
Abstract
There is copious evidence of abnormalities in resting-state functional network connectivity states, grey and white matter pathology and impaired cerebral perfusion in patients afforded a diagnosis of multiple sclerosis, major depression or chronic fatigue syndrome (CFS) (myalgic encephalomyelitis). Systemic inflammation may well be a major element explaining such findings. Inter-patient and inter-illness variations in neuroimaging findings may arise at least in part from regional genetic, epigenetic and environmental variations in the functions of microglia and astrocytes. Regional differences in neuronal resistance to oxidative and inflammatory insults and in the performance of antioxidant defences in the central nervous system may also play a role. Importantly, replicated experimental findings suggest that the use of high-resolution SPECT imaging may have the capacity to differentiate patients afforded a diagnosis of CFS from those with a diagnosis of depression. Further research involving this form of neuroimaging appears warranted in an attempt to overcome the problem of aetiologically heterogeneous cohorts which probably explain conflicting findings produced by investigative teams active in this field. However, the ionising radiation and relative lack of sensitivity involved probably preclude its use as a routine diagnostic tool.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Many patients with multiple sclerosis (MS), a neuroinflammatory illness, major depressive disorder (MDD), a neuroprogressive condition and chronic fatigue syndrome (CFS) (myalgic encephalomyelitis), a phenotype of no fixed aetiology, share a range of underlying abnormalities [1, 2]. These include mitochondrial dysfunction; systemic inflammation; oxidative stress; a range of autoimmune phenomena; hypothalamic-pituitary axis dysfunction and evidence of structural and functional brain change obtained by neuroimaging using magnetic resonance imaging (MRI), functional MRI (fMRI), magnetic resonance spectroscopy (MRS), positron emission tomography (PET) and single-photon emission computerised tomography (SPECT) (also known as single-photon emission tomography (SPET)) and cognitive symptoms [1,2,3]. Neuroimaging abnormalities in MS are well recognised, and there are numerous excellent reviews on the subject [4,5,6]. There are also many excellent reviews of neuroimaging abnormalities in MDD utilising voxel-based morphometry (VBM) which is widely used within neuropsychiatry [7,8,9,10,11]. Such neuroimaging abnormalities are routinely seen in many patients afforded a diagnosis of CFS according to international consensus criteria [3]. Such abnormalities have been discerned via MRI [12,13,14], fMRI [15,16,17], MRS [17,18,19] and PET [20].
Despite the existence of such data, however, there would appear to be no comprehensive review of neuroimaging abnormalities in patients afforded such a diagnosis which includes the most recent evidence obtained via VBM analysis. There have been few attempts to compare the transdiagnostic neuroimaging data obtained for patients with MS and MDD [2] although there have been several studies comparing neuroimaging abnormalities seen in MS patients with or without co-morbid depressive symptoms which appear to demonstrate changes in global and regional patterns of white and grey matter reduction [21,22,23]. Similarly, there have been few attempts to compare neuroimaging data in MS, MDD and CFS [2, 3]. We aim to remedy this by engaging in such an in-depth review. We have chosen to focus on data examining resting-state functional connectivity, white and grey matter structure and volume and cerebral perfusion for two main reasons. First, this renders the analysis manageable in terms of length. Second, and perhaps more importantly, these parameters are strongly influenced by the presence of systemic inflammation [24,25,26,27,28,29,30,31]. Hence, by comparing and contrasting neuroimaging data in these domains, we may be in a position to comment on mechanisms driving neuroimaging abnormalities in these illnesses which might be similar or different, which in turn could speak to differences in the pathogenesis of these conditions. To this end, we will review data on resting-state functional connectivity, white and grey matter abnormalities and abnormalities in cerebral perfusion in patients with various subtypes of MS before examining data relating to the same parameters in MDD and CFS.
Neuroimaging Abnormalities in MS
Resting-State Functional Connectivity in MS
Functional dissociation or disconnection between large-scale resting-state neural networks is a characteristic feature of relapsing-remitting MS (RRMS) in the advanced stage of the disease [32, 33]. Moreover, the degree of dissociation correlates with the severity of disability in multiple dimensions and with the extent of T2-lesion load [32, 33]. The clinical significance of these abnormalities is emphasised by data demonstrating that functional connectivity between networks is significantly stronger in patients with preserved cognitive function and correlates with lower lesion load and other structural abnormalities [34].
Cortical reorganisation in patterns of resting-state network connectivity is evident in patients with a clinically isolated syndrome [35] and is typically apparent as increased functional connectivity which is widely regarded as an adaptive modification in an attempt to mitigate the deleterious effects on brain function by disease processes [35, 36]. This pattern of reorganisation involves several networks and has been most commonly reported in the executive control network, sensory-mode network and default-mode network (DMN) [32, 37].
Some authors report an increase in the functional connectivity within resting-state networks in patients with RRMS [38,39,40] while others report a decrease [32, 41]. Specific resting networks such as the DMN and resting-state motor networks display increased or decreased connectivity with some other networks, which suggests a general reorganisation of connectivity between and within neural networks rather than a simple pattern of increase or decrease [39, 41].
Several research teams have also reported correlations between abnormal intra-network functional connectivity and clinical parameters, but these relationships are far from straightforward [42,43,44]. For example, while more studies report an inverse relationship between functional connectivity and clinical disability [45,46,47], other studies indicate a positive association between functional connectivity and clinical disability [39].
The same variation in patterns persists for specified dimensions of disability such as impaired motor function where reduced and increased connectivity within the network is associated with impaired motor performance [44, 48]. Increased connectivity within the dorsal premotor cortex correlates with increasing clinical disability as measured by the Expanded Disability Status Scale (EDSS), and this has been held once again to be an adaptive reorganisation aimed at preserving motor function [43]. However, this team of authors have also reported that impaired regional connectivity in the left cerebellar hemisphere, a region of the brain which regulates multiple aspects of motor performance, is associated with increasing clinical disability [39]. The latter pattern appears to predominate in advanced stages of disease where impaired connectivity throughout the motor network is associated with a higher burden of symptoms [48]. In this context, it is noteworthy that recovery from acute relapse appears to be associated with changes in resting-state motor network connectivity [49]. It has also been proposed that reduced activity in the medial prefrontal cortex or the anterior cingulate cortex (ACC) may in fact function as a state marker for the disease [50].
The picture is if anything more complex in studies investigating the resting-state functional connectivity associated with cognitive dysfunction within the DMN, as impaired and enhanced conductivity between and within sub-regions in the DMN have been repeatedly observed [51, 52]. However, once again, decreased connectivity within this region of the brain and associated impairment of cognitive performance appears to be the most common pattern with advancing disease severity [42, 47].
When considered as a whole, the data would suggest that in early or mild disease, there is a compensatory positive reorganisation within and between resting-state networks which eventually are overcome by disease progression [35, 53, 54]. This would explain data illustrating changes in the patterns of connectivity with increasing disease severity but not how such patterns are modified by gender, which seems to be an issue worthy of research [55]. Finally, impaired neural connectivity between the hippocampus and the subgenual cingulate, parietal and prefrontal regions correlates with the severity of depression [54]. Interestingly, such defects in connectivity are provoked by activated microglia in the hippocampal region [54].
White Matter Abnormalities in MS
Conventional structural MRI of MS patients is characterised by the existence of cortical demyelination and focal and diffuse white matter (WM) lesions [56,57,58]. Researchers have reported a plethora of variations in T1 and T2 signals which may well reflect considerable intra-patient variability in underlying lesion pathology [59, 60]. Several research teams using MR approaches such as magnetisation transfer ratio (MTR), magnetic resonance spectroscopy (MRS) and diffusion tensor imaging (DTI) have reported a wide range of abnormalities in seemingly normal-appearing WM (NAWM) [61,62,63]. These include changes in the diffusion properties of water, reduced MTR, abnormal T1 and T2 relaxation times and a range of metabolic abnormalities including reduced N-acetyl aspartate, increased glutamate/glutamine and reduced choline [64,65,66,67,68,69,70,71]. In addition, prolonged T1 and T2 times have also been reported in diffusely abnormal WM (DAWM), which is suggestive of decreased myelin density, axonal loss and chronic fibrillary gliosis [72, 73]. It is also noteworthy that NAWM abnormalities revealed by MTR histogram analysis worsen over the disease course when compared with age- and sex-matched healthy controls [74, 75].
Grey Matter Abnormalities in MS
Grey matter (GM) pathology is evident in the hippocampus, basal ganglia, hypothalamus, cerebellum and spinal cord [76]. GM atrophy is seen in the earliest stage of the disease, and its extent is predictive of the development of cognitive disability, physical disability (as indexed by the EDSS) and overall disease prognosis (reviewed [77, 78]). It is also of interest that GM atrophy increases over time as the disease worsens, which is a phenomenon which is not apparent in WM atrophy [79]. While there is scant post-mortem evidence of an inflammatory origin of GM pathology [80], biopsies taken from patients at early disease stages reveal a different picture—there is ample evidence of T-cell infiltration, activated microglia and astrocytes together with other markers of chronic inflammation associated with cortical lesions and diffuse injury [81, 82].
The reasons for the discrepancy between in vivo and post-mortem studies are not clear but may stem from re-myelination processes which are far more active in GM than WM (reviewed [83]). However, it should be noted that there is evidence of neurodegenerative as well as inflammatory damage in the GM of RRMS patients and current debate centres on whether such neurodegeneration is independent of inflammation or has an independent origin [79, 84]. There is evidence suggesting that neurodegenerative damage is secondary to mitochondrial dysfunction and impaired ion channel activity [85, 86]. Such data are difficult to interpret however as several authors have concluded that mitochondrial dysfunction seen in the central nervous system of MS patients is in fact driven by chronic inflammation in general and oxidative bursts in particular [87, 88]. Moreover, an intriguing study conducted by Frischer and fellow workers established an inverse relation between neuronal integrity in the GM and the magnitude of inflammation rather suggesting that neurodegenerative processes are not independent of inflammatory drivers [87]. Once again, given the relationship between the development of systemic inflammation and neuroinflammation, it seems reasonable to conclude that chronic inflammation in the periphery makes a significant contribution to the development of central GM pathology.
Cerebral Perfusion in MS
There is a considerable and accruing body of research demonstrating globally reduced cerebral perfusion and blood volume abnormalities in MS patients obtained via the use of SPECT [89]; PET [90] and, more recently, dynamic susceptibility contrast-enhanced perfusion MRI [91,92,93]. Interestingly, this state of chronically impaired cerebral blood flow (CBF) appears to exist in all subtypes of the illness [94, 95], develops in patients with a clinically isolated syndrome and in very early disease [93, 96] and would seem to affect NAWN before deep GM changes [93].
Several research teams have reported relationships between abnormal patterns of perfusion and various elements involved in the pathophysiology or pathogenesis of the illness [94, 97,98,99]. For example, Taghizadeh and colleagues examined the brains of 25 RRMS patients via 99mTc-labelled SPECT and reported a positive association of the degree of perfusional impairment between disease duration and disease severity indexed by the EDSS [97]. Interestingly, these authors also reported a significant increase in perfusion following 20 sessions of hyperbaric oxygen treatment but unfortunately, this improvement was not accompanied by any improvement in disease status as measured by any objective or subjective parameter [97]. Another team of authors utilising dynamic susceptibility contrast-perfusion MRI reported a significant decrease in cerebral hypoperfusion in 22 RRMS patients, compared with an equal number of sex-matched controls, whose extent correlated with the severity of their self-reported fatigue [98]. There is also considerable evidence associating reduced CBF in GM and WM with the development of cognitive dysfunction in multiple domains [100,101,102,103].
Neuroimaging Abnormalities in MDD
Resting-State Functional Connectivity in MDD
Several studies have revealed dysfunction of functional connectivity within or between large-scale functional resting-state neural networks such as the DMN in patients with MDD using resting-state fMRI [104]. Abnormalities in two large-scale neuronal networks—the frontoparietal central executive network (CEN) and the medial prefrontal-medial parietal DMN—are consistent findings in depression and potential therapeutic targets for transcranial magnetic stimulation [105]. Resting-state functional conductivity is also altered by some psychotropics efficacious in depression such as ketamine, lithium and antidepressants [106, 107].
Intra-network changes are exemplified by enhanced connectivity within the anterior DMN and abnormal connectivity between the anterior and posterior DMN. Inter-network abnormalities are evidenced by increased connectivity between the anterior DMN and salience network and diminished connectivity between the posterior DMN and the CEN (review [108]). MDD network research has largely focused on the DMN which is understandable given that this network incorporates the anterior cingulate and an extensive area of the medial prefrontal cortex extending into the orbitofrontal cortices considered to play a major if not dominant role in the pathophysiology of the illness [109]. Anterior regions of the DMN, particularly the medial prefrontal cortex, mediate self-referential processing while posterior regions are associated with the formation and retrieval of episodic memories (review [109]).
Dissociation of functional connectivity between the anterior and posterior areas of this region and dissociation between the DMN and other large-scale resting networks are characteristic of patients with MDD [110, 111]. Zhu and fellow workers reported that a pattern of dissociation between anterior and posterior functional connectivity in resting-state DMNs of first-episode, treatment-naïve young adults with MDD is evidenced by increased functional connectivity in anterior medial cortical regions (especially the medial prefrontal cortex and ACC) and decreased functional connectivity in posterior medial cortical regions (especially the posterior cingulate cortex/precuneus) in MDD patients compared with control subjects [110]. In the depressed group, the increased functional connectivity in the anterior medial cortex correlated positively with rumination score, while the decreased functional connectivity in the posterior medial cortex correlated negatively with over-general autobiographical memory test scores [110]. More recent data have established a relationship between the patterns of functional dissociation in connectivity between the precuneus (PN), a region within the DMN, and the clinical presentation and/or severity of MDD [111, 112]. For example, the magnitude of hyperconnectivity between the PN and the dorsal medial prefrontal/ACC region is associated with increased severity while reduced connectivity between the PN and fusiform areas is associated with the variable symptoms seen in the illness.
While alterations in brain connectivity centring on the DMN continue to be a focus of research, abnormal patterns of connectivity are also seen within and between the affective network and the cognitive control network (CCN) [113, 114]. In fact, several trait- and state-dependent abnormalities in patterns of functional connectivity have been reported in the cerebellum, ACC, lingual gyrus, dorsolateral prefrontal cortex (DLPFC), middle frontal gyrus, amygdala and insula [113].
Successful therapy is associated with changes in connectivity within the DMN and corticolimbic areas, and it is noteworthy that changes in the latter areas correlate with the degree of clinical improvement while changes in the DMN do not [114]. The success or otherwise of transcranial magnetic stimulation would also appear to depend on the positive modulation of functional connectivity in this instance both between and within the CEN and DMN [105].
White Matter Abnormalities in MDD
DTI utilising tract-based spatial statistics (TBSS) [115] has revealed disrupted WM tracts and WM degeneration in the prefrontal cortex, the temporal cortex and limbic system, the uncinate fasciculus, the internal capsule and the superior longitudinal fasciculus (review [116, 117]). The geographical extent of these WM tract abnormalities has prompted some authors to describe MDD as a disconnection syndrome [118]. DTI studies have reported microstructural abnormalities, albeit of a somewhat different pattern, in WM integrity characterised by low fractional anisotropy (FA) values associated with MDD in elderly patients [119, 120], middle-aged patients [121], young treatment-naïve first-episode adolescent patients [122, 123] (review [124]) and even disease-free individuals with a familial vulnerability to the development of the illness [125, 126]. A recent meta-analysis conducted by Chen and fellow workers using TBSS concluded that there was consistent evidence of WM architecture and integrity disruption in the frontal and thalamic regions, and the anterior limb of the internal capsule and importantly that WM reduction in the corpus callosum is associated with more severe disease [127].
Interestingly, Le Win and others reported similar findings using a DTI TBSS-based approach in adolescents with MDD [128]. Lower FA values in the WM associated with the corpus callosum and the cortical-striatal-thalamic circuit were detected, as well as lower FA values in the superior and anterior corona radiata [128]. Very similar findings in adolescent MDD have been reported by Hendeson and others in a study aiming to investigate possible associations between patterns of WM abnormalities and symptom severity in this patient group [129]. In this study, MDD severity was once again correlated with reduced WM integrity in the corpus callosum, but also with reduced integrity in the anterior cingulum, anterior thalamic radiation and sagittal stratum [129]. They also reported that the presence of anhedonia and irritability was associated with disrupted WM tracts in the anterior limb of the internal capsule and in projection fibres to the orbitofrontal cortex and impaired integrity in the anterior corona radiata, sagittal stratum and in efferent tracts leading to temporal and prefrontal cortices [129]. It is also noteworthy that Bessette et al. reported a unique pattern of FA abnormalities in the genu of the corpus callosum, thalamus, midbrain and internal and external capsule tracts, which have yet to be reported in adult-onset MDD [123].
Yuan and fellow workers reported whole-brain reductions in participants with first-episode geriatric MDD in remission, characterised by low FA values in the right superior frontal gyrus, left middle temporal gyrus, left inferior frontal gyrus, right middle occipital gyrus, right inferior parietal lobule, left lingual gyrus, right putamen and right caudate [119]. Taylor et al. also reported widespread decreases in WM FA values in participants with late-onset depression consistent with a state of disconnection between frontal and limbic regions [120]. However, these authors also detected increases in FA values in the frontal gyri and ACC which was predictive of resistance to the selective serotonin re-uptake inhibitor antidepressant sertraline [120]. Interestingly, the same team reported that those with late-onset MDD in remission following antidepressant therapy displayed WM changes which were almost identical to normal patterns of WM integrity seen in never-depressed patients [130].
The weight of evidence indicates that compared with healthy controls, those with midlife-onset MDD display decreased FA values in the left anterior limb of the internal capsule, left posterior cingulate cortex, right cingulate cortex, bilateral parahippocampal gyri, hippocampus, pons, right and left frontal lobes, sagittal striatum and cerebellum [117, 131]. There is also evidence that FA values in the left anterior limb of the internal capsule correlate negatively with MDD severity [117].
Huang and others reported lower FA values in the splenium of the corpus callosum, left cingulum, inferior fronto–occipital fasciculi, superior longitudinal fasciculi and uncinate in patients than in controls, and that this measure might serve as a vulnerability marker for the illness [126]. Xiao et al. reported a virtually identical pattern of WM pathology in the cerebral peduncle, the anterior and posterior limbs of the internal capsule, the external capsule and the cingulum in patients with chronic depression and individuals at high familial risk of developing the illness, suggesting that whole-brain WM disruption precedes the development of active disease [125]. It is also notable that several authors reported significant differences between people with MDD in an acute episode compared with those in remission [132, 133], as well as negative correlations between FA and current illness severity.
Widespread decreases in FA values in the cingulum, superior and inferior longitudinal fascicule and corpus callosum compared with first-episode patients are characteristic of resistant to antidepressant therapy [116]. It is also notable that FA values in the ventromedial prefrontal region in this group are lower even in relation to MDD subjects with a history of remission and recurrence [134]. Finally, recent meta-analyses concluded that compromised WM integrity in the sub-callosal cingulate cortex and in cortical-limbic or cortical-subcortical circuits was most commonly associated with the development of treatment-resistant depression [111, 116].
Not all research teams have reported significant WM abnormalities in MDD patients however [121, 135]. The reasons for such discrepant results are not entirely clear, but differences in technique and evidence that depression is an aetiologically heterogeneous syndrome rather than a single discrete neurobiological entity [136] is likely also relevant.
Grey Matter Abnormalities in MDD
Several research teams have detected severe or moderate GM volume loss in the amygdala, the thalamus and many cortical brain regions, including the pre- and post-central gyri, orbitofrontal cortex, ACC and temporal cortex and lateral prefrontal cortex [137,138,139,140]. Relatively recent meta-analyses presented convergent findings for the ACC and many orbitofrontal cortical structures but divergent findings for limbic and striatal regions [138, 141,142,143]. Readers interested in a more detailed consideration of the variable findings regarding the patterns of GM pathology in MDD are referred to excellent discussions by [142, 144].
From a treatment perspective, several research teams have reported that patients with treatment-resistant depression display greater reductions in GM volume in the left temporal cortex and right medial frontal cortex compared with age- and sex-matched controls or patients in remission [145, 146]. Reduced GM volume is also a replicated finding in treatment-naïve first-episode adolescent patients [147,148,149] and indeed may even precede the first episode [143]. The significance of such reductions from a clinical perspective was recently investigated by Luby et al. who reported that the rate of GM loss in adolescent patients correlated with the severity of depression, probably stemming from aggressive synaptic pruning [150].
The pattern of GM structural changes in young first-episode people with MDD is not straightforward, however, with decreases in GM volume in the left lingual gyrus and left putamen and increases in GM volume in the left posterior cingulate cortex and inferior cingulate gyrus, being reported by Yang and others [151]. This abnormal pattern of GM volume change has also been reported by others. For example, Kong and fellow authors reported decreased GM volume in the DLPFC and left frontal gyrus but GM volume increases in the left thalamus and right insula in treatment-naïve patients with first-episode depression [152]. Larger left hippocampal bilateral posterior cingulate volumes appear to be predictive of benefit from antidepressant therapy while low volumes in the hippocampal area in particular are predictive of resistance to antidepressants and disease recurrence. Reductions in hippocampal volume are associated with non-remitting depression, and it is interesting that GM volume change accelerates with duration of illness, concordant with there being an active process of neuroprogression in the disorder [153,154,155].
Cerebral Perfusion in MDD
The earliest data were obtained from studies using PET and SPECT [156, 157] based on operator-dependent analysis and revealed a general pattern of reduced regional CBF (rCBF) in the DLPFC, ACC and medial prefrontal cortex [156, 158]. However, the use of techniques such as resting-state perfusion with arterial spin labelling (ASL) perfusion MRI has revealed complex and variable patterns of regional hyperperfusion and hypoperfusion compared with unaffected controls [10, 159,160,161,162]. For example, Duhameau et al. reported statistically significant hyperperfusion regions in a depressed patient group compared with a healthy control group in the pallidum, amygdala, putamen, ACC and left prefrontal dorsomedial cortex [161]. On the other hand, another research team using the same imaging techniques combined with whole-brain voxel-based analyses in a study involving medication-naïve adolescent patients and matched healthy controls reported significant hypoperfusion in the amygdala together with paralimbic, frontal and cingulate regions and a pattern of hyperperfusion in the subcallosal cingulate, fusiform gyrus superior insula and putamen [10]. Another imaging technique previously used in cardiac imaging is 320-slice computed-tomography (CT), which was used to assess rCBF in MDD. There was a reduction in the pulsatility index in the MDD group as well as lower rCBF in the left GM than the right in those with MDD [163].
Overall, the most common observations are hyperperfusion in the sub-colossal cingulate [164,165,166] and hypoperfusion in frontal regions [167, 168], but there is also copious evidence of abnormal perfusion levels in numerous regions of the brain in MDD patients. A recent meta-analysis by Chen and others of studies of resting CBF in medication-free MDD patients obtained by PET, SPECT and ASL revealed elevated values in the right caudate and right thalamus and reduced values in the right ACC relative to healthy individuals [169]. This meta-analysis also revealed somewhat inconsistent changes in rCBF in the frontal–limbic–thalamic–striatal circuits and the precuneus [169].
There are significant differences in the rCBF abnormalities between patients with refractory versus non-refractory MDD ascertained by ASL [161, 164]. One of the largest such studies was of 24 individuals with refractory MDD (RM), 37 patients with non-refractory MDD (NRM) and 42 healthy controls. Individuals with NRM displayed impaired perfusion in the left prefrontal cortex compared with the controls and elevated perfusion largely confined to limbic-striatal areas [164]. RM patients on the other hand displayed impaired perfusion mainly in the bilateral thalamic and frontal regions. Importantly, NRM patients displayed higher CBF in the limbic striatal areas, left and right hippocampal areas and right lentiform nucleus compared to RM patients and healthy controls [164].
Several research teams have also reported an improvement or reversal of rCBF abnormalities following successful antidepressant therapy [162, 170]. For example, Kaichi et al. reported significantly impaired CBF, in the middle and inferior frontal gyri, left inferior temporal gyri and the subgenual anterior cingulate, detected via ASL, which normalised following a 6-week course of the selective serotonin re-uptake inhibitor escitalopram [162]. In addition, Ota and others reported decreased CBF values in the inferior frontal cortex and ACC which reversed following successful antidepressant therapy [170]. This latter finding is of particular interest as there is some evidence that impaired CBF in the ACC and indeed the thalamus may be the cause of the dysfunction seen in MDD patients in these regions of the brain [10]. In support of this view, there are replicated data demonstrating increased glucose metabolism in these areas following successful antidepressant therapy [171, 172] and that the impairment of glucose metabolism seen in active disease may well stem from a decrease in neurones in these brain regions [173].
Neuroimaging Abnormalities in CFS
Resting-State Functional Conductivity in CFS
Several authors have reported abnormal patterns of functional connectivity in patients afforded a diagnosis of CFS according to international criteria, whose magnitude correlates with levels of fatigue and pain [174,175,176,177]. The precise patterns of impairment are highly variable however.
Wortinger and fellow workers reported a significant decrease in functional connectivity of the salience network to the right posterior insula in adolescent CFS patients compared with age-matched healthy controls. Interestingly, this decrease was negatively associated with the severity of fatigue [174]. In addition, this research team reported a positive association between pain severity and salience network functional connectivity to the left middle insula and caudate which differed between the two groups [174]. Kim et al. also reported an association between altered patterns and strength of functional connectivity and fatigue severity in CFS. In this instance, the authors reported increased resting-state connectivity between the posterior cingulate cortex and the rostral and dorsal ACC whose magnitude significantly correlated with fatigue levels measured by the Chalder Fatigue Scale score while controlling for depression [175]. Gay and fellow workers have also reported widespread impairments in functional connectivity, in this case within the lateral prefrontal cortex and between the occipital lobes and other brain regions [176]. These authors also reported impaired connectivity between the left anterior midcingulate cortex and the sensory motor network and between the left posterior cingulate cortex and the salience network [176]. Importantly, the magnitude of each of these connective impairments correlated with the severity of reported fatigue. On the other hand, Boissoneault and others reported a pattern of increased functional connectivity within and between brain regions in their trial cohort compared with healthy controls. In particular, their CFS participants displayed increased connectivity in the bilateral superior frontal gyrus, ACC, precuneus, thalamus, supplementary motor area, right postcentral gyrus and posterior cingulate gyrus [177]. This contrasted markedly with healthy participants, who displayed greater functional connectivity in the left parahippocampal gyrus, right insula, right precentral gyrus and hippocampus. It is noteworthy however that the relatively reduced level of connectivity in the left parahippocampal gyrus seen in CFS correlated with fatigue severity [177].
This team also examined the patterns of dynamic and static functional connectivity in CFS compared with unaffected controls during a fatiguing cognitive task and reported major group differences. Specifically, Boissoneault and others reported decreased dynamic connectivity between the hippocampus and right superior parietal lobule and increased static inferior frontal gyrus connectivity to occipital and temporal structures and to the cerebellum [178]. It is also noteworthy that healthy controls displayed greater dynamic connectivity increases than did CFS patients between the temporo-occipital regions and insula structures and between thalamus/striatum and the precuneus [178]. Moreover, the degree of static or dynamic connectivity impairment correlated with the self-reported fatigue in CFS [178]. It is difficult to determine a typical pattern of impaired functional connectivity in CFS by a consideration of these studies. It would appear to be different from those displayed in MS and MDD, but clearly, more studies are needed to confirm this, and a study examining resting functional state abnormalities containing CFS, MDD and MS patients allowing direct comparisons to be made could be very informative. Factors likely to affect functional connectivity include peripheral inflammation, disease severity, disease progression and genetic heterogeneity.
White Matter Abnormalities in CFS
Early research investigating potential neuroanatomical abnormalities using MRI focused on WM using observer-dependent analysis [14, 179,180,181]. Natelson and others reported subcortical WM hyperintensities in 52 subjects fulfilling the original CDC selection criteria [182] compared with a control group who had undergone MRI investigation for head trauma [179]. On the other hand, Greco and others reported widespread demyelination in many of their CFS participants but at a group level, WM hyperintensities were not significantly different from healthy controls [180]. However, Lange and others also reported significantly increased WM hyperintensities in their CFS group who were free of comorbid anxiety or depression compared with healthy controls and CFS subjects with co-morbid psychopathology [183]. Interestingly, when the data obtained from the two CFS groups were combined, the levels of WM hyperintensities were not significantly different from the control group [183]. In another large study involving 48 CFS patients, Cook and others reported widespread WM abnormalities in their cohort whose extent correlated with symptom severity [14]. It should also be noted that a more recent MRI study using observer-dependent methods failed to detect any WM abnormalities in their CFS cohort although these patients were not recruited according to international consensus guidelines [184].
Recent studies based on T1- and T2-weighted MRI using VBM have universally reported significantly increased WM abnormalities in CFS subjects selected according to the “Fukuda” CDC criteria compared with healthy participants [185,186,187,188]. Puri and others reported reduced WM voxel volumes in the left occipital lobe while Barnden and others reported reduced WM voxel volumes in the frontal cortex, caudate and hypothalamus [185, 186]. Pertinently, Shan and others reported decreased WM volume in the left frontal occipital lobe and the fasciculus whose extent increased with self and clinically assessed symptom severity and also worsened over time [188]. Finally, Barnden and others reported reduced WM volume in the prefrontal cortex, the ventrolateral thalamus and internal capsule even following correction for the potential effects of anxiety and depression [187].
Grey Matter Abnormalities in CFS
Several research teams utilising VBM have reported reduced global or regional GM volumes in those with CFS [185, 188,189,190,191]. Puri and fellow workers in a study consisting of 26 CFS participants and 26 age- and sex-matched healthy controls reported significantly reduced GM volume in the left and right occipital poles, left lateral occipital cortex (superior division), left supracalcrine cortex, the posterior division of the left parahippocampal gyrus and the right angular gyrus [185]. Okada and fellow workers on the other hand reported reduced GM volume in the right prefrontal cortex [189]. It is also worthy of note that these authors also reported that the extent of GM volume reduction in the right prefrontal cortex correlated with the severity of reported fatigue [189]. However, it should also be noted that this was a relatively small study consisting of just 16 CFS participants and 16 healthy controls [189].
The results reported by deLange and others appear to be particularly interesting as their studies were specifically designed to examine potential relationships between the severity of symptoms in CFS and cerebral abnormalities and the effect of cognitive-behavioural therapy (CBT) on those cerebral abnormalities respectively [190, 191]. In the first relatively large study consisting of 28 CFS patients and 28 healthy controls, these authors reported significant reductions in global GM volume in CFS and that the extent of such reductions correlated with the degree of physical disability [190]. In the later study, these authors also reported a global reduction in GM volume which increased significantly, albeit slightly (approximately 6%), following a course of CBT [191]. Finally, in their longitudinal MRI study, Shan and others have recently reported a progressive decrease in WM volumes in the left inferior fronto–occipital fasciculus in a cohort of 15 CFS participants (unchanged in 10 normal controls), with decreased regional WM volumes in adjacent regions and GM volume reduction in contralateral regions over a 6-year period; these regional WM and GM volumes once again correlated with illness severity [188].
Abnormalities in Cerebral Perfusion in CFS
Most of the SPECT studies investigating potential CBF abnormalities in CFS have reported areas of significant cerebral hypoperfusion, at either a global [20, 192] or regional level [193, 194] compared with unaffected controls. However, a minority of researchers reported no significant CBF impairments in comparison with unaffected controls or MDD participants [195, 196]. The reasons for such discrepant findings are unclear, but fewer participants and differences in selection methods may be relevant. The much larger studies conducted by Ichise, Schwartz, Costa and Goldstein, and their colleagues [20, 192,193,194] appear worthy of particular consideration as they examined multiple brain regions and perhaps even more importantly, apart from Ichise and others [192], examined differences in patterns of abnormal perfusion between participants with CFS and MDD, which is currently a contentious area.
Ischise et al. compared rCBF in 60 CFS participants and 14 healthy controls and reported significant reductions in cortical/cerebellar rCBF ratios in the frontal, temporal, parietal and occipital lobes, providing objective evidence of widespread functional impairment of the brain in the vast majority of their CFS subjects [192]. Schwartz et al. examined SPECT data differences in terms of regional perfusional deficits and mid-cerebral uptake indices between 45 CFS participants, 14 MDD participants, 27 participants with AIDS dementia complex (ADC) and 38 healthy controls [20]. These authors noted more areas of impaired frontal and temporal perfusion in the ADC group compared with those with CFS or MDD. However, the mid-cerebral uptake index was lower in patients with ADC and CFS than in the MDD and healthy controls groups [20].
The largest study examining potential 99mTc-HMPAO SPECT differences between CFS and MDD patients was carried out by Costa and others [193]. It included 40 healthy participants, 67 CFS participants (both with and without psychiatric co-morbidity) and 29 early- and late-onset MDD participants [193]. These authors reported significantly greater global and brain-stem hypoperfusion in CFS participants free of psychiatric symptoms compared with controls and MDD participants, which was not apparent in CFS participants displaying symptoms of anxiety and depression [193]. Goldstein and others compared high-resolution 99mTc-HMPAO SPECT imaging patterns of rCBF in 33 CFS subjects (55 ± 10 years), 26 age-matched MDD participants and 19 healthy participants in an attempt to investigate the potential pathophysiology of CFS [194]. Importantly, they reported dorsofrontal hypoperfusion as the dominant profile in CFS while hypoperfusion seen in MDD was predominantly in the right orbitofrontal lobe, left temporal lobe and, in particular, the left anterior frontal lobes [194].
More recent research using xenon-computed tomography, ASL and high-resolution SPECT reported consistently impaired CBF at a global and regional level at rest and during exercise [197,198,199,200]. Yoshiuchi and others reported reduced CBF in the right and left hemisphere in CFS participants free of psychiatric comorbidity compared with healthy controls but also noted that hypoperfusion in CFS patients with psychiatric comorbidity appeared to be exclusively limited to the left hemisphere [198]. These results have been broadly replicated by Biswal and others who reported impaired regional and global CBF in the vast majority of their participants with evidence of regional hyperperfusion in a minority [197]. Evidence of hyperperfusion in some CFS subjects has also been supplied by Machale and others who reported significantly increased hyperperfusion in the left thalamus in CFS compared with a pattern of hyperperfusion in the left prefrontal cortex of MDD [200]. Finally, Patrick-Neary and others reported significantly decreased CBF in CFS participants during maximal exercise compared with healthy controls which they suggested as a cause of the profound disability experienced by many patients [199].
The potential capacity of SPECT to differentiate CFS from MDD may be highly significant as CFS is a purely descriptive diagnosis reliant on phenotype only, and there is accumulating evidence that the diagnosis identifies a clinically and aetiologically heterogeneous syndrome rather than a discrete biological entity even when such a diagnosis is made via the application of international guidelines [1, 3, 201, 202]. Unsurprisingly, research in this area has been bedevilled by a lack of reproducibility which is also the case in many other areas where diagnostic categories are descriptive and homogeneity of causation is assumed (reviewed in [203]). This issue in ‘CFS research’ is even more complex because of the frequent use of unvalidated criteria which only mandate the presence of chronic mental and physical tiredness [204] or where a diagnosis is based on the presence of intermittent symptoms based on a population study of fatigue in a localised population [205]. Hence, a method for increasing the homogeneity of a study population and excluding patients with MDD is to be welcomed. The question of heterogeneity is also relevant to the next section of this paper which briefly examines mechanisms whereby systemic inflammation could be a major element initiating and maintaining neuropathology in different illnesses even though investigation with an array of neuroimaging techniques produces very different and often illness-specific results.
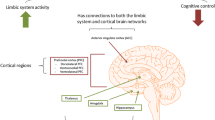
Factors Affecting Severity and Specificity of Neuropathology
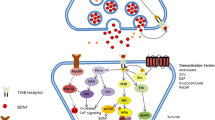
There is widespread agreement that the relationship between the presence of systemic inflammation and various dimensions of neuropathology is established via the conveyance of inflammatory signals to the brain through a range of humoral and neural routes culminating in the activation of microglia and astrocytes [206, 207]. Once activated, these glial cells secrete a range of inflammatory molecules such as pro-inflammatory cytokines, reactive oxygen species, reactive nitrogen species, prostaglandins and quinolinic acid [208, 209]. These are well-documented effects and have been described in a number of detailed reviews [210, 211].
Another source of neuropathology is more indirect however and stems from the loss or corruption of the regulatory functions exerted by these glial cells via a sophisticated communication system with neurones and oligodendrocytes founded on the secretion of exosomes containing mRNAs, miRNAs, caspases and P2X7 (reviewed in [212]). This is a highly complex area, but the key point from the perspective of this paper is that abnormally expressed microglial exosomal miRNAs appear to be independent contributors to the development maintenance and illness-specific patterns of neuroinflammation seen in many different neurodegenerative and neuroprogressive disorders [213]. Thus, microglial activation following prolonged systemic inflammation could produce an illness-specific or at least a range of characteristic WM abnormalities depending on a particular signature of abnormally expressed miRNAs. The mechanisms underpinning this phenomenon are currently unknown but may lie in the recently discovered capacity of at least some miRNAs to activate toll-like receptors and are another mechanism underpinning ‘sterile’ immune activation [214, 215].
Microglia also display distinct regional differences in density and transcriptional identity particularly in bioenergetic and immunoregulatory pathways [216, 217]. This underpins observations of region-specific sensitivities to microglial dysfunction and in particular responses to different pathogen-associated molecular patterns [216, 217]. Hence, this provides another mechanism whereby systemic inflammation could produce geographically variable neuropathology. Another highly pertinent example of the role of microglial genetics in creating unique patterns of abnormalities can be found from recent data in MS, where microglia deficient in the production of caspase-8 are considered to be the source of necroptotic markers seen in active plaques of MS patients. These glial cells are likely one of the main drivers of neuronal and oligodendrocyte destruction seen in this illness [218, 219]. The heterogeneous responses of microglia to external or internal stimuli are also under epigenetic regulation involving DNA methylation, histone acetylation and the expression of a wide range of miRNAs [220]. It is also noteworthy that different genomic patterns of DNA methylation within and outside glial cells are associated with different patterns of neuropathology in distinct brain regions [221].
Predictably, astrocytes also display extensive variability in biochemical, biophysical and immunoregulatory properties in distinct brain regions (reviewed in [222, 223]). The signalling pathways orchestrated by astrocytes are also strongly influenced by genetics [224]. Importantly, some of the neuroprotective functions of astrocytes are also under epigenetic control. Reactive microglia also compromise astrocytic function and survival in an inflammatory environment with the latter property resulting from a decrease of histone acetylation in astrocytes and a silencing of Nrf-2-mediated antioxidant defences [225].
Antioxidant defence systems also demonstrate pronounced regional differences in composition and efficacy. For example, thioredoxin and thioredoxin reductase levels vary in different brain regions [226]. This is also true of glutathione transferase, glutathione reductase and glutathione peroxidase, with the former two enzymes being reduced in the striatum, neocortex, cerebellum and hippocampus, whereas expression of the last enzyme appears to be reduced in the striatum, cerebellum, cortex and corpus callosum [227, 228]. The expression and activity of NADPH oxidases also displays considerable variation in different brain regions [229]. Selenium distribution across different brain regions is also heterogeneous, with the highest concentrations being in the parietal inferior lobule, putamen and occipital cortex and the lowest levels in the cerebellum and medulla [230].
Different discrete neuronal populations existing in different brain regions display differential susceptibility to neurodegenerative stressors which is a phenomenon often described as selective neuronal vulnerability (SNV). Well-documented examples include neurones in the entorhinal cortex, frontal cortex, hippocampal CA1 region and amygdala, which are the most susceptible to neurodegenerative death in populations of neurones most sensitive to the neurodegeneration associated with Alzheimer’s disease [231, 232]. The increased susceptibility of dopaminergic neurones of the substantia nigra to neurodegenerative destruction in Parkinson’s disease is equally well documented [233, 234]. It should be stressed that the pattern and extent of neural damage seen in different neurodegenerative disease is not just dependent on SNV but also stems from specific factors unique to the aetiology of the illness (reviewed in [235]). SNV also exists within populations in the same brain region. For example, the hippocampal CA1 neurones are much more vulnerable than are CA3 neurones to global cerebral ischaemia [236, 237] and oxidative stress [238, 239].
Brain volume and structure are strongly influenced by genetic factors [240,241,242] and resting-state functional connectivity [243, 244], GM and WM volume [245, 246] and CBF [247, 248] are sensitive to environmentally induced epigenetic changes in gene methylation and/or acetylation and/or changes in the expression of a wide range of miRNAs. These factors may also produce heterogeneity in neuroimaging phenotypes between individuals with the same illness or between illnesses even if systemic inflammation is a major driver of neuropathology in each instance.
Conclusions and Future Directions
We begin by briefly summarising the key neuroimaging findings.
In MS, there is evidence of impaired neural connectivity and functional dissociation on resting-state fMRI, which correlates with the severity of disability. Cortical demyelination and reduced cerebral perfusion also occur. Early GM atrophy appears to be predictive of the subsequent level of disability and of the prognosis.
MDD is associated with dysfunction of functional connectivity and both microstructural and macroscopic WM abnormalities, including disrupted tracts. There is evidence of GM volume loss in several regions.
CFS is associated with cerebral hypoperfusion, while large VBM studies have shown evidence of GM and WM changes.
The evidence given in this paper points to the likelihood that systemic inflammation may be a major underlying cause of structural and functional neuroimaging abnormalities reported in seemingly diverse disorders including MS, MDD and CFS. Furthermore, reported inter-patient and inter-illness differences may be a function of regional genetic, epigenetic and environmental variations in microglial and astrocytic functioning.
While SPECT appears to be able to differentiate CFS from MDD, its use of ionising radiation makes it unlikely to be a neuroimaging modality of choice in the future for this purpose. Rather, efforts are likely to be undertaken to develop corresponding functional magnetic resonance-based techniques instead. Not only are magnetic resonance techniques free of the use of ionising radiation, they also afford a higher resolution than does SPECT (even high-resolution SPECT). In this vein, a further development is likely to be the use of machine learning classification methodologies, which may, for example, be able in the near future to differentiate different disorders and, within a given disorder, be able to classify patients with a given diagnosis. Finally, there is also a strong case for further combining neuroimaging techniques with investigative modalities from the fields of biochemistry, neurophysiology, environmental medicine, immunology and epigenetics.
References
Morris G, Berk M (2015) The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med 13(1):68
Morris G, Berk M, Walder K, Maes M (2015) Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med 13(1):28
Morris G, Maes M (2013) Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med 11(1):205
Katdare A, Ursekar M (2015) Systematic imaging review: multiple sclerosis. Ann Indian Acad Neurol 18(5):24–29. doi:10.4103/0972-2327.164821
Sicotte NL (2011) Neuroimaging in multiple sclerosis: neurotherapeutic implications. Neurother J Am Soc Exp NeuroTher 8(1):54–62. doi:10.1007/s13311-010-0008-y
Rashid W, Miller DH (2008) Recent advances in neuroimaging of multiple sclerosis. Semin Neurol 28(1):46–55. doi:10.1055/s-2007-1019127
Wise T, Cleare AJ, Herane A, Young AH, Arnone D (2014) Diagnostic and therapeutic utility of neuroimaging in depression: an overview. Neuropsychiatr Dis Treat 10:1509–1522. doi:10.2147/NDT.S50156
Kempton M, Salvador Z, Munafo M, Geddes J, Simmons A, Frangou S (2011) Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Arch Gen Psychiatry 68:675–690
Rao NP, Venkatasubramanian G, Gangadhar BN (2011) Proton magnetic resonance spectroscopy in depression. Indian J Psychiatry 53(4):307–311. doi:10.4103/0019-5545.91903
Ho T, Wu J, Shin D, Liu T, Tapert S, Yang G (2013) Altered cerebral perfusion in executive, affective, and motor networks during adolescent depression. J Am Acad Child Adolesc Psychiatry 52:1076–1091
MacMaster FP, Carrey N, Langevin LM, Jaworska N, Crawford S (2014) Disorder-specific volumetric brain difference in adolescent major depressive disorder and bipolar depression. Brain Imaging Behav 8(1):119–127. doi:10.1007/s11682-013-9264-x
Buchwald D, Cheney PR, Peterson DL, Henry B, Wormsley SB, Geiger A, Ablashi DV, Salahuddin Z, et al. (1992) A chronic illness characterized by fatigue, neurologic and immunologic disorders, and active human herpesvirus type 6 infection. Ann Intern Med 116. doi:10.7326/0003-4819-116-2-103
de Lange FP, Kalkman JS, Bleijenberg G, Hagoort P, van der Meer JW, Toni I (2005) Gray matter volume reduction in the chronic fatigue syndrome. NeuroImage 26(3):777–781. doi:10.1016/j.neuroimage.2005.02.037
Cook DB, Lange G, DeLuca J, Natelson BH (2001) Relationship of brain MRI abnormalities and physical functional status in chronic fatigue syndrome. Intern J Neurosci 107. doi:10.3109/00207450109149754
Tanaka M, Sadato N, Okada T, Mizuno K, Sasabe T, Tanabe HC, Saito DN, Onoe H, et al. (2006) Reduced responsiveness is an essential feature of chronic fatigue syndrome: a fMRI study. BMC Neurol 6. doi:10.1186/1471-2377-6-9
de Lange FP, Kalkman JS, Bleijenberg G, Hagoort P, van der Werf SP, van der Meer JW, Toni I (2004) Neural correlates of the chronic fatigue syndrome—an fMRI study. Brain 127(Pt 9):1948–1957. doi:10.1093/brain/awh225
Puri BK (2006) Proton and 31-phosphorus neurospectroscopy in the study of membrane phospholipids and fatty acid intervention in schizophrenia, depression, chronic fatigue syndrome (myalgic encephalomyelitis) and dyslexia. Int Rev Psychiatry (Abingdon, England) 18(2):145–147. doi:10.1080/09540260600581852
Cox IJ, Puri BK (2004) In vivo MR spectroscopy in diagnosis and research of neuropsychiatric disorders. Prostaglandins Leukot Essent Fatty Acids 70(4):357–360. doi:10.1016/j.plefa.2003.12.010
Mathew S, Mao X, Keegan K, Levine S, Smith E, Heier L (2009) Ventricular cerebrospinal fluid lactate is increased in chronic fatigue syndrome compared with generalized anxiety disorder: an in vivo 3.0T (1)H MRS imaging study. NMR Biomed 22:251–258
Schwartz R, Garada B, Komaroff A, Tice H, Gleit M, Jolesz F, Holman B (1994) Detection of intracranial abnormalities in patients with chronic fatigue syndrome: comparison of MR imaging and SPECT. AJR Am J Roentgenol 162:935–941
Sanfilipo MP, Benedict RH, Weinstock-Guttman B, Bakshi R (2006) Gray and white matter brain atrophy and neuropsychological impairment in multiple sclerosis. Neurology 66(5):685–692. doi:10.1212/01.wnl.0000201238.93586.d9
Politte LC, Huffman JC, Stern TA (2008) Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry 10(4):318–324
Reipert B (2004) Multiple sclerosis: a short review of the disease and its differences between men and women. J Men’s Health Gend 1(4):334–340. doi:10.1016/j.jmhg.2004.10.005
Labrenz F, Wrede K, Forsting M, Engler H, Schedlowski M, Elsenbruch S, Benson S (2016) Alterations in functional connectivity of resting state networks during experimental endotoxemia—an exploratory study in healthy men. Brain Behav Immun 54:17–26. doi:10.1016/j.bbi.2015.11.010
Lekander M, Karshikoff B, Johansson E, Soop A, Fransson P, Lundstrom JN, Andreasson A, Ingvar M et al (2016) Intrinsic functional connectivity of insular cortex and symptoms of sickness during acute experimental inflammation. Brain Behav Immun 56:34–41. doi:10.1016/j.bbi.2015.12.018
Marsland AL, Gianaros PJ, Kuan DC, Sheu LK, Krajina K, Manuck SB (2015) Brain morphology links systemic inflammation to cognitive function in midlife adults. Brain Behav Immun 48:195–204. doi:10.1016/j.bbi.2015.03.015
Sankowski R, Mader S, Valdés-Ferrer SI (2015) Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosci 9:28. doi:10.3389/fncel.2015.00028
Riverol M, Becker J, Lopez O, Raji C, Thompson P, Carmichael O, Gach H, Longstreth WT et al (2012) Systemic inflammatory markers, cognition and brain structure among cognitively normal elderly (P02.061). Neurology 78(1 Supplement):P02.061
Adam N, Kandelman S, Mantz J, Chretien F, Sharshar T (2013) Sepsis-induced brain dysfunction. Expert review of anti-infective therapy 11(2):211–221. doi:10.1586/eri.12.159
Sonneville R, Verdonk F, Rauturier C, Klein IF, Wolff M, Annane D, Chretien F, Sharshar T (2013) Understanding brain dysfunction in sepsis. Ann Intensive Care 3(1):15. doi:10.1186/2110-5820-3-15
Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, Miller AH (2015) Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. doi:10.1038/mp.2015.168
Rocca MA, Valsasina P, Martinelli V, Misci P, Falini A, Comi G, Filippi M (2012) Large-scale neuronal network dysfunction in relapsing-remitting multiple sclerosis. Neurology 79(14):1449–1457. doi:10.1212/WNL.0b013e31826d5f10
He Y, Dagher A, Chen Z, Charil A, Zijdenbos A, Worsley K, Evans A (2009) Impaired small-world efficiency in structural cortical networks in multiple sclerosis associated with white matter lesion load. Brain 132(12):3366–3379. doi:10.1093/brain/awp089
Louapre C, Perlbarg V, Garcia-Lorenzo D, Urbanski M, Benali H, Assouad R, Galanaud D, Freeman L et al (2014) Brain networks disconnection in early multiple sclerosis cognitive deficits: an anatomofunctional study. Hum Brain Mapp 35(9):4706–4717. doi:10.1002/hbm.22505
Rocca MA, Amato MP, De Stefano N, Enzinger C, Geurts JJ, Penner IK, Rovira A, Sumowski JF et al (2015) Clinical and imaging assessment of cognitive dysfunction in multiple sclerosis. Lancet Neurol 14(3):302–317. doi:10.1016/s1474-4422(14)70250-9
Roosendaal SD, Schoonheim MM, Hulst HE, Sanz-Arigita EJ, Smith SM, Geurts JJ, Barkhof F (2010) Resting state networks change in clinically isolated syndrome. Brain 133(Pt 6):1612–1621. doi:10.1093/brain/awq058
Sacco R, Bonavita S, Esposito F, Tedeschi G, Gallo A (2013) The contribution of resting state networks to the study of cortical reorganization in MS. Mult Scler Int 2013:857807. doi:10.1155/2013/857807
Dogonowski AM, Andersen KW, Madsen KH, Sorensen PS, Paulson OB, Blinkenberg M, Siebner HR (2014) Multiple sclerosis impairs regional functional connectivity in the cerebellum. Neuroimage Clin 4:130–138. doi:10.1016/j.nicl.2013.11.005
Dogonowski AM, Siebner HR, Soelberg Sorensen P, Paulson OB, Dyrby TB, Blinkenberg M, Madsen KH (2013) Resting-state connectivity of pre-motor cortex reflects disability in multiple sclerosis. Acta Neurol Scand 128(5):328–335. doi:10.1111/ane.12121
Liu Y, Liang P, Duan Y, Jia X, Yu C, Zhang M, Wang F, Zhang M et al (2011) Brain plasticity in relapsing-remitting multiple sclerosis: evidence from resting-state fMRI. J Neurol Sci 304(1–2):127–131. doi:10.1016/j.jns.2011.01.023
Tona F, Petsas N, Sbardella E, Prosperini L, Carmellini M, Pozzilli C, Pantano P (2014) Multiple sclerosis: altered thalamic resting-state functional connectivity and its effect on cognitive function. Radiology 271(3):814–821. doi:10.1148/radiol.14131688
Gamboa OL, Tagliazucchi E, von Wegner F, Jurcoane A, Wahl M, Laufs H, Ziemann U (2014) Working memory performance of early MS patients correlates inversely with modularity increases in resting state functional connectivity networks. NeuroImage 94:385–395. doi:10.1016/j.neuroimage.2013.12.008
Dogonowski AM, Siebner HR, Sorensen PS, Wu X, Biswal B, Paulson OB, Dyrby TB, Skimminge A et al (2013) Expanded functional coupling of subcortical nuclei with the motor resting-state network in multiple sclerosis. Mult Scler 19(5):559–566. doi:10.1177/1352458512460416
Rocca MA, Valsasina P, Absinta M, Riccitelli G, Rodegher ME, Misci P, Rossi P, Falini A et al (2010) Default-mode network dysfunction and cognitive impairment in progressive MS. Neurology 74(16):1252–1259. doi:10.1212/WNL.0b013e3181d9ed91
Faivre A, Rico A, Zaaraoui W, Reuter F, Confort-Gouny S, Guye M, Pelletier J, Ranjeva J-P et al (2015) Brain functional plasticity at rest and during action in multiple sclerosis patients. J Clin Neurosci 22(9):1438–1443. doi:10.1016/j.jocn.2015.02.033
Faivre A, Rico A, Zaaraoui W, Crespy L, Reuter F, Wybrecht D, Soulier E, Malikova I et al (2012) Assessing brain connectivity at rest is clinically relevant in early multiple sclerosis. Mult Scler J 18(9):1251–1258. doi:10.1177/1352458511435930
Cruz-Gomez AJ, Ventura-Campos N, Belenguer A, Avila C, Forn C (2014) The link between resting-state functional connectivity and cognition in MS patients. Mult Scler 20(3):338–348. doi:10.1177/1352458513495584
Janssen AL, Boster A, Patterson BA, Abduljalil A, Prakash RS (2013) Resting-state functional connectivity in multiple sclerosis: an examination of group differences and individual differences. Neuropsychologia 51(13):2918–2929. doi:10.1016/j.neuropsychologia.2013.08.010
Dogonowski A-M, Blinkenberg M, Paulson OB, Sellebjerg F, Sørensen PS, Siebner HR, Madsen KH (2015) Recovery from an acute relapse is associated with changes in motor resting-state connectivity in multiple sclerosis. J Neurol Neurosurg Psychiatry. doi:10.1136/jnnp-2015-311375
Bonavita S, Gallo A, Sacco R, Corte MD, Bisecco A, Docimo R, Lavorgna L, Corbo D et al (2011) Distributed changes in default-mode resting-state connectivity in multiple sclerosis. Mult Scler J 17(4):411–422. doi:10.1177/1352458510394609
Basile B, Castelli M, Monteleone F, Nocentini U, Caltagirone C, Centonze D, Cercignani M, Bozzali M (2013) Functional connectivity changes within specific networks parallel the clinical evolution of multiple sclerosis. Mult Scler 20(8):1050–1057. doi:10.1177/1352458513515082
Zhou F, Zhuang Y, Gong H, Wang B, Wang X, Chen Q, Wu L, Wan H (2014) Altered inter-subregion connectivity of the default mode network in relapsing remitting multiple sclerosis: a functional and structural connectivity study. PLoS One 9(7):e101198. doi:10.1371/journal.pone.0101198
Schoonheim M, Geurts J, Wiebenga O, De Munck J, Polman C, Stam C, Barkhof F, Wink A (2013) Changes in functional network centrality underlie cognitive dysfunction and physical disability in multiple sclerosis. Mult Scler 20(8):1058–1065. doi:10.1177/1352458513516892
Colasanti A, Guo Q, Giannetti P, Wall MB, Newbould RD, Bishop C, Onega M, Nicholas R et al Hippocampal neuroinflammation, functional connectivity, and depressive symptoms in multiple sclerosis. Biol Psychiatry. doi:10.1016/j.biopsych.2015.11.022
Koenig KA, Lowe MJ, Lin J, Sakaie KE, Stone L, Bermel RA, Beall EB, Rao SM et al (2013) Sex differences in resting-state functional connectivity in multiple sclerosis. AJNR Am J Neuroradiol 34(12):2304–2311. doi:10.3174/ajnr.A3630
Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE et al (2005) Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128(Pt 11):2705–2712. doi:10.1093/brain/awh641
Bar-Zohar D, Agosta F, Goldstaub D, Filippi M (2008) Magnetic resonance imaging metrics and their correlation with clinical outcomes in multiple sclerosis: a review of the literature and future perspectives. Mult Scler 14(6):719–727. doi:10.1177/1352458507088102
Frohman EM, Racke MK, Raine CS (2006) Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med 354(9):942–955. doi:10.1056/NEJMra052130
Larsson HB, Frederiksen J, Petersen J, Nordenbo A, Zeeberg I, Henriksen O, Olesen J (1989) Assessment of demyelination, edema, and gliosis by in vivo determination of T1 and T2 in the brain of patients with acute attack of multiple sclerosis. Magn Reson Med 11(3):337–348
Sinnecker T, Mittelstaedt P, Dorr J, Pfueller CF, Harms L, Niendorf T, Paul F, Wuerfel J (2012) Multiple sclerosis lesions and irreversible brain tissue damage: a comparative ultrahigh-field strength magnetic resonance imaging study. Arch Neurol 69(6):739–745. doi:10.1001/archneurol.2011.2450
Miller DH, Thompson AJ, Filippi M (2003) Magnetic resonance studies of abnormalities in the normal appearing white matter and grey matter in multiple sclerosis. J Neurol 250(12):1407–1419. doi:10.1007/s00415-003-0243-9
Ciccarelli O, Werring DJ, Barker GJ, Griffin CM, Wheeler-Kingshott CA, Miller DH, Thompson AJ (2003) A study of the mechanisms of normal-appearing white matter damage in multiple sclerosis using diffusion tensor imaging—evidence of Wallerian degeneration. J Neurol 250(3):287–292. doi:10.1007/s00415-003-0992-5
Ge Y, Grossman RI, Babb JS, He J, Mannon LJ (2003) Dirty-appearing white matter in multiple sclerosis: volumetric MR imaging and magnetization transfer ratio histogram analysis. AJNR Am J Neuroradiol 24(10):1935–1940
Rigotti DJ, Gass A, Achtnichts L, Inglese M, Babb JS, Naegelin Y, Hirsch J, Amann M et al (2012) Multiple sclerosis severity scale and whole-brain N-acetylaspartate concentration for patients’ assessment. Mult Scler (Houndmills, Basingstoke, England) 18(1):98–107. doi:10.1177/1352458511415142
Achtnichts L, Gonen O, Rigotti DJ, Babb JS, Naegelin Y, Penner IK, Bendfeldt K, Hirsch J et al (2013) Global N-acetylaspartate concentration in benign and non-benign multiple sclerosis patients of long disease duration. Eur J Radiol 82(12):e848–e852. doi:10.1016/j.ejrad.2013.08.037
Filippi M, Campi A, Dousset V, Baratti C, Martinelli V, Canal N, Scotti G, Comi G (1995) A magnetization transfer imaging study of normal-appearing white matter in multiple sclerosis. Neurology 45(3 Pt 1):478–482
Davie CA, Barker GJ, Thompson AJ, Tofts PS, McDonald WI, Miller DH (1997) 1H magnetic resonance spectroscopy of chronic cerebral white matter lesions and normal appearing white matter in multiple sclerosis. J Neurol Neurosurg Psychiatry 63(6):736–742
Droogan AG, Clark CA, Werring DJ, Barker GJ, McDonald WI, Miller DH (1999) Comparison of multiple sclerosis clinical subgroups using navigated spin echo diffusion-weighted imaging. Magn Reson Imaging 17(5):653–661
Stevenson VL, Parker GJ, Barker GJ, Birnie K, Tofts PS, Miller DH, Thompson AJ (2000) Variations in T1 and T2 relaxation times of normal appearing white matter and lesions in multiple sclerosis. J Neurol Sci 178(2):81–87
Leary SM, Silver NC, Stevenson VL, Barker GJ, Miller DH, Thompson AJ (1999) Magnetisation transfer of normal appearing white matter in primary progressive multiple sclerosis. Mult Scler 5(5):313–316
Gustafsson M, Dahlqvist O, Jaworski J, Lundberg P, Landtblom A (2007) Low choline concentrations in normal-appearing white matter of patients with multiple sclerosis and normal MR imaging brain scans. AJNR Am J Neuroradiol 28:1306–1312
Seewann A, Vrenken H, van der Valk P, Blezer EL, Knol DL, Castelijns JA, Polman CH, Pouwels PJ et al (2009) Diffusely abnormal white matter in chronic multiple sclerosis: imaging and histopathologic analysis. Arch Neurol 66(5):601–609. doi:10.1001/archneurol.2009.57
Vrenken H, Seewann A, Knol DL, Polman CH, Barkhof F, Geurts JJ (2010) Diffusely abnormal white matter in progressive multiple sclerosis: in vivo quantitative MR imaging characterization and comparison between disease types. AJNR Am J Neuroradiol 31(3):541–548. doi:10.3174/ajnr.A1839
Richert ND, Ostuni JL, Bash CN, Duyn JH, McFarland HF, Frank JA (1998) Serial whole-brain magnetization transfer imaging in patients with relapsing-remitting multiple sclerosis at baseline and during treatment with interferon beta-1b. AJNR Am J Neuroradiol 19(9):1705–1713
van Buchem MA, McGowan JC, Kolson DL, Polansky M, Grossman RI (1996) Quantitative volumetric magnetization transfer analysis in multiple sclerosis: estimation of macroscopic and microscopic disease burden. Magn Reson Med 36(4):632–636
Geurts JJ, Barkhof F (2008) Grey matter pathology in multiple sclerosis. Lancet Neurol 7(9):841–851. doi:10.1016/s1474-4422(08)70191-1
Jacobsen CO, Farbu E (2014) MRI evaluation of grey matter atrophy and disease course in multiple sclerosis: an overview of current knowledge. Acta Neurol Scand 129:32–36. doi:10.1111/ane.12234
Radu EW, Bendfeldt K, Mueller-Lenke N, Magon S, Sprenger T (2013) Brain atrophy: an in-vivo measure of disease activity in multiple sclerosis. Swiss Med Wkly 143:w13887. doi:10.4414/smw.2013.13887
Haider L, Simeonidou C, Steinberger G, Hametner S, Grigoriadis N, Deretzi G (2014) Multiple sclerosis deep grey matter: the relation between demyelination, neurodegeneration, inflammation and iron. J Neurol Neurosurg Psychiatry 85:1386–1395
Bo L, Vedeler CA, Nyland HI, Trapp BD, Mork SJ (2003) Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol 62(7):723–732
Lucchinetti CF, Popescu BFG, Bunyan RF, Moll NM, Roemer SF, Lassmann H, Brück W, Parisi JE et al (2011) Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 365(23):2188–2197. doi:10.1056/NEJMoa1100648
Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, Serafini B, Aloisi F et al (2011) Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 134(Pt 9):2755–2771. doi:10.1093/brain/awr182
Stadelmann C, Bruck W (2008) Interplay between mechanisms of damage and repair in multiple sclerosis. J Neurol 255(Suppl 1):12–18. doi:10.1007/s00415-008-1003-7
Calabrese M, Magliozzi R, Ciccarelli O, Geurts JJG, Reynolds R, Martin R (2015) Exploring the origins of grey matter damage in multiple sclerosis. Nat Rev Neurosci 16(3):147–158. doi:10.1038/nrn3900
Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL, Mutani R, Giordana MT et al (2007) Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 66(8):732–739. doi:10.1097/nen.0b013e31812571b0
Witte M, Nijland P, Drexhage J, Gerritsen W, Geerts D, van Het Hof B (2013) Reduced expression of PGC-1alpha partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathol 125:231–243
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS et al (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132(5):1175–1189. doi:10.1093/brain/awp070
Lassman H, van Horssen J (2011) The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett 585:3715–3723
Lycke J, Wikkelso C, Bergh AC, Jacobsson L, Andersen O (1993) Regional cerebral blood flow in multiple sclerosis measured by single photon emission tomography with technetium-99m hexamethylpropyleneamine oxime. Eur Neurol 33(2):163–167
Sun X, Tanaka M, Kondo S, Okamoto K, Hirai S (1998) Clinical significance of reduced cerebral metabolism in multiple sclerosis: a combined PET and MRI study. Ann Nucl Med 12(2):89–94
Law M, Saindane AM, Ge Y, Babb JS, Johnson G, Mannon LJ, Herbert J, Grossman RI (2004) Microvascular abnormality in relapsing-remitting multiple sclerosis: perfusion MR imaging findings in normal-appearing white matter. Radiology 231(3):645–652. doi:10.1148/radiol.2313030996
Adhya S, Johnson G, Herbert J, Jaggi H, Babb J, Grossman R, Inglese M (2006) Pattern of hemodynamic impairment in multiple sclerosis: dynamic susceptibility contrast perfusion MR imaging at 3.0 T. NeuroImage 33:1029–1035
Varga AW, Johnson G, Babb JS, Herbert J, Grossman RI, Inglese M (2009) White matter hemodynamic abnormalities precede sub-cortical gray matter changes in multiple sclerosis. J Neurol Sci 282(1–2):28–33. doi:10.1016/j.jns.2008.12.036
D’Haeseleer M, Hostenbach S, Peeters I, Sankari SE, Nagels G, De Keyser J, D’Hooghe MB (2015) Cerebral hypoperfusion: a new pathophysiologic concept in multiple sclerosis? J Cereb Blood Flow Metab 35(9):1406–1410. doi:10.1038/jcbfm.2015.131
D’Haeseleer M, Cambron M, Vanopdenbosch L, De Keyser J (2011) Vascular aspects of multiple sclerosis. Lancet Neurol 10(7):657–666. doi:10.1016/s1474-4422(11)70105-3
Papadaki EZ, Mastorodemos VC, Amanakis EZ, Tsekouras KC, Papadakis AE, Tsavalas ND, Simos PG, Karantanas AH et al (2012) White matter and deep gray matter hemodynamic changes in multiple sclerosis patients with clinically isolated syndrome. Magn Reson Med 68(6):1932–1942. doi:10.1002/mrm.24194
Taghizadeh Asl M, Nemati R, Chabi N, Salimipour H, Nabipour I, Assadi M (2016) Brain perfusion imaging with voxel-based analysis in secondary progressive multiple sclerosis patients with a moderate to severe stage of disease: a boon for the workforce. BMC Neurol 16(1):79. doi:10.1186/s12883-016-0605-4
Inglese M, Park S, Johnson G, Babb J, Miles L, Jaggi H (2007) Deep gray matter perfusion in multiple sclerosis: dynamic susceptibility contrast perfusion magnetic resonance imaging at 3T. Arch Neurol 64:196–202
Debernard L, Melzer T, Van Stockum S, Graham C, Wheeler-Kingshott C, Dalrymple-Alford J (2013) Reduced grey matter perfusion without volume loss in early relapsing-remitting multiple sclerosis. J Neurol Neurosurg Psychiatry 85:544–551
D’Haeseleer M, Steen C, Hoogduin JM, van Osch MJ, Fierens Y, Cambron M, Koch MW, De Keyser J (2013) Performance on paced auditory serial addition test and cerebral blood flow in multiple sclerosis. Acta Neurol Scand 128(5):e26–e29. doi:10.1111/ane.12129
Inglese M, Adhya S, Johnson G, Babb JS, Miles L, Jaggi H, Herbert J, Grossman RI (2008) Perfusion magnetic resonance imaging correlates of neuropsychological impairment in multiple sclerosis. J Cereb Blood Flow Metab 28(1):164–171. doi:10.1038/sj.jcbfm.9600504
Francis PL, Jakubovic R, O’Connor P, Zhang L, Eilaghi A, Lee L, Carroll TJ, Mouannes-Srour J et al (2013) Robust perfusion deficits in cognitively impaired patients with secondary-progressive multiple sclerosis. AJNR Am J Neuroradiol 34(1):62–67. doi:10.3174/ajnr.A3148
Aviv RI, Francis PL, Tenenbein R, O’Connor P, Zhang L, Eilaghi A, Lee L, Carroll TJ et al (2012) Decreased frontal lobe gray matter perfusion in cognitively impaired patients with secondary-progressive multiple sclerosis detected by the bookend technique. AJNR Am J Neuroradiol 33(9):1779–1785. doi:10.3174/ajnr.A3060
Wei M, Qin J, Yan R, Li H, Yao Z, Lu Q (2013) Identifying major depressive disorder using Hurst exponent of resting-state brain networks. Psychiatry Res 214(3):306–312. doi:10.1016/j.pscychresns.2013.09.008
Liston C, Chen AC, Zebley BD, Drysdale AT, Gordon R, Leuchter B, Voss HU, Casey BJ et al (2014) Default mode network mechanisms of transcranial magnetic stimulation in depression. Biol Psychiatry 76(7):517–526. doi:10.1016/j.biopsych.2014.01.023
Nugent AC, Robinson SE, Coppola R, Zarate CA Jr (2016) Preliminary differences in resting state MEG functional connectivity pre- and post-ketamine in major depressive disorder. Psychiatry Res 254:56–66. doi:10.1016/j.pscychresns.2016.06.006
Cullen KR, Klimes-Dougan B, Vu DP, Westlund Schreiner M, Mueller BA, Eberly LE, Camchong J, Westervelt A et al (2016) Neural correlates of antidepressant treatment response in adolescents with major depressive disorder. J Child Adolesc Psychopharmacol 26(8):705–712. doi:10.1089/cap.2015.0232
Mulders PC, van Eijndhoven PF, Schene AH, Beckmann CF, Tendolkar I (2015) Resting-state functional connectivity in major depressive disorder: a review. Neurosci Biobehav Rev 56:330–344. doi:10.1016/j.neubiorev.2015.07.014
Greicius MD, Flores BH, Menon V, Glover GH, Solvason HB, Kenna H, Reiss AL, Schatzberg AF (2007) Resting-state functional connectivity in major depression: abnormally increased contributions from Subgenual cingulate cortex and thalamus. Biol Psychiatry 62(5):429–437. doi:10.1016/j.biopsych.2006.09.020
Zhu X, Wang X, Xiao J, Liao J, Zhong M, Wang W, Yao S (2012) Evidence of a dissociation pattern in resting-state default mode network connectivity in first-episode, treatment-naive major depression patients. Biol Psychiatry 71(7):611–617. doi:10.1016/j.biopsych.2011.10.035
Peng D, Liddle EB, Iwabuchi SJ, Zhang C, Wu Z, Liu J, Jiang K, Xu L et al (2015) Dissociated large-scale functional connectivity networks of the precuneus in medication-naïve first-episode depression. Psychiatry Res Neuroimaging 232(3):250–256. doi:10.1016/j.pscychresns.2015.03.003
De Kwaasteniet BP, Rive MM, Ruhé EHG, Schene AH, Veltman DJ, Fellinger L, van Wingen GA, Denys D (2015) Decreased resting-state connectivity between neurocognitive networks in treatment resistant depression. Front Psych 6. doi:10.3389/fpsyt.2015.00028
Dutta A, McKie S, Deakin JF (2014) Resting state networks in major depressive disorder. Psychiatry Res 224(3):139–151. doi:10.1016/j.pscychresns.2014.10.003
Gudayol-Ferre E, Pero-Cebollero M, Gonzalez-Garrido AA, Guardia-Olmos J (2015) Changes in brain connectivity related to the treatment of depression measured through fMRI: a systematic review. Front Hum Neurosci 9:582. doi:10.3389/fnhum.2015.00582
Smith SM, Johansen-Berg H, Jenkinson M, Rueckert D, Nichols TE, Miller KL, Robson MD, Jones DK et al (2007) Acquisition and voxelwise analysis of multi-subject diffusion data with tract-based spatial statistics. Nat Protoc 2(3):499–503. doi:10.1038/nprot.2007.45
Serafini G, Pompili M, Borgwardt S, Giuffra E, Howes O, Girardi P, Amore M (2015) The role of white matter abnormalities in treatment-resistant depression: a systematic review. Curr Pharm Des 21(10):1337–1346
Zhu X, Wang X, Xiao J, Zhong M, Liao J, Yao S (2011) Altered white matter integrity in first-episode, treatment-naive young adults with major depressive disorder: a tract-based spatial statistics study. Brain Res 1369:223–229. doi:10.1016/j.brainres.2010.10.104
Liao Y, Huang X, Wu Q, Yang C, Kuang W, Du M, Lui S, Yue Q et al (2013) Is depression a disconnection syndrome? Meta-analysis of diffusion tensor imaging studies in patients with MDD. J Psychiatry Neurosci JPN 38(1):49–56. doi:10.1503/jpn.110180
Yuan Y, Zhang Z, Bai F, Yu H, Shi Y, Qian Y, Zang Y, Zhu C et al (2007) White matter integrity of the whole brain is disrupted in first-episode remitted geriatric depression. Neuroreport 18(17):1845–1849. doi:10.1097/WNR.0b013e3282f1939f
Taylor WD, Kuchibhatla M, Payne ME, Macfall JR, Sheline YI, Krishnan KR, Doraiswamy PM (2008) Frontal white matter anisotropy and antidepressant remission in late-life depression. PLoS One 3(9):e3267. doi:10.1371/journal.pone.0003267
Abe O, Yamasue H, Kasai K, Yamada H, Aoki S, Inoue H, Takei K, Suga M et al (2010) Voxel-based analyses of gray/white matter volume and diffusion tensor data in major depression. Psychiatry Res 181(1):64–70. doi:10.1016/j.pscychresns.2009.07.007
Aghajani M, Veer IM, van Lang ND, Meens PH, van den Bulk BG, Rombouts SA, Vermeiren RR, van der Wee NJ (2014) Altered white-matter architecture in treatment-naive adolescents with clinical depression. Psychol Med 44(11):2287–2298. doi:10.1017/s0033291713003000
Bessette KL, Nave AM, Caprihan A, Stevens MC (2014) White matter abnormalities in adolescents with major depressive disorder. Brain Imaging Behav 8(4):531–541. doi:10.1007/s11682-013-9274-8
Cole J, Chaddock CA, Farmer AE, Aitchison KJ, Simmons A, McGuffin P, Fu CH (2012) White matter abnormalities and illness severity in major depressive disorder. Br J Psychiatry 201(1):33–39. doi:10.1192/bjp.bp.111.100594
Xiao J, He Y, McWhinnie CM, Yao S (2015) Altered white matter integrity in individuals with cognitive vulnerability to depression: a tract-based spatial statistics study. Sci Rep 5:9738. doi:10.1038/srep09738
Huang H, Fan X, Williamson DE, Rao U (2011) White matter changes in healthy adolescents at familial risk for unipolar depression: a diffusion tensor imaging study. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 36(3):684–691. doi:10.1038/npp.2010.199
Chen G, Hu X, Li L, Huang X, Lui S, Kuang W, Ai H, Bi F et al (2016) Disorganization of white matter architecture in major depressive disorder: a meta-analysis of diffusion tensor imaging with tract-based spatial statistics. Sci Rep 6:21825. doi:10.1038/srep21825
LeWinn KZ, Connolly CG, Wu J, Drahos M, Hoeft F, Ho TC, Simmons AN, Yang TT (2014) White matter correlates of adolescent depression: structural evidence for frontolimbic disconnectivity. J Am Acad Child Adolesc Psychiatry 53(8):899–909, 909.e891-897. doi:10.1016/j.jaac.2014.04.021
Henderson SE, Johnson AR, Vallejo AI, Katz L, Wong E, Gabbay V (2013) A preliminary study of white matter in adolescent depression: relationships with illness severity, anhedonia, and irritability. Front Psychiatry 4:152. doi:10.3389/fpsyt.2013.00152
Taylor WD, Macfall JR, Boyd B, Payne ME, Sheline YI, Krishnan RR, Murali Doraiswamy P (2011) One-year change in anterior cingulate cortex white matter microstructure: relationship with late-life depression outcomes. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry 19(1):43–52. doi:10.1097/JGP.0b013e3181e70cec
Kieseppa T, Eerola M, Mantyla R, Neuvonen T, Poutanen VP, Luoma K, Tuulio-Henriksson A, Jylha P et al (2010) Major depressive disorder and white matter abnormalities: a diffusion tensor imaging study with tract-based spatial statistics. J Affect Disord 120(1–3):240–244. doi:10.1016/j.jad.2009.04.023
Bae JN, MacFall JR, Krishnan KR, Payne ME, Steffens DC, Taylor WD (2006) Dorsolateral prefrontal cortex and anterior cingulate cortex white matter alterations in late-life depression. Biol Psychiatry 60(12):1356–1363. doi:10.1016/j.biopsych.2006.03.052
Alexopoulos GS, Murphy CF, Gunning-Dixon FM, Latoussakis V, Kanellopoulos D, Klimstra S, Lim KO, Hoptman MJ (2008) Microstructural white matter abnormalities and remission of geriatric depression. Am J Psychiatry 165(2):238–244. doi:10.1176/appi.ajp.2007.07050744
de Diego-Adelino J, Pires P, Gomez-Anson B, Serra-Blasco M, Vives-Gilabert Y, Puigdemont D, Martin-Blanco A, Alvarez E et al (2014) Microstructural white-matter abnormalities associated with treatment resistance, severity and duration of illness in major depression. Psychol Med 44(6):1171–1182. doi:10.1017/s003329171300158x
Choi KS, Holtzheimer PE, Franco AR, Kelley ME, Dunlop BW, Hu XP, Mayberg HS (2014) Reconciling variable findings of white matter integrity in major depressive disorder. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 39(6):1332–1339. doi:10.1038/npp.2013.345
Paris J (2014) The mistreatment of major depressive disorder. Can J Psychiatry 59(3):148–151
Grieve S, Korgaonkar M, Koslow S, Gordon E, Williams L (2013) Widespread reductions in gray matter volume in depression. Neuroimage Clin 3:332–339
Egger K, Schocke M, Weiss E, Auffinger S, Esterhammer R, Goebel G, Walch T, Mechtcheriakov S et al (2008) Pattern of brain atrophy in elderly patients with depression revealed by voxel-based morphometry. Psychiatry Res Neuroimaging 164(3):237–244. doi:10.1016/j.pscychresns.2007.12.018
Ballmaier M, Sowell ER, Thompson PM, Kumar A, Narr KL, Lavretsky H, Welcome SE, DeLuca H et al (2004) Mapping brain size and cortical gray matter changes in elderly depression. Biol Psychiatry 55(4):382–389. doi:10.1016/j.biopsych.2003.09.004
Taki Y, Kinomura S, Awata S, Inoue K, Sato K, Ito H, Goto R, Uchida S et al (2005) Male elderly subthreshold depression patients have smaller volume of medial part of prefrontal cortex and precentral gyrus compared with age-matched normal subjects: a voxel-based morphometry. J Affect Disord 88(3):313–320. doi:10.1016/j.jad.2005.08.003
Lai CH (2013) Gray matter volume in major depressive disorder: a meta-analysis of voxel-based morphometry studies. Psychiatry Res 211(1):37–46. doi:10.1016/j.pscychresns.2012.06.006
Bora E, Fornito A, Pantelis C, Yucel M (2012) Gray matter abnormalities in major depressive disorder: a meta-analysis of voxel based morphometry studies. J Affect Disord 138(1–2):9–18. doi:10.1016/j.jad.2011.03.049
Tang Y, Wang F, Xie G, Liu J, Li L, Su L, Liu Y, Hu X et al (2007) Reduced ventral anterior cingulate and amygdala volumes in medication-naive females with major depressive disorder: a voxel-based morphometric magnetic resonance imaging study. Psychiatry Res 156(1):83–86. doi:10.1016/j.pscychresns.2007.03.005
Johnston BA, Steele JD, Tolomeo S, Christmas D, Matthews K (2015) Structural MRI-based predictions in patients with treatment-refractory depression (TRD). PLoS One 10(7):e0132958. doi:10.1371/journal.pone.0132958
Serra-Blasco M, Portella MJ, Gómez-Ansón B, de Diego-Adeliño J, Vives-Gilabert Y, Puigdemont D, Granell E, Santos A et al (2013) Effects of illness duration and treatment resistance on grey matter abnormalities in major depression. Br J Psychiatry 202(6):434–440. doi:10.1192/bjp.bp.112.116228
Haubold A, Peterson BS, Bansal R (2012) Annual research review: progress in using brain morphometry as a clinical tool for diagnosing psychiatric disorders. J Child Psychol Psychiatry Allied Disciplines 53(5):519–535. doi:10.1111/j.1469-7610.2012.02539.x
Pannekoek JN, van der Werff SJ, van den Bulk BG, van Lang ND, Rombouts SA, van Buchem MA, Vermeiren RR, van der Wee NJ (2014) Reduced anterior cingulate gray matter volume in treatment-naive clinically depressed adolescents. Neuroimage Clin 4:336–342. doi:10.1016/j.nicl.2014.01.007
Zou K, Deng W, Li T, Zhang B, Jiang L, Huang C, Sun X, Sun X (2010) Changes of brain morphometry in first-episode, drug-naive, non-late-life adult patients with major depression: an optimized voxel-based morphometry study. Biol Psychiatry 67(2):186–188. doi:10.1016/j.biopsych.2009.09.014
Ma N, Li L, Shu N, Liu J, Gong G, He Z, Li Z, Tan L et al (2007) White matter abnormalities in first-episode, treatment-naive young adults with major depressive disorder. Am J Psychiatry 164(5):823–826. doi:10.1176/ajp.2007.164.5.823
Luby JL, Belden AC, Jackson JJ et al (2016) Early childhood depression and alterations in the trajectory of gray matter maturation in middle childhood and early adolescence. JAMA Psychiatry 73(1):31–38. doi:10.1001/jamapsychiatry.2015.2356
Yang X, Ma X, Huang B, Sun G, Zhao L, Lin D, Deng W, Li T et al (2015) Gray matter volume abnormalities were associated with sustained attention in unmedicated major depression. Compr Psychiatry 63:71–79. doi:10.1016/j.comppsych.2015.09.003
Kong L, Wu F, Tang Y, Ren L, Kong D, Liu Y, Xu K, Wang F (2014) Frontal-subcortical volumetric deficits in single episode, medication-naive depressed patients and the effects of 8 weeks fluoxetine treatment: a VBM-DARTEL study. PLoS One 9(1):e79055. doi:10.1371/journal.pone.0079055
Li CT, Lin CP, Chou KH, Chen IY, Hsieh JC, Wu CL, Lin WC, Su TP (2010) Structural and cognitive deficits in remitting and non-remitting recurrent depression: a voxel-based morphometric study. NeuroImage 50(1):347–356. doi:10.1016/j.neuroimage.2009.11.021
Cheng YQ, Xu J, Chai P, Li HJ, Luo CR, Yang T, Li L, Shan BC et al (2010) Brain volume alteration and the correlations with the clinical characteristics in drug-naive first-episode MDD patients: a voxel-based morphometry study. Neurosci Lett 480(1):30–34. doi:10.1016/j.neulet.2010.05.075
Moylan S, Maes M, Wray NR, Berk M (2013) The neuroprogressive nature of major depressive disorder: pathways to disease evolution and resistance, and therapeutic implications. Mol Psychiatry 18(5):595–606. doi:10.1038/mp.2012.33
Bench CJ, Friston KJ, Brown RG, Scott LC, Frackowiak RS, Dolan RJ (1992) The anatomy of melancholia—focal abnormalities of cerebral blood flow in major depression. Psychol Med 22(3):607–615
Ebmeier KP, Cavanagh JT, Moffoot AP, Glabus MF, O’Carroll RE, Goodwin GM (1997) Cerebral perfusion correlates of depressed mood. Br J Psychiatry 170:77–81
Bench CJ, Friston KJ, Brown RG, Frackowiak RS, Dolan RJ (1993) Regional cerebral blood flow in depression measured by positron emission tomography: the relationship with clinical dimensions. Psychol Med 23(3):579–590
Haller S, Zaharchuk G, Thomas DL, Lovblad K-O, Barkhof F, Golay X (2016) Arterial spin labeling perfusion of the brain: emerging clinical applications. Radiology 281(2):337–356. doi:10.1148/radiol.2016150789
Colloby SJ, Firbank MJ, He J, Thomas AJ, Vasudev A, Parry SW, O’Brien JT (2012) Regional cerebral blood flow in late-life depression: arterial spin labelling magnetic resonance study. Br J Psychiatry 200(2):150–155. doi:10.1192/bjp.bp.111.092387
Duhameau B, Ferre JC, Jannin P, Gauvrit JY, Verin M, Millet B, Drapier D (2010) Chronic and treatment-resistant depression: a study using arterial spin labeling perfusion MRI at 3Tesla. Psychiatry Res 182(2):111–116. doi:10.1016/j.pscychresns.2010.01.009
Kaichi Y, Okada G, Takamura M, Toki S, Akiyama Y, Higaki T, Matsubara Y, Okamoto Y et al (2016) Changes in the regional cerebral blood flow detected by arterial spin labeling after 6-week escitalopram treatment for major depressive disorder. J Affect Disord 194:135–143. doi:10.1016/j.jad.2015.12.062
Wang Y, Zhang H, Tang S, Liu X, O’Neil A, Turner A, Chai F, Chen F et al (2014) Assessing regional cerebral blood flow in depression using 320-slice computed tomography. PLoS One 9(9):e107735. doi:10.1371/journal.pone.0107735
Lui S, Parkes LM, Huang X, Zou K, Chan RCK, Yang H, Zou L, Li D et al (2009) Depressive disorders: focally altered cerebral perfusion measured with arterial spin-labeling MR imaging. Radiology 251(2):476–484. doi:10.1148/radiol.2512081548
Pizzagalli DA (2011) Frontocingulate dysfunction in depression: toward biomarkers of treatment response. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 36(1):183–206. doi:10.1038/npp.2010.166
Lozano AM, Mayberg HS, Giacobbe P, Hamani C, Craddock RC, Kennedy SH (2008) Subcallosal cingulate gyrus deep brain stimulation for treatment-resistant depression. Biol Psychiatry 64(6):461–467. doi:10.1016/j.biopsych.2008.05.034
Smith DJ, Cavanagh JT (2005) The use of single photon emission computed tomography in depressive disorders. Nucl Med Commun 26(3):197–203
Mayberg HS (2003) Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull 65:193–207
Chen ZQ, Du MY, Zhao YJ, Huang XQ, Li J, Lui S, Hu JM, Sun HQ et al (2015) Voxel-wise meta-analyses of brain blood flow and local synchrony abnormalities in medication-free patients with major depressive disorder. J Psychiatry Neurosci JPN 40(6):401–411
Ota M, Noda T, Sato N, Hattori K, Teraishi T, Hori H (2014) Characteristic distributions of regional cerebral blood flow changes in major depressive disorder patients: a pseudo-continuous arterial spin labeling (pCASL) study. J Affect Disord 165:59–63
Kennedy SH, Evans KR, Kruger S, Mayberg HS, Meyer JH, McCann S, Arifuzzman AI, Houle S et al (2001) Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry 158(6):899–905. doi:10.1176/appi.ajp.158.6.899
Brody AL, Saxena S, Stoessel P, Gillies LA, Fairbanks LA, Alborzian S, Phelps ME, Huang SC et al (2001) Regional brain metabolic changes in patients with major depression treated with either paroxetine or interpersonal therapy: preliminary findings. Arch Gen Psychiatry 58(7):631–640
Young KA, Holcomb LA, Yazdani U, Hicks PB, German DC (2004) Elevated neuron number in the limbic thalamus in major depression. Am J Psychiatry 161(7):1270–1277. doi:10.1176/appi.ajp.161.7.1270
Wortinger LA, Endestad T, Melinder AM, Oie MG, Sevenius A, Bruun Wyller V (2016) Aberrant resting-state functional connectivity in the salience network of adolescent chronic fatigue syndrome. PLoS One 11(7):e0159351. doi:10.1371/journal.pone.0159351
Kim BH, Namkoong K, Kim JJ, Lee S, Yoon KJ, Choi M, Jung YC (2015) Altered resting-state functional connectivity in women with chronic fatigue syndrome. Psychiatry Res 234(3):292–297. doi:10.1016/j.pscychresns.2015.10.014
Gay CW, Robinson ME, Lai S, O’Shea A, Craggs JG, Price DD, Staud R (2016) Abnormal resting-state functional connectivity in patients with chronic fatigue syndrome: results of seed and data-driven analyses. Brain Connect 6(1):48–56. doi:10.1089/brain.2015.0366
Boissoneault J, Letzen J, Lai S, O’Shea A, Craggs J, Robinson ME, Staud R (2016) Abnormal resting state functional connectivity in patients with chronic fatigue syndrome: an arterial spin-labeling fMRI study. Magn Reson Imaging 34(4):603–608. doi:10.1016/j.mri.2015.12.008
Boissoneault J, Letzen J, Lai S, Robinson ME, Staud R (2016) Static and dynamic functional connectivity in patients with chronic fatigue syndrome: use of arterial spin labelling fMRI. Clin Physiol Funct Imaging. doi:10.1111/cpf.12393
Natelson B, Cohen J, Brassloff I, Lee H (1993) A controlled study of brain magnetic resonance imaging in patients with the chronic fatigue syndrome. J Neurol Sci 120:213–217
Greco A, Tannock C, Brostoff J, Costa DC (1997) Brain MR in chronic fatigue syndrome. Am J Neuroradiol 18(7):1265–1269
Lange G, DeLuca J, Maldjian JA, Lee H, Tiersky LA, Natelson BH (1999) Brain MRI abnormalities exist in a subset of patients with chronic fatigue syndrome. J Neurol Sci 171(1):3–7
Holmes G, Kaplan J, Gantz N, Komaroff A, Schonberger L, Straus S (1988) Chronic fatigue syndrome: a working case definition. Ann Intern Med 108:387–389
Lange G, DeLuca J, Maldjian J, Lee H, Tiersky L, Natelson B (2005) Brain MRI abnormalities exist in a subset of patients with chronic fatigue syndrome. J Neurol Sci 171:3–7
Perrin R, Embleton K, Pentreath V, Jackson A (2010) Longitudinal MRI shows no cerebral abnormality in chronic fatigue syndrome. Br J Radiol 83:419–423
Puri BK, Jakeman PM, Agour M, Gunatilake KDR, Fernando KAC, Gurusinghe AI, Treasaden IH, Waldman AD et al (2012) Regional grey and white matter volumetric changes in myalgic encephalomyelitis (chronic fatigue syndrome): a voxel-based morphometry 3 T MRI study. Br J Radiol 85(1015):e270–e273. doi:10.1259/bjr/93889091
Barnden L, Crouch B, Kwiatek R, Burnet R, Mernone A, Chryssidis S (2011) A brain MRI study of chronic fatigue syndrome: evidence of brainstem dysfunction and altered homeostasis. NMR Biomed 24:1302–1312
Barnden LR, Crouch B, Kwiatek R, Burnet R, Del Fante P (2015) Evidence in chronic fatigue syndrome for severity-dependent upregulation of prefrontal myelination that is independent of anxiety and depression. NMR Biomed 28(3):404–413. doi:10.1002/nbm.3261
Shan ZY, Kwiatek R, Burnet R, Del Fante P, Staines DR, Marshall-Gradisnik SM, Barnden LR (2016) Progressive brain changes in patients with chronic fatigue syndrome: a longitudinal MRI study. J Magn Reson Imaging. doi:10.1002/jmri.25283
Okada T, Tanak M, Kuratsune H, Watanabe Y, Sadato N (2004) Mechanisms underlying fatigue: a voxel-based morphometric study of chronic fatigue syndrome. BMC Neurol 4:14
de Lange F, Kalkman J, Bleijenberg G, Hagoort P, van der Meer J, Toni I (2005) Gray matter volume reduction in the chronic fatigue syndrome. NeuroImage 26:777–781
de Lange F, Koers A, Kalkman J, Bleijenberg G, Hagoort P, van der Meer J (2008) Increase in prefrontal cortical volume following cognitive behavioural therapy in patients with chronic fatigue syndrome. Brain 131:2172–2180
Ichise M, Salit I, Abbey S, Chung D, Gray B, Kirsh J (1992) Assessment of regional cerebral perfusion by 99Tcm-HMPAO SPECT in chronic fatigue syndrome. Nucl Med Commun 13:767–772
Costa D, Tannock C, Brostoff J (1995) Brainstem perfusion is impaired in chronic fatigue syndrome. QJM 88:767–773
Goldstein JA, Mena I, Jouanne E, Lesser I (1995) The assessment of vascular abnormalities in late life chronic fatigue syndrome by brain SPECT. J Chronic Fatigue Syndr 1(1):55–79. doi:10.1300/J092v01n01_05
Fischler B, D’Haenen H, Cluydts R, Michiels V, Demets K, Bossuyt A, Kaufman L, De Meirleir K (1996) Comparison of 99m Tc HMPAO SPECT scan between chronic fatigue syndrome, major depression and healthy controls: an exploratory study of clinical correlates of regional cerebral blood flow. Neuropsychobiology 34:175–183
Peterson PK, Sirr SA, Grammith FC, Schenck CH, Pheley AM, Hu S, Chao CC (1994) Effects of mild exercise on cytokines and cerebral blood flow in chronic fatigue syndrome patients. Clin Diagn Lab Immunol 1(2):222–226
Biswal B, Kunwar P, Natelson BH (2011) Cerebral blood flow is reduced in chronic fatigue syndrome as assessed by arterial spin labeling. J Neurol Sci 301(1–2):9–11. doi:10.1016/j.jns.2010.11.018
Yoshiuchi K, Farkas J, Natelson B (2006) Patients with chronic fatigue syndrome have reduced absolute cortical blood flow. Clin Physiol Funct Imaging 26:83–86
Patrick Neary J, Roberts AD, Leavins N, Harrison MF, Croll JC, Sexsmith JR (2008) Prefrontal cortex oxygenation during incremental exercise in chronic fatigue syndrome. Clin Physiol Funct Imaging 28(6):364–372. doi:10.1111/j.1475-097X.2008.00822.x
Machale S, Lawrie S, Cavanagh J, Glabus M, Murray C, Goodwin G (2000) Cerebral perfusion in chronic fatigue syndrome and depression. Br J Psychiatry 176:550–556
Fukuda K (1994) The chronic fatigue syndrome: a comprehensive approach to its definition and study. Ann Intern Med 121(12):953. doi:10.7326/0003-4819-121-12-199412150-00009
Jason LA, Corradi K, Torres-Harding S, Taylor RR, King C (2005) Chronic fatigue syndrome: the need for subtypes. Neuropsychol Rev 15(1):29–58
Siniscalco D, Cirillo A, Bradstreet JJ, Antonucci N (2013) Epigenetic findings in autism: new perspectives for therapy. Int J Environ Res Public Health 10(9):4261–4273. doi:10.3390/ijerph10094261
Sharpe M, Archard L, Banatvala J, Borysiewicz L, Clare A, David A (1991) A report-chronic fatigue syndrome: guidelines for research. J R Soc Med 84:118–121
Reeves WC, Wagner D, Nisenbaum R, Jones JF, Gurbaxani B, Solomon L, Papanicolaou DA, Unger ER et al (2005) Chronic fatigue syndrome—a clinically empirical approach to its definition and study. BMC Med 3:19. doi:10.1186/1741-7015-3-19
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9(1):46–56. doi:10.1038/nrn2297
Morris G, Berk M, Galecki P, Walder K, Maes M (2015) The neuro-immune pathophysiology of central and peripheral fatigue in systemic immune-inflammatory and neuro-immune diseases. Mol Neurobiol 53(2):1195–1219. doi:10.1007/s12035-015-9090-9
Morris G, Maes M (2014) Oxidative and nitrosative stress and immune-inflammatory pathways in patients with myalgic encephalomyelitis (ME)/chronic fatigue syndrome (CFS). Curr Neuropharmacol 12:168–185
Morris G, Maes M (2012) A neuro-immune model of myalgic encephalomyelitis/chronic fatigue syndrome. Metab Brain Dis 28(4):523–540. doi:10.1007/s11011-012-9324-8
Morris G, Anderson G, Galecki P, Berk M, Maes M (2013) A narrative review on the similarities and dissimilarities between myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and sickness behavior. BMC Med 11:64
Morris G, Berk M, Walder K, Maes M (2015) Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med 13(1):1–23. doi:10.1186/s12916-014-0259-2
Brites D, Fernandes A (2015) Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front Cell Neurosci 9:476. doi:10.3389/fncel.2015.00476
Brites D, Vaz AR (2014) Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci 8:117. doi:10.3389/fncel.2014.00117
Paschon V, Takada SH, Ikebara JM, Sousa E, Raeisossadati R, Ulrich H, Kihara AH (2016) Interplay between exosomes, microRNAs and toll-like receptors in brain disorders. Mol Neurobiol 53(3):2016–2028. doi:10.1007/s12035-015-9142-1
Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P et al (2012) MicroRNAs bind to toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 109(31):E2110–E2116. doi:10.1073/pnas.1209414109
Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM et al (2016) Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 19(3):504–516. doi:10.1038/nn.4222 http://www.nature.com/neuro/journal/v19/n3/abs/nn.4222.html#supplementary-information
Doorn KJ, Brevé JJP, Drukarch B, Boddeke HW, Huitinga I, Lucassen PJ, van Dam A-M (2015) Brain region-specific gene expression profiles in freshly isolated rat microglia. Front Cell Neurosci 9:84. doi:10.3389/fncel.2015.00084
Dhib-Jalbut S, Kalvakolanu DV (2015) Microglia and necroptosis: the culprits of neuronal cell death in multiple sclerosis. Cytokine 76(2):583–584. doi:10.1016/j.cyto.2015.06.004
Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X et al (2015) Activation of necroptosis in multiple sclerosis. Cell Rep 10(11):1836–1849. doi:10.1016/j.celrep.2015.02.051
Gardner C, Magliozzi R, Durrenberger PF, Howell OW, Rundle J, Reynolds R (2013) Cortical grey matter demyelination can be induced by elevated pro-inflammatory cytokines in the subarachnoid space of MOG-immunized rats. Brain 136(Pt 12):3596–3608. doi:10.1093/brain/awt279
Lu H, Liu X, Deng Y, Qing H (2013) DNA methylation, a hand behind neurodegenerative diseases. Front Aging Neurosci 5:85. doi:10.3389/fnagi.2013.00085
Schitine C, Nogaroli L, Costa MR, Hedin-Pereira C (2015) Astrocyte heterogeneity in the brain: from development to disease. Front Cell Neurosci 9. doi:10.3389/fncel.2015.00076
Höft S, Griemsmann S, Seifert G, Steinhäuser C (2014) Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: insights from the thalamus. Philos Trans R Soc B Biol Sci 369(1654). doi:10.1098/rstb.2013.0602
Kang W, Hebert JM (2011) Signaling pathways in reactive astrocytes, a genetic perspective. Mol Neurobiol 43(3):147–154. doi:10.1007/s12035-011-8163-7
Garden GA (2013) Epigenetics and the modulation of neuroinflammation. Neurother J Am Soc Exp NeuroTher 10(4):782–788. doi:10.1007/s13311-013-0207-4
Akterin S, Cowburn RF, Miranda-Vizuete A, Jimenez A, Bogdanovic N, Winblad B, Cedazo-Minguez A (2005) Involvement of glutaredoxin-1 and thioredoxin-1 in [beta]-amyloid toxicity and Alzheimer’s disease. Cell Death Differ 13(9):1454–1465
Kaminsky YG, Beloushko EE, Kosenko EA (2014) Antioxidant defense in the rat brain cortex, cerebellum, hippocampus, and striatum and its alterations during portacaval shunting. Neurochem J 8(4):289–294. doi:10.1134/s1819712414030064
Brannan TS, Maker HS, Raes IP (1981) Regional distribution of catalase in the adult rat brain. J Neurochem 36(1):307–309. doi:10.1111/j.1471-4159.1981.tb02411.x
Infanger DW, Sharma RV, Davisson RL (2006) NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal 8(9–10):1583–1596. doi:10.1089/ars.2006.8.1583
Ramos P, Santos A, Pinto NR, Mendes R, Magalhaes T, Almeida A (2015) Anatomical regional differences in selenium levels in the human brain. Biol Trace Elem Res 163(1–2):89–96. doi:10.1007/s12011-014-0160-z
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30(4):572–580. doi:10.1002/ana.410300410
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39(6):889–909
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122(Pt 8):1437–1448
Wang X, Michaelis E (2010) Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2(12). doi:10.3389/fnagi.2010.00012
Olsson T, Wieloch T, Smith ML (2003) Brain damage in a mouse model of global cerebral ischemia. Effect of NMDA receptor blockade. Brain Res 982(2):260–269
Schmidt-Kastner R, Freund TF (1991) Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 40(3):599–636
Wang X, Pal R, Chen XW, Limpeanchob N, Kumar KN, Michaelis EK (2005) High intrinsic oxidative stress may underlie selective vulnerability of the hippocampal CA1 region. Brain Res Mol Brain Res 140(1–2):120–126. doi:10.1016/j.molbrainres.2005.07.018
Wilde GJ, Pringle AK, Wright P, Iannotti F (1997) Differential vulnerability of the CA1 and CA3 subfields of the hippocampus to superoxide and hydroxyl radicals in vitro. J Neurochem 69(2):883–886
Blokland GAM, de Zubicaray GI, McMahon KL, Wright MJ (2012) Genetic and environmental influences on neuroimaging phenotypes: a meta-analytical perspective on twin imaging studies. Twin Res Hum Genet Off J Int Soc Twin Stud 15(3):351–371. doi:10.1017/thg.2012.11
Rijsdijk FV, Viding E, De Brito S et al (2010) Heritable variations in gray matter concentration as a potential endophenotype for psychopathic traits. Arch Gen Psychiatry 67(4):406–413. doi:10.1001/archgenpsychiatry.2010.20
van der Schot AC, Vonk R, Brans RG, van Haren NE, Koolschijn PC, Nuboer V, Schnack HG, van Baal GC et al (2009) Influence of genes and environment on brain volumes in twin pairs concordant and discordant for bipolar disorder. Arch Gen Psychiatry 66(2):142–151. doi:10.1001/archgenpsychiatry.2008.541
Muehlhan M, Kirschbaum C, Wittchen HU, Alexander N (2015) Epigenetic variation in the serotonin transporter gene predicts resting state functional connectivity strength within the salience-network. Hum Brain Mapp 36(11):4361–4371. doi:10.1002/hbm.22923
Fornito A, Zalesky A, Bassett DS, Meunier D, Ellison-Wright I, Yucel M, Wood SJ, Shaw K et al (2011) Genetic influences on cost-efficient organization of human cortical functional networks. J Neurosci 31(9):3261–3270. doi:10.1523/jneurosci.4858-10.2011
Dannlowski U, Kugel H, Redlich R, Halik A, Schneider I, Opel N, Grotegerd D, Schwarte K et al (2014) Serotonin transporter gene methylation is associated with hippocampal gray matter volume. Hum Brain Mapp 35(11):5356–5367. doi:10.1002/hbm.22555
Swain JE, Perkins SC, Dayton CJ, Finegood ED, Ho SS (2012) Parental brain and socioeconomic epigenetic effects in human development. Behav Brain Sci 35(5):378–379. doi:10.1017/S0140525X12001112
Hu Z, Zhong B, Tan J, Chen C, Lei Q, Zeng L (2016) The emerging role of epigenetics in cerebral ischemia. Mol Neurobiol 1-19. doi:10.1007/s12035-016-9788-3
Schweizer S, Meisel A, Marschenz S (2013) Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab 33(9):1335–1346. doi:10.1038/jcbfm.2013.93
Acknowledgements
MB is supported by a NHMRC Senior Principal Research Fellowship 1059660.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Authorships
All authors contributed equally to the writing up of the paper.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Morris, G., Berk, M. & Puri, B.K. A Comparison of Neuroimaging Abnormalities in Multiple Sclerosis, Major Depression and Chronic Fatigue Syndrome (Myalgic Encephalomyelitis): is There a Common Cause?. Mol Neurobiol 55, 3592–3609 (2018). https://doi.org/10.1007/s12035-017-0598-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0598-z