. https://doi.org/10.1016/j.pnpbp.2004.05.008
Krivoy A, Hochman E, Sendt K-V et al (2017) Association between serum levels of glutamate and neurotrophic factors and response to clozapine treatment. Schizophr Res. https://doi.org/10.1016/j.schres.2017.05.040
Nikolac Perkovic M, Nedic Erjavec G, Zivkovic M et al (2014) Association between the brain-derived neurotrophic factor Val66Met polymorphism and therapeutic response to olanzapine in schizophrenia patients. Psychopharmacology (Berlin) 231:3757–3764. https://doi.org/10.1007/s00213-014-3515-4
Barthel H, Schroeter ML, Hoffmann KT, Sabri O (2015) PET/MR in dementia and other neurodegenerative diseases. Semin Nucl Med 45:224–233
Agrawal M, Biswas A (2015) Molecular diagnostics of neurodegenerative disorders. Front Mol Biosci 2. https://doi.org/10.3389/fmolb.2015.00054
Zhu L, Ploessl K, Kung HF (2014) PET/SPECT imaging agents for neurodegenerative diseases. Chem Soc Rev 43:6683–6691. https://doi.org/10.1039/C3CS60430F
Jové M, Portero-Otín M, Naudí A et al (2014) Metabolomics of human brain aging and age-related neurodegenerative diseases. J Neuropathol Exp Neurol 73:640–657. https://doi.org/10.1097/NEN.0000000000000091
Olsson B, Lautner R, Andreasson U et al (2016) CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 15:673–684
Parnetti L, Castrioto A, Chiasserini D et al (2013) Cerebrospinal fluid biomarkers in Parkinson disease. Nat Rev Neurol 9:131–140. https://doi.org/10.1038/nrneurol.2013.10
Gan KJ, Silverman MA (2015) Dendritic and axonal mechanisms of Ca2+ elevation impair BDNF transport in a oligomer-treated hippocampal neurons. Mol Biol Cell 26:1058–1071. https://doi.org/10.1091/mbc.E14-12-1612
Tejeda GS, Díaz-Guerra M (2017) Integral characterization of defective BDNF/TrkB Signalling in neurological and psychiatric disorders leads the way to new therapies. Int J Mol Sci 18:268. https://doi.org/10.3390/ijms18020268
Rosas-Vidal LE, Do-Monte FH, Sotres-Bayon F, Quirk GJ (2014) Hippocampal–prefrontal BDNF and memory for fear extinction. Neuropsychopharmacology 39:2161–2169. https://doi.org/10.1038/npp.2014.64
Levada OA, Cherednichenko NV, Trailin AV, Troyan AS (2016) Plasma brain-derived neurotrophic factor as a biomarker for the main types of mild neurocognitive disorders and treatment efficacy: a preliminary study. Dis Markers 2016:4095723. https://doi.org/10.1155/2016/4095723
Gezen-Ak D, Dursun E, Hanağası H et al (2013) BDNF, TNFα, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J Alzheimer’s Dis 37:185–195. https://doi.org/10.3233/JAD-130497
Laske C, Stellos K, Hoffmann N et al (2011) Higher BDNF serum levels predict slower cognitive decline in Alzheimer’s disease patients. Int J Neuropsychopharmacol 14:399–404. https://doi.org/10.1017/S1461145710001008
Buchman AS, Yu L, Boyle PA et al (2016) Higher brain BDNF gene expression is associated with slower cognitive decline in older adults. Neurology 86:735–741. https://doi.org/10.1212/WNL.0000000000002387
Town T, Tan J, Flavell RA, Mullan M (2005) T-cells in Alzheimer’s disease. NeuroMolecular Med 7:255–264. https://doi.org/10.1385/NMM:7:3:255
Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477. https://doi.org/10.1038/nri3705
Zheng C, Zhou X-W, Wang J-Z (2016) The dual roles of cytokines in Alzheimer’s disease: update on interleukins, TNF-α, TGF-β and IFN-γ. Transl Neurodegener 5:7. https://doi.org/10.1186/s40035-016-0054-4
Iwashyna TJ, Ely EW, Smith DM, Langa KM (2010) Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304:1787. https://doi.org/10.1001/jama.2010.1553
Holmes C, Cunningham C, Zotova E et al (2009) Systemic inflammation and disease progression in Alzheimer disease. Neurology 73:768–774. https://doi.org/10.1212/WNL.0b013e3181b6bb95
Garcez ML, Mina F, Bellettini-Santos T et al (2017) Minocycline reduces inflammatory parameters in the brain structures and serum and reverses memory impairment caused by the administration of amyloid β (1-42) in mice. Prog Neuro-Psychopharmacol Biol Psychiatry 77:23–31. https://doi.org/10.1016/j.pnpbp.2017.03.010
Biscaro B, Lindvall O, Tesco G et al (2012) Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer’s disease. Neurodegener Dis 9:187–198. https://doi.org/10.1159/000330363
Koivunen J, Scheinin N, Virta JR et al (2011) Amyloid PET imaging in patients with mild cognitive impairment: a 2-year follow-up study. Neurology 76:1085–1090. https://doi.org/10.1212/WNL.0b013e318212015e
Okello A, Edison P, Archer HA et al (2009) Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 72:56–62. https://doi.org/10.1212/01.wnl.0000338622.27876.0d
Edison P, Archer HA, Gerhard A et al (2008) Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis 32:412–419. https://doi.org/10.1016/j.nbd.2008.08.001
Prakash A, Kumar A (2014) Role of nuclear receptor on regulation of BDNF and neuroinflammation in hippocampus of β-amyloid animal model of Alzheimer’s disease. Neurotox Res 25:335–347. https://doi.org/10.1007/s12640-013-9437-9
Chen J-H, Ke K-F, Lu J-H et al (2015) Protection of TGF-β1 against neuroinflammation and neurodegeneration in Aβ1-42-induced Alzheimer’s disease model rats. PLoS One 10:e0116549. https://doi.org/10.1371/journal.pone.0116549
Dursun E, Gezen-Ak D, Hanağası H et al (2015) The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J Neuroimmunol 283:50–57. https://doi.org/10.1016/j.jneuroim.2015.04.014
Lynch MA (2015) Neuroinflammatory changes negatively impact on LTP: a focus on IL-1β. Brain Res 1621:197–204. https://doi.org/10.1016/j.brainres.2014.08.040
Tong L, Prieto GA, Kramár EA et al (2012) Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J Neurosci 32:17714–17724. https://doi.org/10.1523/JNEUROSCI.1253-12.2012
Balestrino R, Martinez-Martin P (2017) Neuropsychiatric symptoms, behavioural disorders, and quality of life in Parkinson’s disease. J Neurol Sci 373:173–178
Leroi I, Ahearn DJ, Andrews M et al (2011) Behavioural disorders, disability and quality of life in Parkinson’s disease. Age Ageing 40:614–621. https://doi.org/10.1093/ageing/afr078
Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18:101–113. https://doi.org/10.1038/nrn.2016.178
Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715–725
Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501:45–51. https://doi.org/10.1038/nature12481
Janakiraman U, Manivasagam T, Justin Thenmozhi A et al (2017) Chronic mild stress augments MPTP induced neurotoxicity in a murine model of Parkinson’s disease. Physiol Behav 173:132–143. https://doi.org/10.1016/j.physbeh.2017.01.046
Howells DWW, Porritt MJJ, Wong JYFY et al (2000) Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol 166:127–135. https://doi.org/10.1006/exnr.2000.7483
Porritt MJ, Batchelor PE, Howells DW (2005) Inhibiting BDNF expression by antisense oligonucleotide infusion causes loss of nigral dopaminergic neurons. Exp Neurol 192:226–234. https://doi.org/10.1016/j.expneurol.2004.11.030
Ventriglia M, Zanardini R, Bonomini C et al (2013) Serum brain-derived neurotrophic factor levels in different neurological diseases. Biomed Res Int 2013:901082. https://doi.org/10.1155/2013/901082
Scalzo P, Kümmer A, Bretas TL et al (2010) Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson’s disease. J Neurol 257:540–545. https://doi.org/10.1007/s00415-009-5357-2
Marinova-Mutafchieva L, Sadeghian M, Broom L et al (2009) Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: a time course study in a 6-hydroxydopamine model of Parkinson’s disease. J Neurochem 110:966–975. https://doi.org/10.1111/j.1471-4159.2009.06189.x
Depino AM, Earl C, Kaczmarczyk E et al (2003) Microglial activation with atypical proinflammatory cytokine expression in a rat model of Parkinson’s disease. Eur J Neurosci 18:2731–2742. https://doi.org/10.1046/j.1460-9568.2003.03014.x
Main BS, Zhang M, Brody KM et al (2016) Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson’s disease. Glia 64:1590–1604. https://doi.org/10.1002/glia.23028
Femminella GD, Ninan S, Atkinson R et al (2016) Does microglial activation influence hippocampal volume and neuronal function in Alzheimer’s disease and Parkinson’s disease dementia? J Alzheimer’s Dis 51:1275–1289. https://doi.org/10.3233/JAD-150827
Gerhard A, Pavese N, Hotton G et al (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21:404–412. https://doi.org/10.1016/j.nbd.2005.08.002
Sawada M, Imamura K, Nagatsu T (2006) Role of cytokines in inflammatory process in Parkinson’s disease. J Neural Transm Suppl 70:373–381
Jensen FE (2011) Epilepsy as a spectrum disorder: implications from novel clinical and basic neuroscience. Epilepsia 52:1–6. https://doi.org/10.1111/j.1528-1167.2010.02904.x
Lee B, Dziema H, Lee KH et al (2007) CRE-mediated transcription and COX-2 expression in the pilocarpine model of status epilepticus. Neurobiol Dis 25:80–91. https://doi.org/10.1016/j.nbd.2006.08.015
Li L, Deng J, Liu C et al (2016) Altered expression of neuropeptide Y receptors caused by focal cortical dysplasia in human intractable epilepsy. Oncotarget 7:15329–15338. https://doi.org/10.18632/oncotarget.7855
Wang Y, Qi J-S, Kong S et al (2009) BDNF-TrkB signaling pathway mediates the induction of epileptiform activity induced by a convulsant drug cyclothiazide. Neuropharmacology 57:49–59. https://doi.org/10.1016/j.neuropharm.2009.04.007
Martínez-Levy GA, Rocha L, Rodríguez-Pineda F, et al (2017) Increased expression of brain-derived neurotrophic factor transcripts I and VI, cAMP response element binding, and glucocorticoid receptor in the cortex of patients with temporal lobe epilepsy. Mol Neurobiol. https://doi.org/10.1007/s12035-017-0597-0
Martínez-Levy GA, Rocha L, Lubin FD et al (2016) Increased expression of BDNF transcript with exon VI in hippocampi of patients with pharmaco-resistant temporal lobe epilepsy. Neuroscience 314:12–21. https://doi.org/10.1016/j.neuroscience.2015.11.046
Unsain N, Montroull LE, Mascó DH (2009) Brain-derived neurotrophic factor facilitates TrkB down-regulation and neuronal injury after status epilepticus in the rat hippocampus. J Neurochem 111:428–440. https://doi.org/10.1111/j.1471-4159.2009.06342.x
Otsuka S, Ohkido T, Itakura M et al (2016) Dual mechanisms of rapid expression of anxiety-related behavior in pilocarpine-treated epileptic mice. Epilepsy Res 123:55–67. https://doi.org/10.1016/j.eplepsyres.2016.04.007
de Souza Bernardino TC, Teixeira AL, Miranda AS et al (2015) Wistar Audiogenic Rats (WAR) exhibit altered levels of cytokines and brain-derived neurotrophic factor following audiogenic seizures. Neurosci Lett 597:154–158. https://doi.org/10.1016/j.neulet.2015.04.046
Carvalho AL, Caldeira MV, Santos SD, Duarte CB (2008) Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br J Pharmacol 153(Suppl):S310–S324. https://doi.org/10.1038/sj.bjp.0707509
Binder DK, Croll SD, Gall CM, Scharfman HE (2001) BDNF and epilepsy: too much of a good thing? Trends Neurosci 24:47–53. https://doi.org/10.1016/S0166-2236(00)01682-9
Liu G, Gu B, He X-P et al (2013) Transient inhibition of TrkB kinase after status epilepticus prevents development of temporal lobe epilepsy. Neuron 79:31–38. https://doi.org/10.1016/j.neuron.2013.04.027
Liu G, Kotloski RJ, McNamara JO (2014) Antiseizure effects of TrkB kinase inhibition. Epilepsia 55:1264–1273. https://doi.org/10.1111/epi.12671
Vezzani A, French J, Bartfai T, Baram TZ (2011) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40. https://doi.org/10.1038/nrneurol.2010.178
Benson MJ, Manzanero S, Borges K (2015) Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia 56:895–905. https://doi.org/10.1111/epi.12960
Lehtimäki KA, Peltola J, Koskikallio E et al (2003) Expression of cytokines and cytokine receptors in the rat brain after kainic acid-induced seizures. Brain Res Mol Brain Res 110:253–260
Zheng H, Zhu W, Zhao H et al (2009) Kainic acid-activated microglia mediate increased excitability of rat hippocampal neurons in vitro and in vivo: crucial role of interleukin-1beta. Neuroimmunomodulation 17:31–38. https://doi.org/10.1159/000243083
Kairisalo M, Korhonen L, Sepp M et al (2009) NF-κB-dependent regulation of brain-derived neurotrophic factor in hippocampal neurons by X-linked inhibitor of apoptosis protein. Eur J Neurosci 30:958–966. https://doi.org/10.1111/j.1460-9568.2009.06898.x
Mattson MP, Camandola S (2001) NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 107:247–254
Lubin FD, Ren Y, Xu X, Anderson AE (2007) Nuclear factor-κB regulates seizure threshold and gene transcription following convulsant stimulation. J Neurochem 103:1381–1395. https://doi.org/10.1111/j.1471-4159.2007.04863.x
Nagahara AH, Tuszynski MH (2011) Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov 10:209–219. https://doi.org/10.1038/nrd3366
Pan W, Banks WA, Fasold MB et al (1998) Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 37:1553–1561
Nagahara AH, Merrill DA, Coppola G et al (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15:331–337. https://doi.org/10.1038/nm.1912
Somoza R, Juri C, Baes M et al (2010) Intranigral transplantation of epigenetically induced BDNF-secreting human mesenchymal stem cells: implications for cell-based therapies in Parkinson’s disease. Biol Blood Marrow Transplant 16:1530–1540. https://doi.org/10.1016/j.bbmt.2010.06.006
Sirianni RW, Olausson P, Chiu AS et al (2010) The behavioral and biochemical effects of BDNF containing polymers implanted in the hippocampus of rats. Brain Res 1321:40–50. https://doi.org/10.1016/j.brainres.2010.01.041
Géral C, Angelova A, Lesieur S (2013) From molecular to nanotechnology strategies for delivery of neurotrophins: emphasis on brain-derived neurotrophic factor (BDNF). Pharmaceutics 5:127–167
Scharfman H, Goodman J, Macleod A et al (2005) Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp Neurol 192:348–356. https://doi.org/10.1016/j.expneurol.2004.11.016
Devi L, Ohno M (2012) 7,8-Dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 37:434–444. https://doi.org/10.1038/npp.2011.191
Razgado-Hernandez LF, Espadas-Alvarez AJ, Reyna-Velazquez P et al (2015) The transfection of BDNF to dopamine neurons potentiates the effect of dopamine D3 receptor agonist recovering the striatal innervation, dendritic spines and motor behavior in an aged rat model of Parkinson’s disease. PLoS One 10:e0117391. https://doi.org/10.1371/journal.pone.0117391
Del Arco A, Segovia G, de Blas M et al (2011) Prefrontal cortex, caloric restriction and stress during aging: studies on dopamine and acetylcholine release, BDNF and working memory. Behav Brain Res 216:136–145. https://doi.org/10.1016/j.bbr.2010.07.024
Vaynman S, Ying Z, Gomez-Pinilla F (2004) Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci 20:2580–2590. https://doi.org/10.1111/j.1460-9568.2004.03720.x
Vedovelli K, Silveira E, Velho E et al (2011) Effects of increased opportunity for physical exercise and learning experiences on recognition memory and brain-derived neurotrophic factor levels in brain and serum of rats. Neuroscience 199:284–291. https://doi.org/10.1016/j.neuroscience.2011.08.012
Farmer J, Zhao X, van Praag H et al (2004) Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 124:71–79. https://doi.org/10.1016/j.neuroscience.2003.09.029
Huang T, Larsen KT, Ried-Larsen M et al (2014) The effects of physical activity and exercise on brain-derived neurotrophic factor in healthy humans: a review. Scand J Med Sci Sports 24:1–10. https://doi.org/10.1111/sms.12069
Zoladz JA, Pilc A (2010) The effect of physical activity on the brain derived neurotrophic factor: from animal to human studies. J Physiol Pharmacol 61:533–541. https://doi.org/10.1523/JNEUROSCI.6251-09.2010
Hoffman JR, Ostfeld I, Kaplan Z et al (2015) Exercise enhances the behavioral responses to acute stress in an animal model of PTSD. Med Sci Sports Exerc 47:2043–2052. https://doi.org/10.1249/MSS.0000000000000642
Hutton CP, Déry N, Rosa E et al (2015) Synergistic effects of diet and exercise on hippocampal function in chronically stressed mice. Neuroscience 308:180–193. https://doi.org/10.1016/j.neuroscience.2015.09.005
Jae KH, Kil SB, So B et al (2014) Increase of circulating BDNF levels and its relation to improvement of physical fitness following 12 weeks of combined exercise in chronic patients with schizophrenia: a pilot study. Psychiatry Res 220:792–796. https://doi.org/10.1016/j.psychres.2014.09.020
Kirshenbaum GS, Burgess CR, Déry N et al (2014) Attenuation of mania-like behavior in Na+,K+-ATPase α3 mutant mice by prospective therapies for bipolar disorder: melatonin and exercise. Neuroscience 260:195–204. https://doi.org/10.1016/j.neuroscience.2013.12.011
García-Mesa Y, Pareja-Galeano H, Bonet-Costa V et al (2014) Physical exercise neuroprotects ovariectomized 3xTg-AD mice through BDNF mechanisms. Psychoneuroendocrinology 45:154–166. https://doi.org/10.1016/j.psyneuen.2014.03.021
Tuon T, Valvassori SS, Dal Pont GC et al (2014) Physical training prevents depressive symptoms and a decrease in brain-derived neurotrophic factor in Parkinson’s disease. Brain Res Bull 108:106–112. https://doi.org/10.1016/j.brainresbull.2014.09.006
Coelho FG d M, Vital TM, Stein AM et al (2014) Acute aerobic exercise increases brain-derived neurotrophic factor levels in elderly with Alzheimer’s disease. J Alzheimer’s Dis 39:401–408. https://doi.org/10.3233/JAD-131073
Dao AT, Zagaar MA, Levine AT et al (2013) Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Curr Alzheimer Res 10:507–515
Noble EE, Billington CJ, Kotz CM, Wang C (2011) The lighter side of BDNF. Am J Physiol Regul Integr Comp Physiol 300:R1053–R1069. https://doi.org/10.1152/ajpregu.00776.2010
Schwartz E, Mobbs CV (2012) Hypothalamic BDNF and obesity: found in translation. Nat Med 18:496–497. https://doi.org/10.1038/nm.2716
Kernie SG, Liebl DJ, Parada LF (2000) BDNF regulates eating behavior and locomotor activity in mice. EMBO J 19:1290–1300. https://doi.org/10.1093/emboj/19.6.1290
Sharma S, Fulton S (2013) Diet-induced obesity promotes depressive-like behaviour that is associated with neural adaptations in brain reward circuitry. Int J Obes 37:382–389. https://doi.org/10.1038/ijo.2012.48
Beilharz JE, Maniam J, Morris MJ (2014) Short exposure to a diet rich in both fat and sugar or sugar alone impairs place, but not object recognition memory in rats. Brain Behav Immun 37:134–141. https://doi.org/10.1016/j.bbi.2013.11.016
Noble EE, Hsu TM, Kanoski SE (2017) Gut to brain dysbiosis: mechanisms linking western diet consumption, the microbiome, and cognitive impairment. Front Behav Neurosci 11:9. https://doi.org/10.3389/fnbeh.2017.00009
Duan W, Lee J, Guo Z, Mattson MP (2001) Dietary restriction stimulates BDNF production in the brain and thereby protects neurons against excitotoxic injury. J Mol Neurosci 16:1–12. https://doi.org/10.1385/JMN:16:1:1
Duan W, Guo Z, Jiang H et al (2003) Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A 100:2911–2916. https://doi.org/10.1073/pnas.0536856100\r0536856100
Wu A, Ying Z, Gomez-Pinilla F (2004) Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J Neurotrauma 21:1457–1467. https://doi.org/10.1089/neu.2004.21.1457
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lima Giacobbo, B., Doorduin, J., Klein, H.C. et al. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol Neurobiol 56, 3295–3312 (2019). https://doi.org/10.1007/s12035-018-1283-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1283-6