
Abstract
Background
‘Encephalomyelitis disseminata’ (multiple sclerosis) and myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) are both classified as diseases of the central nervous system by the World Health Organization. This review aims to compare the phenomenological and neuroimmune characteristics of MS with those of ME/CFS.
Discussion
There are remarkable phenomenological and neuroimmune overlaps between both disorders. Patients with ME/CFS and MS both experience severe levels of disabling fatigue and a worsening of symptoms following exercise and resort to energy conservation strategies in an attempt to meet the energy demands of day-to-day living. Debilitating autonomic symptoms, diminished cardiac responses to exercise, orthostatic intolerance and postural hypotension are experienced by patients with both illnesses. Both disorders show a relapsing-remitting or progressive course, while infections and psychosocial stress play a large part in worsening of fatigue symptoms. Activated immunoinflammatory, oxidative and nitrosative (O+NS) pathways and autoimmunity occur in both illnesses. The consequences of O+NS damage to self-epitopes is evidenced by the almost bewildering and almost identical array of autoantibodies formed against damaged epitopes seen in both illnesses. Mitochondrial dysfunctions, including lowered levels of ATP, decreased phosphocreatine synthesis and impaired oxidative phosphorylation, are heavily involved in the pathophysiology of both MS and ME/CFS. The findings produced by neuroimaging techniques are quite similar in both illnesses and show decreased cerebral blood flow, atrophy, gray matter reduction, white matter hyperintensities, increased cerebral lactate and choline signaling and lowered acetyl-aspartate levels.
Summary
This review shows that there are neuroimmune similarities between MS and ME/CFS. This further substantiates the view that ME/CFS is a neuroimmune illness and that patients with MS are immunologically primed to develop symptoms of ME/CFS.
Similar content being viewed by others

Background
‘Encephalomyelitis disseminata’/multiple sclerosis (MS) and myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) are both classified as diseases of the central nervous system by the World Health Organization (WHO). MS exhibits an almost bewildering radiological, clinical, and pathological heterogeneity. Evidence reveals that different processes, such as autoimmunity, inflammation and virus infection, may induce the pathology characteristic of the disease and suggests that MS is an illness involving the presence of different causative mechanisms. Distinct patterns of demyelination have been repeatedly documented. Two patterns bear a very close resemblance to autoimmune encephalomyelitis induced either by T cells alone or T and B cells in combination. The other patterns are highly indicative of virus infection or demyelination generated by exposure to environmental toxins rather overt autoimmune mechanisms. Pathological, biochemical and immunological data indicate that different pathways are generating the distinct pathology visible in different MS patients [1]. Research repeatedly demonstrates a proinflammatory milieu in MS patients as reflected by elevated levels of proinflammatory cytokines [2, 3].
Patients with ME/CFS experience disabling levels of fatigue, as do people with MS, and they also have a wide range of neurological signs. The latter involve neurocognitive and autonomic symptoms, for example, postural hypotension and orthostatic intolerance [4, 5] and a wide range of abnormalities on brain scans indicating elevated levels of lactate, cerebral hypoperfusion, and glucose hypometabolism [5]. Evidence shows that different trigger factors, such as infections and (auto)immune disorders, may be associated with the onset of ME/CFS [6]. ME/CFS patients display numerous immune abnormalities indicating an activated but dysregulated immune system, including chronically elevated levels of cytokines, signs of immune activation, loss of T cell homeostasis, decreased natural killer cell activity and autoimmune responses [6].
This review aims to compare the diseases of MS and ME/CFS on several different dimensions. These dimensions will include phenomenological similarities, including symptoms and course, elevated oxidative and nitrosative stress (O+NS), the existence of autoimmunity, cell mediated immunity and cytokine abnormalities and abnormalities in T cell activation and homeostasis, and a comparison of brain imaging findings. It is worth noting at the onset, however, that although CFS is recognized as an alternative term to myalgic encephalomyelitis there are many instances in the literature where the term is used as a synonym for fatigue of a psychiatric or idiopathic origin and patients are selected who only experience fatigue [7]. In this review we only consider data from studies where patients are recruited using CDC criteria and eschew studies where patients are selected because they meet arbitrary criteria produced by unvalidated or invalidated symptom questionnaires or generic fatigue scales. Trying to synthesize results where selection criteria are major confounding variable is virtually impossible, and in any event can lead to false conclusions [8].
Discussion
Phenomenological similarities between MS and ME/CFS
Many patients with MS have symptoms that are characteristic for ME/CFS. We will first discuss the typical symptoms of ME/CFS and then show that many patients with MS also have ME/CFS symptoms in conjunction with typical neurological deficits. People with ME/CFS can have a wide range of symptoms [9]. Typical symptoms include chronic fatigue, hyperalgesia, migraine-type headaches, unrefreshing sleep or even sleep/wake cycle reversal. Patients also present with symptoms consistent with a chronic influenza-like syndrome. These symptoms include unrelenting severe disabling fatigue in the physical and mental domains combined with incapacitating levels of muscle fatigability. Problems with memory retrieval and formation are also frequently observed. Problems with word retrieval mean that patients are frequently unable to finish sentences. These symptoms are all made worse by increases in cognitive and or physical activity. An inability to tolerate even trivial increases in physical or mental activity above individual norms is the hallmark symptom of ME/CFS [5]. This intolerance manifests itself in disease exacerbation, which may be short lived or prolonged [10, 11]. ME/CFS patients display abnormalities in parameters appertaining to sympathetic and parasympathetic nervous system activity [12, 13]. Orthostatic intolerance, and neurally mediated hypotension are commonly reported cardiovascular symptoms [4, 14]. Postural orthostatic tachycardia syndrome (POTS) is another common finding. Exaggerated postural tachycardia and enhanced sympathetic activity have been reported [12, 15]. Autonomic symptoms also include an intolerance of wide temperature fluctuations and grossly impaired thermostatic stability. A diminished cardiac response to exercise has also been demonstrated [16]. Resting sympathetic overactivity coupled with reduced vagal modulation appears to be a reproducible finding in ME/CFS [17]. De Becker et al. [18] reported a sympathetic drive increased heart rate (HR) on tilt compared to controls. Another study demonstrated impaired HR responses indicating the existence of attenuated cardiac sympathetic responsiveness. These lower HR responses could not be reconciled with an explanation based on patient deconditioning or prolonged inactivity [19]. Those authors reported that hemodynamic responses in patients with ME/CFS were significantly impaired during exercise compared to healthy controls. A number of other workers have reported autonomic dysregulation in people with ME/CFS [20–27]. Patients with ME/CFS typically use energy conservation strategies or pacing as a method of minimizing the effects of their fatigue on daily living [28].
Around 53% to 92% of MS patients experience disabling levels of fatigue [29]. Patients with MS frequently report that their fatigue is unremitting and relentless [30–32], and causes daytime sleepiness [33] and an irresistible urge to rest [32]. Clinically, fatigue presents as exhaustion, loss of energy, daytime somnolence, or exacerbation of symptoms. Activity usually acts to increase fatigue levels in MS [34]. Thus, the experience of permanent exhaustion is magnified by exacerbations leading to complete absence of energy after physical or cognitive activity [35, 36]. Rapid exhaustion and markedly reduced exercise tolerance is a source of profound disability in patients with MS [36]. People with MS not only report a lack of energy, but also a profound intolerance of even minor physical activities [31, 33, 37]. MS patients often have concentration difficulties or an inability to complete mental tasks [30, 33, 37]. Reports of malaise are also commonplace [32, 37].
The most frequent symptoms of autonomic dysfunction in MS patients are impotence, gastrointestinal dysfunction, sleep disturbances, disordered micturition and orthostatic intolerance [38, 39]. Patients may develop POTS as a result of their underlying autonomic dysfunction [40]. A number of different groups have reported orthostatic dysregulation, neurocardiogenic syncope and cardiac dysrhythmias [41–44].
People with MS also use pacing strategies to minimize the effects of their fatigue on daily living. Such strategies include planning daily routines, ensuring that periods of higher activity take place in the mornings and budgeting time for rest or even sleep between such periods of increased activity [37]. A number of studies have empirically examined the effectiveness of pacing by enrolling patients in a course where they were given instructions in various pacing techniques [45–47]. These courses resulted in significant reductions in fatigue.
In summary (see Table 1), patients with ME/CFS and MS both experience severe levels of disabling fatigue and a worsening of symptoms following exercise and resort to energy conservation strategies in an attempt to meet the energy demands of day-to-day living. Debilitating autonomic symptoms are experienced by people with both illnesses. Diminished cardiac responses to exercise are a common finding as are reports of orthostatic intolerance and postural hypotension. It appears, however, that ME/CFS patients may be more sensitive to physical or cognitive activities than patients with MS.
Similarities in the course and other disease characteristics of MS and ME/CFS
There are four MS types, outlined below. (1) Patients with relapsing-remitting type MS endure relapses or episodes of acute impairment of neurologic function. Relapses may be replaced by times of partial or complete remissions without further exacerbations. This is easily the most common presentation of MS and people with relapsing-remitting MS make up about 85% of the MS population. (2) Patients with primary-progressive type MS endure a continuous deterioration of their disease typically, but not exclusively, without a pattern of relative relapse or remission. This phenotype is relatively rare comprising about 10% of the MS population. (3) Patients with secondary-progressive type MS endure an initial pattern of relapsing-remitting disease followed by a progressive deterioration of disease activity amidst a pattern of minor relapses and remissions. Around half of those with the relapsing-remitting course of disease will go on to develop secondary-progressive MS in the absence of treatment. (4) Patients with progressive-relapsing type MS experience a progressive worsening of symptoms from onset coupled with acute exacerbations with or without recovery. There is no period of remission in this form of MS. The frequency of this phenotype is approximately 5% of the total MS population.
In addition, ME/CFS is a chronic, relapsing-remitting disease involving the waxing and waning of symptoms [48–51]. The majority of studies examining longitudinal changes in disease activity using internationally agreed diagnostic criteria report a relapsing-remitting or progressive pattern of disease in the patients monitored [52–55]. Recovery from ME/CFS is extremely rare [53–55]. The average figure reported in studies using 1988 or 1994 Centers for Disease Control and Prevention recruitment criteria is some 4% [53, 55, 56]. Peterson et al. [57] reported a relative remission in some 40% of patients monitored over 12 months while 20% of patients demonstrated progressive worsening of their disease and 40% remained stable. Unfortunately, none recovered. Saltzstein et al. [58] reported very similar statistics.
Different trigger factors such as infections and other environmental factors play a role in the onset of MS and ME/CFS. Infections have been implicated in relapse of MS. Activation of immune pathways, mostly via cytokines, is believed to be responsible for the occurrence of severe relapses during or following infection [59]. Stress also increases the frequency of relapses or a general worsening of symptoms [60]. Stress leads to elevated cytokines and increases in general proinflammatory states, which may well create the environment fostering increased disease activity [61, 62].
The vast majority of ME/CFS patients endure multiple persistent bacterial and viral infections [63–70]. The existence of these infections correlates positively with the total number of symptoms and the severity of those symptoms including the neurological symptoms [66]. Concurrent infections appear to worsen symptoms globally [71]. Stress is also related to worsening symptoms of ME/CFS [72, 73].
The incidence and prevalence of MS demonstrates considerable geographic variability [74, 75]. High frequency areas (prevalence of 60 per 100,000 or more) include Europe, the area encompassing the northern USA and Southern Canada, the antipodes including all of New Zealand, the south eastern part of Australia. In the US, the prevalence is 100 per 100,000. At a rate of 300/100,000, the residents of the Orkney Islands in the UK have a disproportionately heavy burden.
Estimating the prevalence of ME/CFS is a difficult exercise however. Although the disease was first reported in 1934, the introduction of new descriptive terminology in 1988 following an outbreak of ME in the USA led many physicians to equate the disease with idiopathic chronic fatigue. The presence of chronic fatigue in a population is common, ranging from just under 3,000 to just over 6,000 cases per 100,000 [76]. While disabling fatigue is certainly present in most people it is just one of an array of disabling neurological and neuroendocrine symptoms experienced by patients with ME/CFS. With that proviso in mind, the figures would indicate that some 0.2% of the people in the USA have ME/CFS [76–80], which is twice the prevalence of MS. As in many autoimmune disorders, ME/CFS and MS are more prevalent in women than in men [9, 81].
Table 1 shows the phenomenological similarities between both disorders. Both MS and ME/CFS show a chronic and/or relapsing-remitting course, while infections and stress appear to play a large part in worsening of symptoms in both illnesses. Immune activation following infection is believed to trigger relapses in MS and ME/CFS. Patients with ME/CFS have more concomitant infections and the number of different infections correlates with the severity of symptoms. Stress is related to worsening of symptoms of fatigue in both MS and ME/CFS.
Oxidative and nitrosative stress
Accumulating data demonstrates that O+NS plays a significant role in the pathophysiology of MS [82, 83]. Studies have revealed the existence of lipid peroxidation in MS as evidenced by elevated levels of the alkanes pentane and ethane, hydrocarbons produced by peroxidation of unsaturated fatty acids [84]. O+NS likely make a major contribution to the pathophysiology of lesions in patients with MS [85]. Peroxinitrite for example is capable of modifying lipid, protein, DNA and mitochondrial structures and functions via the production of oxidizing and nitrating free radicals. Evidence supporting the presence of O+NS in demyelinating and inflammatory lesions includes the existence of nitrotyrosine together with lipid and protein peroxides [86, 87]. Increased nitric oxide (NO) and inducible nitric oxide synthase (iNOS) production have been detected in peripheral blood mononuclear cells caused by raised levels of oxidative stress [87, 88]. Isoprostanes, isomers of prostaglandins, are generated by peroxidation of fatty acids and can be detected in the urine as well as in the plasma from people with MS [89–91]. Elevated levels of isoprostane 8-epi-prostaglandin are found in the cerebrospinal fluid (CSF) of patients with MS [92].
Many studies using peripheral blood measures have shown increased O+NS in patients with ME/CFS, including increased levels of malondialdehyde (MDA), isoprostane, 8-OH-deoxyguanosine, 2,3 diphosphoglyceric acid, thiobutyric acid, and protein carbonyls [93–101]. The production of iNOS is significantly increased in ME/CFS patients as compared with normal controls [102]. As we will discuss below, there is also evidence that there is a chronic hyperproduction of NO [93]. Raised oxidative stress levels also occur in response to exercise in ME/CFS [103] potentially explaining one of the mechanisms underlying post-exertional malaise. In ME/CFS patients, exercise induces striking changes in the excitability of muscle membranes [104]. Skeletal muscle oxidative imbalance contributes to increased muscle fatigability [105]. Several authors have reported that O+NS measures demonstrate a significant and positive correlation with symptom severity [93–96, 99, 101, 106, 107].
Both MS and ME/CFS are accompanied by significantly depressed levels of crucial antioxidants and antioxidant enzymes. Syburra and Passi [108] reported low levels of vitamin E, ubiquinone (coenzyme Q10) and glutathione (GSH) peroxidase in MS. de Bustos et al. [109], however, did not find significant differences in serum levels of coenzyme Q10 between patients with MS and controls. There are reports on lowered levels of glutathione in the brains of MS patients [110]. Lowered zinc levels have been observed in MS [111].
Lowered levels of zinc, coenzyme Q10 and glutathione have been reported in ME/CFS [95, 107, 112]. A recent proton magnetic resonance spectroscopy study reported decreased cortical glutathione levels in the brain in ME/CFS that inversely correlated with lactate levels [107]. A study examining blood vitamin E levels showed that amelioration of oxidative stress occurs when ME/CFS patients enter remission [100].
Table 2 shows the similarities in O+NS pathways between both disorders. Markers of elevated O+NS are found in both illnesses. Moreover, the abnormalities reported are virtually identical in both illnesses and include elevated levels of peroxinitrite, NO and iNOS. Evidence of nitrosatively modified amino acids and proteins and oxidatively modified lipids are found in both illnesses. Reduced levels of antioxidants, including vitamin E, zinc and glutathione are also found in both diseases. While coenzyme Q10 is clearly related to fatigue in ME/CFS, the findings in MS are less evident.
Cytokine levels in MS and ME/CFS
Studies have repeatedly reported elevated levels of the proinflammatory cytokines, IL-1β [113–115], TNFα [115, 116] and IL-6 [117, 118] in the CSF and plasma of people with MS. Moreover, the cytokine pattern observed in the CSF of patients with relapsing-remitting MS varies depending on the stage of the disease [119]. Th1-like cytokines, such as IL-2, and interferon (IFN), and interleukin (IL)-12, are increased during active disease. T helper (Th)2 cytokines, such as the anti-inflammatory IL-10, transforming growth factor (TGF)-β and IL-4, are elevated during relative remission [120–123]. Th1 and Th2 cytokines are present in the CSF and lesions [124, 125] during relapse and remission. The high concentrations of tumor necrosis factor (TNF)α and IL-10 (both in serum and CSF), which accompany an MS attack, suggest a concomitant expression of Th1 and Th2 cytokines and not to the sequential expression of Th1 cytokines followed by Th2 cytokines [126].
A number of studies have found increased levels of the major proinflammatory cytokines TNFα and IL-1β in ME/CFS (for a review see Maes et al. [127]). Recent evidence has challenged the view that patients with ME/CFS display an activated Th2 dominated immune system [5, 128]. Proinflammatory and anti-inflammatory cytokines are known to coexist also in ME/CFS, although in many patients proinflammatory cytokines are dominant [127, 129, 130]. Studies examining the Th cytokine profiles in people with ME/CFS also show a large number of different findings almost certainly for methodological inconsistencies, including patient selection [5]. Rose et al. [131] reported that there was a significant upregulation of cyclo-oxygenase 2 (COX2), usually accompanied by increased iNOS, in MS lesions and opined that COX2 promoted excitotoxic death and damage of oligodendrocytes by coupling with iNOS. The involvement of COX2 in oligodendrocyte death was confirmed by Carslon et al. [132] using histopathological techniques. Upregulation of nuclear factor (NFκB in lesion-based macrophages amplifies the inflammatory reaction by stimulating the production of adhesion molecules and proinflammatory cytokines [133]. Activated NFκB is found at high levels in microglia of active lesions [134]. These authors proposed that high NFκB levels explains the relative rarity of oligodendrocyte death in MS. Generally it seems that upregulation of NFκB in neurons is protective but activation of NFκB in microglia stimulates neuronal degeneration [135, 136]. Maes et al. [102] reported significantly elevated levels of COX2 and NFκB in patients with ME/CFS compared to healthy controls. Moreover, the severity of the illness correlated significantly and positively with the elevation in concentrations COX2 and NFκB.
Table 3 displays the similarities in immunoinflammatory pathways between MS and ME/CFS. Overall, proinflammatory cytokines are elevated in MS and ME/CFS but the results of investigative trials depend on methodology and can vary according to the state of the disease. Th1 and Th2 cytokines coexist in both illnesses. COX2 and NFκB are upregulated in both disorders and may play a role in the pathophysiology of both MS and ME/CFS.
Cell mediated immunity in MS and ME/CFS
Neopterin
Levels of neopterin have been reported as being higher in CSF of MS patients during exacerbations in comparison with remissions [136]. Increased urinary neopterin to creatinine ratio is an accurate surrogate marker of cell-mediated immune activation in MS [137]. Relapses and disease activity are related to increased neopterin levels [138–140]. Many studies have detected high levels of serum neopterin in ME/CFS [127, 141–144].
Regulatory T cells
T cells are anergized in patients with MS in remission [145] indicating functioning regulatory T (Treg) cells. The situation in active disease is quite different however. Programmed death 1 (PD-1)-regulatory T cells are elevated in the peripheral blood of relapsing-remitting MS with active disease [146] and these Treg cells appear to be clonally exhausted [146, 147] and only a small fraction express PD-1 receptors which are needed for their suppressive function [146]. An absence of PD-1 expression on forkhead box P3 (FOXP3) + CD4+ T cells greatly reduces their ability to suppress the activity of effector T cells, which is essential if self-tolerance is to be maintained and autoimmunity to be prevented [148]. A number of studies report impaired function of Tregs in active disease [149, 150]. Tregs can react to inflammation by increasing numbers in active disease in an attempt to restore homeostasis [151]. PD-1 also has a key role in the modulation of T cell function during a prolonged viral infection. The functional impairment of T cells that occurs during chronic viral infections is considered to be due to T cell exhaustion promoted by activation of the PD-1 pathway and elevation of Tregs as the infection progresses [148, 152, 153]. Defective Treg function in people with MS is shown by the existence of Th17 lymphocytes in the peripheral circulation and the CNS [154–156]. Patients with relapsing-remitting MS display a Th1/Th17 phenotype in active disease [157, 158]. Reduction in Treg function leads to a Th1/Th17 phenotype resulting from activation of naive T cells [159, 160].
Compared to healthy controls, ME/CFS patients display statistically significant increases in CD4(+)CD25(+) Treg cells and FOXP3 expression [161]. Interestingly, chronically elevated IL-2 expression leads to the exhaustion of FOXP3 expression on CD25 + CD4+ Treg cells over time [159]. IL-2 is found chronically activated in MS [162–165] and ME/CFS [166, 167]. This also indicates that a chronically activated immune system may exist in both diseases caused by a failure of the FOXP3/IL-2 feedback mechanism [168]. The pattern of raised IL-2 levels and chronic immune activation with disrupted homeostasis [169, 170] coupled with raised levels of Treg cells seen in patients with ME/CFS strongly suggests Treg cell exhaustion as a feature of this disease as well. Strong evidence of T cell exhaustion has been reported in ME/CFS patients by many different researchers [171–175].
CD26+ and CD69+ T cells
Further evidence of an activated or dysregulated immune system is provided by a consideration of the data relating to the CD26 and CD69 T cell receptors in both MS and ME/CFS. CD26 is a T cell activation antigen with dipeptidyl peptidase 4 (DPPIV) activity [176]. CD26 as a surrogate marker of T cell activation correlates well with the activity of a number of autoimmune diseases [177, 178]. CD26 is a marker of T cell activation and autoimmunity [179, 180] and a key modulator of immune responsiveness [181]. CD26 expression is associated with Th-17 cells and IL-17 production is related to the CD26 + CD4+ T cell subset [182]. Memory CD4+ T cells with high expression of CD26+ correlate with clinical severity of MS [183, 184]. The likelihood of a relapse is approximately three times higher in MS patients with high CD26 levels [185]. Importantly, elevated CD26 + CD4+ T cell numbers have been reported in people with ME/CFS compared to controls [186].
Another inhibitory regulator of Th17 cell differentiation is CD69, an early activation marker that promotes activation of the signal transducer and activator of transcription 5 (STAT5) pathway [187]. Patients with MS show an increased CD69 expression on T Lymphocytes [188]. The less pronounced IFN-induced effects on CD69 expression in MS versus controls is evidence of a defect in immunoregulation [189]. In ME/CFS, significantly lower CD69 expression on mitogen stimulated T cells has been detected [190]. This decreased expression of CD69 upon stimulation was strongly associated with inflammatory markers and indicates a defect in the initial activation of T lymphocytes and natural killer (NK) cells [112, 190].
NK cells in MS and ME/CFS
Both MS and ME/CFS are accompanied by reduced and impaired activity of NK cells (NKCs). NKCs have a key role in immunoregulation. Crosstalk between NKCs and dendritic cells acts as a rheostat for the immune system and hence NKCs play a pivotal role in maintaining immune homeostasis [191] and in the suppression of T cell responses. In addition, NK CD56 bright cells have also a major role in combating autoimmunity. Benczur et al. [192] reported that NKC function was reduced in people with active disease compared with those in remission. Enhancement of NKC CD56 function leads to an amelioration of symptoms in MS [193]. Longitudinal disease activity, determined both clinically and by serial magnetic resonance imaging (MRI), correlates with natural killer cell activity (NKCA) and phenotype. Mean NKCA is significantly reduced in patients with MS as compared to normal controls. In relapsing-remitting MS, there is a significant association between lowered NKCA and the onset of new lesions on MRI [194]. In patients with MS, CD56 bright NKCs mediate immunoregulation [193]. Takahashi et al. [195] reported that a subset of NKCs was responsible for the maintenance of remission in relapsing-remitting MS and the development of relapses [192, 195] and expansion corresponds with remission [196]. In ME/CFS, reduced and impaired NKC functioning has been a consistent finding reported by many authors [173, 197, 198].
It is tempting to speculate that gender-related differences in immune responsiveness may be associated with the higher prevalence of ME/CFS and MS in women as compared to men. The immunological processes occurring in the effector phase and induction of T cell priming are much stronger in female mice [81].
Summary
Table 3 shows that markers of immune activation are comparable in both illnesses. Elevated neopterin levels and elevated expression of the CD26 antigen on T cells demonstrate chronic immune activation while chronically elevated IL-2 levels indicate dysfunctional T cell activation and disordered homeostasis. Defective functionality of Treg cells is evidenced in both illnesses and clonal exhaustion of T cells is found in patients with MS and ME/CFS. There are, however, also differences between both disorders. Thus, clonal exhaustion may be limited to Treg cells in the active stage of MS, while the T cell exhaustion in patients with ME/CFS may be more global. While MS is characterized by increased in vivo expression of CD69, a decreased ex vivo CD69 expression is found in ME/CFS. Both MS and ME/CFS are accompanied by reduced NKCA.
B cells and autoimmunity in MS and ME/CFS
B cells
Increased numbers of B cells are observed in ME and ME/CFS. CD80+ B cells numbers are increased in relapse phases of MS, relative to the values found in patients in remission or healthy controls [199]. Increases in the number of mature CD19 B cells have been reported in ME/CFS patients [200–202]. Klimas et al. [173] reported elevated numbers of CD20+ and CD21+ B cells in ME/CFS. Activated B cells possess a high capacity to generate inflammatory and regulatory cytokines and have a regulatory function in autoimmune diseases [203].
Autoimmunity
MS is widely considered to be at least in part an autoimmune disorder. Autoimmune reactions are also highly prevalent in ME/CFS. The presence of anti-nuclear antibodies and antibodies directed against cardiolipin and other phospholipids has been reported in some patients with MS [204–206]. Anti-neuronal antibodies, anti-muscle antibodies, anti-ganglioside antibodies [207–209] and anti-serotonin antibodies [210] are also active in MS.
Many individuals with ME/CFS show several indicators of autoimmune responses. Anti-cardiolipin antibodies have been reported in people with ME/CFS [211, 212]. Konstantinov et al. [213] reported the presence of autoantibodies to nuclear envelope antigens. Anti-neuronal antibody levels are elevated in ME/CFS patients with neurologic abnormalities [214]. Other indicators include elevated antibody titers towards phospholipids, gangliosides and serotonin; anti-lamine SS DNA as well as anti-68/48 kDa and microtubule-associated proteone [215–217]. In addition to these increased antibody levels that are also observed in MS, patients with ME/CFS show various other markers of autoimmunity. Thus, autoantibodies against the muscarinic cholinergic receptor, mu-opioid receptor, 5-hydroxytryptamine (5-HT; serotonin) receptor 1A and dopamine receptor D2 have all been detected in ME/CFS patients [218] (for a review see [9]).
Both MS and ME/CFS are also accompanied by autoimmune reactions sometimes described as secondary. These reactions are directed against neoantigenic determinants (neoepitopes), which are created as a result of damage to lipids and proteins by O+NS [93, 219]. Thus, the organisms may mount IgM and IgG mediated autoimmune reactions against oxidatively modified epitopes, such as fatty acids or byproducts of oxidative processes (for example, azelaic acid and malondialdehyde) and nitrosatively modified proteins [93]. Antibody titers against azelaic acid are higher during acute relapses in MS [220]. Anti-oleic acid conjugated antibodies have also been found in the sera of patients with MS when in acute relapse [221]. There is also a considerable amount of direct evidence of elevated protein S-nitrosation and nitrite content together with markedly increased levels of lipid peroxidation in the serum of patients with MS [222, 223]. Specific IgM antibodies towards NO-modified amino acids and azelaic acid and malodialdehyde have been reported in MS patients [219]. The basic principle underpinning these data is that self-epitopes may be damaged by exposure to prolonged O+NS and thus lose their immunogenic tolerance and become a target for the hosts immune system.
The same IgM-related autoimmune responses can be detected in patients with ME/CFS, including autoimmune responses directed against disrupted lipid membrane components (palmitic, myristic and oleic acid), and residue molecules of lipid peroxidation, such as azelaic acid and malondialdehyde, IgM responses against the S-farnesyl-l-cysteine, and amino acids, modified by nitrating species such as nitrotyrosine, nitrophenylalanine, nitrotryptophan, nitroarginine and nitrocysteine, have all been reported [93, 224]. These molecules have been damaged undergoing conformational change because of high levels of O+NS damage and have thus become immunogenic. The levels of these corrupted entities correlate positively and significantly with the severity of the ME/CFS symptoms [93].
Rituximab
The contribution of B cells to pathology in MS has been underlined by the evidence that rituximab, a monoclonal antibody that depletes CD20+ B cells in particular, has proven to be effective in the treatment of MS [225, 226]. Rituximab has demonstrated efficacy in peripheral neurological diseases [227] by producing a sustained depression of pathogenic B cells [228]. In cerebrospinal fluid of MS patients, rituximab reduces not only B cells but also T cells [229]. This suggests that rituximab may well have properties other than as a monoclonal antibody to CD20+ B cells. T lymphocytes from relapsing-remitting MS patients demonstrate an impaired response to antigen stimulation following treatment with rituximab [230]. This finding supports the hypothesis that B cell activity is needed to maintain disease activity in MS [230, 231]. The effectiveness of rituximab is not dependent on secreted antibody, as rituximab does not alter plasma cell frequencies in CSF or serum [232].
Rituximab has also some efficacy in the treatment of ME/CFS [233]. Thus, 30 patients with ME/CFS were randomized to rituximab or placebo in a placebo-controlled study and monitored for a calendar year. Positive responses were seen in 67% of the rituximab treated patients and 13% of the placebo group. The treatment improved symptoms globally. There were no serious side effects. The delayed nature of the responses (beginning from 2 to 7 months following rituximab treatment) lead the authors to conclude that ME/CFS was, at least in part, an autoimmune disease.
Rituximab is gathering momentum as a treatment in a variety of autoimmune diseases [234, 235] especially where the patients are refractory to first and second line therapies [236]. When taken as a whole the trial data reveals that the vast majority of patients respond positively to rituximab. This benefit applies to people with rheumatoid arthritis [237, 238], which is a licensed indication and other autoimmune conditions such as systemic lupus erythematosus [239] and Sjögren’s syndrome [240] where the drug is used off license. Rituximab appears to have a number of additional benefits in addition to CD20 B cell depletion which all act to normalize immune homeostasis. Rituximab has several effects on the immune system. One of the most potentially surprising effects is the reduction of Th17 T cell production [241, 242]. These T cells are the cause of T cell induced autoimmunity and neurotoxicity in autoimmune diseases such as MS. Rituximab has a direct effect on reducing IL-2 levels and thus potentially inactivating a chronically activated immune system [243]. Rituximab achieves this by inhibiting the production of NFκB [244, 245], which is another key mediator of autoimmunity. Rituximab also raises the function of Treg cells [246, 247].
Summary
Table 4 displays the similarities in B cells and autoimmune responses between MS and ME/CFS. In all, the range of autoantibodies produced in both illnesses is vast and once again virtually identical. Antibodies are found against nuclear and neuronal antigens, cardiolipin, phospholipids, serotonin, and gangliosides. IgM responses are detected against oleic, palmitic and myristic acid in patients with ME/CFS, and oleic and palmitic acid in patients with MS. Antibodies towards the byproducts of lipid peroxidation, that is, azelaic acid and MDA, and S-farnesyl-l-cysteine, are found in patients with ME/CFS but only towards azelaic acid and MDA in patients with MS. Autoantibodies to nitrotyrosine are found in both illnesses but a wider range of autoantibodies to other nitrated amino acids are found in ME/CFS which have not been reported in MS. This hints at the fact that O+NS-induced autoimmune responses may be higher in patients with ME/CFS than in those with MS.
Mitochondrial dysfunctions in MS and ME/CFS
Many studies report changes in the levels of circulating compounds related to impaired energy metabolism, elevated O+NS, and impaired antioxidant status occurring in MS [248]. Biochemical abnormalities in levels of pyrimidines, creatine, malondialdehyde ascorbic acid, nitrate and nitrite strongly suggest a profound alteration in redox balance and energy metabolism in patients with MS [248, 249]. Elevated levels of oxypurines in the serum are a product of abnormal purine nucleotide metabolism when adenosine triphosphate (ATP) production is insufficient to meet cellular demand [250, 251]. Nuclear magnetic resonance imaging (NMRI) techniques allow the direct measurement of mitochondrial functioning, rate of glycolysis and availability of energy, and this approach is being increasingly used in MS research. For example, Lazzarino et al. [252] reported a significant degree of central ATP depletion in their MS patients and concluded that an increased energy demand and mitochondrial failure was the cause of that depletion. Mitochondrial damage may be driven by free radicals produced by activated microglia [253]. Damage to mitochondria and the ensuing energy failure are well known drivers for tissue injury [254]. In MS lesions, conformational changes are observed in proteins of the mitochondrial respiratory chain [255, 256]. Deletions in mitochondrial DNA may be observed in neurons [257]. Mitochondrial DNA and proteins are both very vulnerable to damage by O+NS [258]. Therefore, it is likely that O+NS drive injuries to mitochondria and mitochondrial DNA in patients with MS [254, 259, 260].
The evidence that mitochondrial dysfunction and abnormally high lactate levels play a pivotal role in the pathophysiology of ME/CFS is expansive and expanding [261–264]. Vermeulen et al. [265] reported that in two exercise tests held 24 h apart, patients with ME/CFS reached their anaerobic threshold at a significantly lower oxygen consumption than healthy controls in the first test. This finding also applied to their maximal exercise capacity, which was also attained at a much lower oxygen capacity than the control group. This difference was even greater on the subsequent test. The researchers concluded that these findings demonstrated an increase in lactate production and decrease in ATP production relative to controls. Arnold et al. [266] using 31P nuclear magnetic resonance spectroscopy, revealed an abnormal increase in the level of intracellular lactic acid in the exercised forearm of a ME/CFS patient. This was proportional to concomitant changes in high-energy phosphates. Behan [261] reported finding structural mitochondrial abnormalities in the skeletal muscle of ME/CFS patients. ME/CFS patients display a significant increase in intracellular lactate levels following exercise compared to controls [263, 267]. They display a significantly lower ATP resynthesis rate during recovery from exercise than normal controls stemming from impaired oxidative phosphorylation [263]. ME/CFS patients display an abnormal rise in lactate with even minor exercise and an extremely slow recovery from this state [262, 263, 265]. Fatigue can result from an accumulation of reactive oxygen species (ROS) and depletion of available ATP in muscle cells [268]. Patients with ME/CFS reach exhaustion at a much earlier time point than healthy controls. Upon the point of exhaustion, ME/CFS patients also have reduced intracellular levels of ATP indicating a defect of oxidative metabolism combined with an acceleration of glycolysis in the working skeletal muscles [269]. ME/CFS is accompanied by significantly increased ventricular lactate, indicating mitochondrial dysfunctions in the illness [107, 270, 271].
Table 5 displays the similarities in mitochondral dysfunctions between MS and ME/CFS. In all, there is considerable evidence that mitochondrial dysfunction is heavily involved in the pathophysiology of both MS and ME/CFS. Production of ATP is suboptimal and levels are depleted in the brain and/or striated muscles. Decreased phosphocreatine synthesis rates following exercise is indicative of abnormal metabolic responses to exercise in MS and ME/CFS. Impaired oxidative phosphorylation is an issue in both disorders but accelerated glycolysis in muscles has been reported in people with ME/CFS but not in people with MS. Conversely, damage to the mitochondrial respiratory chain in neurons has been reported in MS but not in ME/CFS.
Brain dysfunctions
Single photon emission computed tomography (SPECT), used to examine cerebral perfusion in MS patients, showed significant decreases in blood flow in areas of cortical gray and white matter [272, 273]. Cerebral hypoperfusion in MS patients relative to controls is evident in both the cortex and deep gray matter, and is especially pronounced in the thalamus and caudate nuclei. Generalized reduction in cerebral oxygen utilization and blood flow in white and gray matter correlates with cognitive impairment [274]. Raschid et al. [275] demonstrated reduced perfusion, particularly in the gray matter of MS patients belonging to the primary and secondary-progressive subgroups. The reduction was observed in both deep gray and cortical matter and suggests depressed neuronal metabolic activity or actual neuronal loss [275].
Patients with ME/CFS display a global reduction of brain perfusion, with a characteristic pattern of hypoperfusion in the brainstem [276, 277]. SPECT abnormalities occur significantly more frequently and in greater numbers than MRI abnormalities do in patients with ME/CFS [278]. Using SPECT, a reduced cerebral blood flow was observed in the brain in 80% of ME/CFS patients [279]. Significant brain stem hypoperfusion has also been revealed in ME/CFS patients compared to healthy controls [276, 280]. Fischler et al. [277] demonstrated a positive and significant association between neurocognitive impairments experienced by patients and reduced frontal blood flow.
Positron emission tomography (PET), using a labeled native glucose analogue, that is, [18F]fludeoxyglucose (FDG-PET), has revealed a positive correlation between clinical progression of MS and cerebral glucose metabolism [281, 282]. Paulasu et al. [283] reported lowered glucose metabolism in the basal ganglia and frontal cortex of MS patients reporting severe levels of fatigue. The use of FDG PET in ME/CFS has revealed glucose hypometabolism in various areas of the brain [280, 284].
Traditionally 1.5 T-weighted, T1-weighted and T2-weighted, non-contrast or gadolinium, and enhanced T1-weighted hyperintense MRI images have been used to monitor disease progress in the white matter of MS patients [285]. Both gray matter atrophy [285, 286] and lesions [287, 288] have also been revealed in the cerebral cortex and deep gray matter structures using MRI. Atrophy in the brains of people with MS is related to gray matter hypointensity [289]. However, sensitivity of the conventional MRI methods for gray matter lesions is low compared to white matter lesions [288, 290]. MRI techniques are not sufficiently sensitive to detect purely cortical MS lesions [291]. This sensitivity can be improved using higher field strength [292, 293] or voxel-based morphometry [294, 295].
MRI involving voxel-based morphometry in patients with ME/CFS has revealed gray matter volume reduction [296–298]. These reductions are apparently unrelated to the duration of illness or the age of the person examined. Subcortical white matter hyperintensities have been repeatedly recorded in ME/CFS [299, 300].
The use of proton magnetic resonance spectroscopy (MRS) has revealed abnormally high levels of cerebral lactate in patients with MS [301, 302]. Using choline MRS, abnormally high choline was detected in the basal ganglia of patients [303–305]. Richards [306] using MRS demonstrated elevated concentrations of choline, lactate and lipids. In another study, proton MRS revealed significantly lower N-acetyl aspartate levels in the hippocampal areas of MS patients [307]. MS patients with active disease have high levels of lactate levels in CSF. This elevation in lactate levels may result from anaerobic glycolysis in activated leukocytes during active disease [308].
Brooks et al. [309] examined a cohort of ME/CFS patients using MRI and nuclear MRS. Using proton MRS, significantly reduced N-acetyl aspartate levels were observed in the hippocampal areas of ME/CFS patients. Chaudhuri et al. [310], using the same technique, demonstrated increased choline signaling in the basal ganglia of ME/CFS patients. The choline peaks in the basal ganglia most likely are related to ‘increased cell membrane turnover due reparative gliosis’ [310]. In addition, Puri et al. [311] established a significant choline/creatine signal in the occipital cortex of patients with ME/CFS. In children with ME/CFS, significant increases in the choline/creatine ratio as measured by MRS were observed [312].
Table 5 shows the similarities in brain dysfunctions between MS and ME/CFS. In summary, using SPECT, PET, and MRI, it was found that both disorders display cerebral hypoperfusion, reduced cerebral glucose metabolism and gray matter atrophy. Nuclear MRS has revealed abnormal choline signaling in the basal ganglia in both diseases coupled with elevated levels of lactate and reduced concentrations of N-acetyl aspartate.
Mechanistic explanations of typical ME/CFS symptoms in MS and ME/CFS
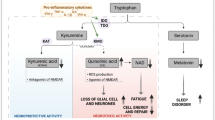
Above, we have already discussed that many patients with MS have typical ME/CFS symptoms, including fatigue and post-exertional malaise. In this section we will discuss the mechanistic explanations of the typical symptoms of ME/CFS that may occur in patients in MS. Fatigue in MS has both central (perception) and peripheral (impaired metabolism) components [313, 314]. Figure 1 shows a diagram that integrates the numerous pathways into a mechanistic model emphasizing the shared and interactive immune signaling and metabolic pathways that explain the symptomatic similarities in both diseases.

Diagram integrating immune signaling and metabolic pathways, which together explain the symptomatic similarities between both multiple sclerosis (MS) and myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Shared pathways are dysfunctions in intracellular signaling pathways, for example, nuclear factor κB (NFκB); immunoinflammatory pathways, for example, T helper (Th) and T regulatory (Treg) cells, cyclo-oxygenase 2 (COX2), and proinflammatory cytokines (PICs); and oxidative and nitrosative stress (O+NS) pathways. These in turn may induce increased damage by O+NS to proteins and lipids, secondary autoimmune responses and mitochondrial defects. There is evidence that these dysfunctions together with brain disorders are associated with the onset of ME/CFS symptoms, which appear in ME/CFS and MS. PEM = post-exertional malaise.
A number of authors have suggested a role for the immunological abnormalities seen in people with MS in the production of the severe fatigue endured by so many patients. Flachenecker et al. [315] found that TNFα levels were significantly higher in people with fatigue than those without fatigue. Further support for this concept is found in studies that posit a mediative role for IL-6 as well as TNFα in the generation of fatigue in MS [316, 317]. Pokryszko-Dragan et al. [318] reported that the severity of fatigue experienced by MS patients is significantly correlated to the stimulated production of IFNγ by T lymphocytes. Increased levels of proinflammatory cytokines are likely involved in the development and maintenance of fatigue in MS [319]. Raised levels of O+NS and mitochondrial dysfunctions could also conspire together to cause fatigue and the post-exertional malaise experienced by people with MS [8, 128].
MS patients demonstrate an exaggerated metabolic response to exercise compared to controls, and thus metabolism appears to be a major contributing factor in creating the excessive muscle fatigue experienced by people with MS [320]. Patients with MS display objective and clinically significant levels of impaired functional capacity indicated by lower maximal oxygen consumption and maximal workload compared to sedentary controls. These objective abnormalities correlate positively and significantly with measures of fatigue [321]. Fatigability of striated muscle not related to central nervous system activity is a frequent manifestation of MS [322]. In addition, it has been shown that individuals with MS have a markedly lowered rate of phosphocreatine resynthesis after depletion compared with controls [323]. The reduced phosphocreatine resynthesis results from impaired oxidative ATP production, probably stemming from impaired oxidative enzyme activities [324]. Muscle in MS patients is significantly smaller than that found in healthy individuals and relies more on anaerobic than aerobic respiration [324]. Several studies have found abnormalities relating to defects in maximal voluntary contraction during exercise or after during the facilitation period in MS patients with muscle weakness [325] and the magnitude of the defects correlates with the degree of nerve damage [326]. Motor evoked potentials (MEPs) in MS tend to be abnormal in MS if people have disabling fatigue [327, 328]. Impaired motor performance has been demonstrated in people with MS [329]. Ng et al. [330] reported that maximal voluntary contraction was 27% lower in MS patients than in the control group. The motor changes however were not related to fatigue but impaired walking ability. It is worthy of note that MS patients with a modest level of disability display gross reductions in exercise capability [331]. Savci et al. [332] noted that weakness in respiratory muscles, impaired lung function and degree of neurological impairment are not factors contributing to lowered functional exercise capability in MS patients.
Functional brain imaging research using SPECT and PET indicate that MS fatigue is connected to global glucose hypometabolism in the prefrontal cortex and the basal ganglia [282, 333–336]. Other researchers [337, 338] demonstrated that hypoperfusion correlates with disease and fatigue severity in MS. PET studies show that hypometabolism of particular brain areas, especially the frontal and subcortical circuits, is associated with fatigue [37, 339, 340]. MRI, PET and functional MRI studies indicate that fatigue is related to gray matter disease, in the thalamus and caudate areas and particularly the cerebral cortex [341].
In addition, the fatigue, fatigability and post-exertional malaise in ME/CFS have central and peripheral components [5, 128]. As explained elsewhere and above, fatigue in ME/CFS is associated with and may be explained by increased levels of proinflammatory cytokines, O+NS and mitochondrial defects [5, 128]. Several studies have found abnormalities relating to defects in maximal voluntary contraction during exercise or after during the post exercise facilitation period in ME/CFS [342]. MEP immediately following a period of exercise was significantly lower in ME/CFS and MEP facilitation 30 minutes after exercise was significantly less than in controls [343, 344]. These parameters were also low in another study [345]. Some authors have proposed the hypothesis that the fatigue in ME/CFS is entirely of neurological origin [346, 347]. This would however seem to be a minority viewpoint at this time.
Schillings et al. [348] reported impaired central activation in ME/CFS during maximal voluntary contraction and reported similar findings in seven out of the nine studies reviewed. Kent-Braun et al. [349] also reported markedly diminished levels of central activation at the end of a muscle contraction period. Schillings et al. [348] reported that the apparent central activation failure at maximal voluntary contraction in ME/CFS is of a similar magnitude to that reported in stroke and Amyotrophic Lateral Sclerosis [350, 351]. A large number of studies demonstrate impaired motor performance in people with ME/CFS [345, 352, 353].
In patients with ME/CFS, Barnden et al. [297] reported that the volume of white matter, as measured using 3 T MRI, correlates significantly and positively with the severity of fatigue experienced by the patients. The authors noted hemodynamic abnormalities in the brainstem, deep frontal white matter, the caudal basal pons and hypothalamus, suggestive of impaired cerebrovascular autoregulation. When taken as a whole, the evidence pointed to astrocyte dysfunction and resetting of homeostatic norms. Astrocyte activity regulates cerebrovascular autoregulation [297] and cerebral blood flow [354, 355]. Astrocyte malfunction is an important cause of mental fatigue [355]. Thus, dysfunctional astrocyte activity reported in ME/CFS would be expected to lead in a breakdown of mechanisms controlling blood flow in the brain. Patients with ME/CFS display a global reduction of brain perfusion, with a characteristic pattern of brainstem hypoperfusion [276, 356]. The severity of disabling fatigue experienced by patients with ME/CFS is associated with the reduction in basal ganglia activation [357]. When taken as a whole, the evidence of gray matter abnormalities and astrocyte dysfunction as contributors to the fatigue experienced by those with both illnesses appears substantive.
In summary, fatigue and post-exertional malaise in MS and ME/CFS may be explained by peripheral and central mechanisms, including increased levels of proinflammatory cytokines, O+NS and mitochondrial dysfunctions. In both disorders, abnormalities relating to defects in maximal voluntary contraction during exercise are detected. Central disorders, including glucose hypometabolism and cerebral hypoperfusion, may contribute to fatigue in both disorders.
Summary
MS and ME/CFS show remarkable levels of similarity in many dimensions. The ever-present disabling fatigue is a burden for both groups of patients to carry. This burden is made heavier by severe levels of exercise intolerance, which induce a worsening of symptoms in people with both illnesses. Both sets of patients resort to ‘pacing’ as an energy conservation strategy in an attempt to meet the energy demands associated with normal living. ME/CFS and MS are more prevalent in women than in men and show a chronic or waxing and waning course. Increased levels of O+NS occur in both illnesses and patients share an almost identical range of empirically determined abnormalities as evidenced by elevated levels of peroxinitrite, NO and iNOS, lipid peroxidation and nitration of amino acids. The consequences of O+NS damage to self-epitopes is evidenced by the almost bewildering and almost identical array of autoantibodies formed against damaged epitopes seen in both illnesses. Reduced levels of antioxidants, including vitamin E, zinc, glutathione are also found in both diseases. Evidence of chronic immune activation coupled with disordered T cell homeostasis is seen in both diseases. Similar abnormalities exist in levels of proinflammatory cytokines, serum neopterin and T cell antigens. Although reduced NKC function in ME/CFS has been emphasized over many years the identical abnormalities in MS has not received such widespread attention. The findings produced by neuroimaging using PET, SPECT and nuclear MRS are similar in both illnesses and in MS the severity of the abnormality in glucose metabolism correlates well with disease activity while MRI findings do not.
There are however also differences in symptomatic and immune profiles between both diagnoses. Thus, patients with ME/CFS seem more sensitive to increases in physical or cognitive activity than patients with MS. ME/CFS patients may have a worse experience with regard to infections, while the number of infections is associated with increasing symptom severity. While MS is characterized by increased expression of CD69, a decreased CD69 expression is seen in ME/CFS. Accelerated glycolysis is reported in ME/CFS but not in MS. Neuronal damage to the respiratory chain has been found in MS but not in ME/CFS. T cell exhaustion seems to be more of an issue in ME/CFS than in MS. When taken together the range of surrogate markers for O+NS and the range of autoantibodies are wider in ME/CFS than in MS and this may be due to an increased severity of O+NS in ME/CFS. While coenzyme Q10 is related to fatigue in ME/CFS, the findings in MS are less evident. Finally, while both ME/CFS and MS are chronic immunoinflammatory diseases, inflammation of the central nervous system is clearly more prominent in MS than in ME/CFS.
Nevertheless, the strong similarities between both disorders in terms of phenomenological, neurobehavior and neuroimmune characteristics further underscore that ME/CFS belongs to the spectrum of neuroimmune disorders. In addition, the data show that the comorbidity between both disorders and the high prevalence of ME/CFS symptoms in patients with MS may be explained by neuroimmune mechanisms.
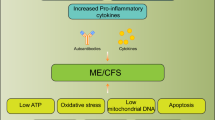
Figure 2 shows that the significant comorbidity between MS and ME/CFS may be based on shared immunoinflammatory, O+NS, autoimmune and mitochondrial pathways and brain dysfunctions.

The significant neuroimmune overlaps in multiple sclerosis (MS) and myalgic encephalomyelitis/chronic fatigue syndrome (MS/CFS) may be based on shared immunoinflammatory, oxidative and nitrosative stress (O+NS), autoimmune and mitochondrial pathways, and brain dysfunctions. CMI = cell-mediated immunity; NKCA: natural killer cell activity; PICs = proinflammatory cytokines; Th = T helper; Treg = T regulatory.
Figure 3 shows a second model that explains the high incidence of typical ME/CFS symptoms in patients with MS. This model suggests that patients with MS are neurologically and immunologically primed for an increased expression of ME/CFS symptoms. Thus, the activation of immunoinflammatory, autoimmune, mitochondrial and O+NS pathways together with brain disorders may prime MS patients for an increased prevalence of ME/CFS symptoms. Other possibilities are that ME/CFS could increase the odds to develop MS or when comorbid with MS could aggravate the severity of MS. It is also possible that there is a junction in immunoinflammatory progression that could explain bifurcation to ME/CFS rather than MS. For example, the initial lesions in ME/CFS could be smaller than in MS but at the expense of greater bioenergetic impairments [5].

A second model that explains the high incidence of characteristic symptoms of myalgic encephalomyelitis/chronic fatigue syndrome (MS/CFS) in multiple sclerosis (MS). This model shows that MS patients are neuroimmunologically primed towards a higher expression of ME/CFS symptoms. Thus, the activation of immunoinflammatory, oxidative and nitrosative (O+NS), autoimmune and mitochondrial pathways, and brain dysfunctions may prime MS patients for an increased prevalence of ME/CFS symptoms. CMI = cell-mediated immunity; PICs = proinflammatory cytokines.
Abbreviations
- COX2:
-
Cyclo-oxygenase 2
- CSF:
-
Cerebrospinal fluid
- DPPIV:
-
Dipeptidyl peptidase 4
- FDG:
-
Fluodeoxyglucose
- HR:
-
Heart rate
- IFN:
-
Interferon
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- ME/CFS:
-
Myalgic encephalomyelitis/chronic fatigue syndrome
- MEP:
-
Motor evoked potentials
- MRI:
-
Magnetic resonance imaging
- MRS:
-
Magnetic resonance spectroscopy
- MS:
-
Multiple sclerosis
- NFκB:
-
Nuclear factor κB
- NKCA:
-
Natural killer cell activity
- NMRI:
-
Nuclear magnetic resonance imaging
- NO:
-
Nitric oxide
- O+NS:
-
Oxidative and nitrosative stress
- PD:
-
Programmed death
- PET:
-
Positron emission tomography
- POTS:
-
Postural orthostatic tachycardia syndrome
- ROS:
-
Reactive oxygen species
- SPECT:
-
Single photon emission computed tomography
- TGF:
-
Transforming growth factor
- Th:
-
T helper
- TNF:
-
Tumor necrosis factor
- Treg:
-
T regulatory.
References
Disanto G, Berlanga AJ, Handel AE, Para AE, Burrell AM, Fries A, Handunnetthi L, De Luca GC, Morahan JM: Heterogeneity in multiple sclerosis: scratching the surface of a complex disease. Autoimm Dis. 2010, 2011: 932351.
Huang YM, Liu X, Steffensen K, Sanna A, Arru G, Fois ML, Rosati G, Sotgiu S, Link H: Immunological heterogeneity of multiple sclerosis in Sardinia and Sweden. Mult Scler. 2005, 11: 16-23.
Hohlfeld R, Wekerle H: Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: from pipe dreams to (therapeutic) pipelines. Proc Natl Acad Sci U S A. 2004, 2004: 14599-14606.
Winkler AS, Blair D, Marsden JT, Peters TJ, Wessely S, Cleare AJ: Autonomic function and serum erythropoietin levels in chronic fatigue syndrome. J Psychosom Res. 2004, 56: 179-183.
Morris G, Maes M: A neuro-immune model of myalgic encephalomyelitis/chronic fatigue syndrome. Metab Brain Dis. 2012, Epub ahead of print
Morris G, Anderson G, Galecki P, Berk M, Maes M: A narrative review on the similarities and dissimilarities between myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and sickness behavior. BMC Med. 2013, 11: 64.
Sharpe MC, Archard LC, Banatvala JE, Borysiewicz LK, Clare AW, David A, Edwards RH, Hawton KE, Lambert HP, Lane RJ: A report-chronic fatigue syndrome: guidelines for research. J R Soc Med. 1991, 84: 118-121.
Morris G, Maes M: Case definitions and diagnostic criteria for myalgic encephalomyelitis and Chronic fatigue Syndrome: from clinical-consensus to evidence-based case definitions. Neuro Endocrinol Lett. 2013, 34: 185-199.
Maes M: Nooit meer moe: CVS ontmaskerd. 2010, Brugge, Belgium: Zorro Uitgevers
Maes M, Twisk FN, Johnson C: Myalgic encephalomyelitis (ME), chronic fatigue syndrome (CFS), and chronic fatigue (CF) are distinguished accurately: Results of supervised learning techniques applied on clinical and inflammatory data. Psychiatr Res. 2012, 200: 754-760.
Carruthers BM, van de Sande MI, De Meirleir KL, Klimas NG, Broderick G, Mitchell T, Staines D, Powles AC, Speight N, Vallings R, Bateman L, Baumgarten-Austrheim B, Bell DS, Carlo-Stella N, Chia J, Darragh A, Jo D, Lewis D, Light AR, Marshall-Gradisbik S, Mena I, Mikovits JA, Miwa K, Murovska M, Pall ML, Stevens S: Myalgic encephalomyelitis: international consensus criteria. J Intern Med. 2011, 270: 327-338.
Freeman R, Komaroff AL: Does the chronic fatigue syndrome involve the autonomic nervous system?. Am J Med. 1997, 102: 357-364.
Allen J, Murrary A, Di Maria C, Newton JL: Chronic fatigue syndrome and impaired peripheral pulse characteristics on orthostasis - a new potential diagnostic biomarker. Physiol Meas. 2010, 33: 231-241.
Newton JL, Okonkwo O, Sutcliffe K, Seth A, Shin J, Jones DEJ: Symptoms of autonomic dysfunction in chronic fatigue syndrome. Q J Med. 2007, 100: 519-526.
Mosqueda Garcia R, Furlan R, Snell M, Jacob G, Harris P: Primary sympathetic hyperadrenergic orthostatic tachycardia syndrome (HOT). Neurology. 1997, 48: A147.
Montague TJ, Marrie TJ, Klassen GA, Bewick DJ, Horacek BM: Cardiac function at rest and with exercise in the chronic fatigue syndrome. Chest. 1989, 95: 779-784.
Pagani M, Lucini D, Mela GS, Langewitz W, Malliani A: Sympathetic overactivity in subjects complaining of unexplained fatigue. Clin Sci. 1994, 87: 655-661.
De Becker P, Dendale P, De Meirleir K, Campine I, Vandenborne K, Hagers Y: Autonomic testing in patients with chronic fatigue syndrome. Am J Med. 1998, 105: 122S-126S.
Soetekouw PM, Lenders JW, Bleijenberg G, Thien T, van der Meer JW: Autonomic function in patients with chronic fatigue syndrome. Clin Auton Res. 1999, 9: 334-340.
Rowe PC, Boo-Holaigh I, Kan JS, Calkins H: Is neurally mediated hypotension an unrecognized cause of chronic fatigue?. Lancet. 1995, 345: 623-624.
Bou-Holaigh I, Rowe PC, Kan JS, Calking H: The relationship between neurally mediated hypotension and the chronic fatigue syndrome. JAMA. 1995, 274: 961-967.
Streeten DH, Anderson GH: The role of delayed orthostatic hypotension in the pathogenesis of chronic fatigue. Clin Auton Res. 1998, 8: 119-124.
Schondorf R, Freeman R: The importance of orthostatic intolerance in the chronic fatigue syndrome. Am J Med Sci. 1999, 317: 117-123.
Axelrod FB, Chelimsky GG, Weese-Mayer DE: Pediatric autonomic disorders. Pediatrics. 2006, 2006: 309-321.
Stewart J, Weldon A, Arlievsky N, Li K, Munoz J: Neurally mediated hypotension and autonomic dysfunction measured by heart rate variability during head-up tilt table testing in children with chronic fatigue syndrome. Clin Auton Res. 1998, 8: 221-230.
Stewart JM, Gewitz MH, Weldon A, Munoz J: Patterns of orthostatic intolerance: the orthostatic tachycardia syndrome and adolescent chronic fatigue. J Pediatr. 1999, 135: 218-225.
Stewart JM: Autonomic nervous system dysfunction in adolescents with postural orthostatic tachycardia syndrome and chronic fatigue syndrome is characterized by attenuated vagal baroreflex and potentiated sympathetic vasomotion. Pediatr Res. 2000, 48: 218-226.
Shepherd C: Pacing and exercise in chronic fatigue syndrome. Physiotherapy. 2001, 87: 395-396.
Iriarte J, de Castro P: Correlation between sympotom fatigue and muscular fatigue in multiple sclerosis. Eur J Neurol. 1998, 5: 579-585.
Barak Y, Achiron A: Cognitive fatigue in multiple sclerosis: findings from a two-wave screening project. J Neurol Sci. 2006, 245: 73-76.
Iriarte J, Subira ML, Castro P: Modalities of fatigue in multiple sclerosis: correlation with clinical and biological factors. Mult Scler. 2000, 6: 124-130.
Olsson M, Lexell J, Soderberg S: The meaning of fatigue for women with multiple sclerosis. J Adv Nurs. 2005, 49: 7-15.
Kos D, Kerckhofs E, Nagels G, D’hooghe B, Ilsbroukx S: Origin of fatigue in multiple sclerosis: review of the literature. Neurorehabil Neural Repair. 2007, 20: 1-10.
Vucic S, Burke D, Kiernan MC: Fatigue in multiple sclerosis: mechanisms and management. Clin Neurophysiol. 2010, 121: 809-817.
Boerio D, Lefaucheur JP, Hogrel JY, Creange A: Pathophysiology and treatment of fatigue in multiple sclerosis. Rev Neurol. 2006, 162: 311-320.
Debouverie M, Pittion S: Fatigue and episodic exhaustion as a feature of multiple sclerosis. Rev Neurol. 2006, 162: 295-297.
Bakshi R: Fatigue associated with multiple sclerosis: diagnosis, impact and management. Mult Scler. 2003, 9: 219-227.
Haensch CA, Jorg J: Autonomic dysfunction in multiple sclerosis. J Neurol. 2006, 253: 13-19.
Lensch E, Jost WH: Autonomic disorders in multiple sclerosis. Autoimmune Dis. 2011, 2011: 803841.
Kanjwal K, Karabin B, Kanjwal Y, Grubb BP: Autonomic dysfunction presenting as postural orthostatic tachycardia syndrome in patients with multiple sclerosis. Int J Med Sci. 2010, 7: 62-67.
McDougall J, McLeod JG: Autonomic nervous system function in multiple sclerosis. J Neurol Sci. 2003, 215: 79-85.
Merkelbach U, Dillmann C, Kolmel J, Holz I, Muller M: Cardiovascular autonomic dysregulation and fatigue in multiple sclerosis. Mult Scler. 2001, 7: 320-326.
de Seze J, Stojkovic T, Gauvrit JY, Devos D, Ayachi M, Cassim F, Saint Michel T, Pruvo JP, Guieu JD, Vermersch P: Autonomic dysfunction in multiple sclerosis: cervical spinal cord atrophy correlates. J Neurol. 2001, 248: 297-303.
Nordenbo AM, Boesen F, Andersen EB: Cardiovascular autonomic function in multiple sclerosis. J Auton Nerv Syst. 1989, 26: 77-84.
Mathiowetz VG, Finlayson ML, Matuska KM, Chen HY, Luo P: Randomized trial of an energy conservation course for persons with multiple sclerosis. Mult Scler. 2005, 11: 592-601.
Matuska K, Mathiowetz V, Finlayson M: Use and perceived effectiveness of energy conservation strategies for managing multiple sclerosis fatigue. Am J Occup Ther. 2007, 61: 62-69.
Vanage SM, Gilbertson KK, Mathiowetz V: Effects of energy conservation course on fatigue impact for persons with progressive multiple sclerosis. Am J Occup Ther. 2003, 57: 315-323.
Papanicolaou DA, Amsterdamb JD, Levine S, McCann SM, Moore RC, Newbrand CH, Allen G, Nisenbaum R, Pfaff DW, Tsokos GC, Vgontzas AN, Kales A: Neuroendocrine Aspects of chronic fatigue syndrome. Neuroimmunomodulation. 2003, 11: 65-74.
Reynolds NL, Brown MM, Jason LA: The relationship of Fennell phases to symptoms among patients with chronic fatigue syndrome. Eval Health Prof. 2009, 32: 264-280.
Nisenbaum R, Jones JF, Unger ER, Reyes M, Reeves WC: A population-based study of the clinical course of chronic. Health Qual Life Outcomes. 2003, 1: 49.
Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A: The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med. 1994, 121: 953-959.
Hinds GME, McCluskey DR: A retrospective study of chronic fatigue syndrome. Proc R Coll Physicians Edinburgh. 1993, 23: 10-14.
Wilson A, Hickie I, Lloyd A, Hadzi-Pavlovic D, Boughton C, Dwyer J, Wakefield D: Longitudinal study of outcome of chronic fatigue syndrome. Br Med J. 1994, 308: 756759.
Tiersky LA, DeLuca J, Hill N, Dhar SK, Johnson SK, Lange G, Rappolt G, Natelson BH: Longitudinal assessment of neuropsychological functioning, psychiatric status, functional disability and employment status in chronic fatigue syndrome. Appl Neuropsychol. 2001, 8: 41-50.
Hill NF, Tiersky LA, Scavalla VR, Lavietes M, Natelson BH: Natural history of severe chronic fatigue syndrome. Arch Phys Med Rehabil. 1999, 80: 1090-1094.
Van der Werf SP, de Vree B, Alberts M, Van der Meer JWM, Bleijenberg G: Natural course and predicting self- reported improvement in patients with chronic fatigue syndrome with a relatively short illness duration. J Psychosomat Res. 2002, 53: 749753.
Peterson PK, Schenck CH, Sherman R: Chronic fatigue syndrome in Minnesota. Minn Med. 1991, 74: 21-26.
Saltzstein BJ, Wyshak G, Hubbuch JT, Perry JC: A naturalistic study of the chronic fatigue syndrome among women in primary care. Gen Hosp Psychiatry. 1998, 20: 307316.
Buljevac D, Flach HZ, Hop WC, Hijdra D, Laman JD, Savelkoul HF, van Der Meche FG, van Doorn PA, Hintzen RQ: Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain. 2002, 125: 952-960.
Buljevac D, Hop WC, Reedeker W, Janssens AC, van der Meche FG, van Doorn PA, Hintzen RQ: Self reported stressful life events and exacerbations in multiple sclerosis: prospective study. BMJ. 2003, 327: 646.
Persoons JH, Schornagel K, Breve J, Berkenbosch F, Kraal G: Acute stress affects cytokines and nitric oxide production by alveolar macrophages differently. Am J Respir Crit Care Med. 1995, 152: 619-624.
Ontaneda D, Rae-Grant A: Management of acute exacerbations in multiple sclerosis. Ann Indian Acad Neurol. 2009, 12: 264-272.
Nicolson GL, Nasralla M, Haier J, Erwin R, Nicolson NL, Ngwenya R: Mycoplasmal infections in chronic illnesses: fibromyalgia and chronic fatigue syndromes, Gulf War illness, HIV-AIDS and rheumatoid arthritis. Med Sentinel. 1999, 4: 172-176.
Nicolson GL, Haier J, Nasralla M, Haier J, Erwin R, Nicolson NL, Ngwenya R: Mycoplasmal infections in chronic fatigue syndrome, fibromyalgia syndrome and Gulf War illness. JCFS. 2000, 6: 23-39.
Nicolson GL, Nasralla M, De Meirleir K, Gan R, Haier J: Evidence for bacterial (Mycoplasma, Chlamydia) and viral (HHV-6) co-infections in chronic fatigue syndrome patients. JCFS. 2003, 11: 7-20.
Nicolson GL, Gan R, Haier J: Multiple co-infections (Mycoplasma, Chlamydia, human herpesvirus-6) in blood of chronic fatigue syndrome patients: association with signs and symptoms. APMIS. 2003, 111: 557-566.
Nicolson GL, Nicolson NL, Haier J: Chronic fatigue syndrome patients subsequently diagnosed with Lyme Disease Borrelia burgdorferi: evidence for mycoplasma species co-infections. JCFS. 2008, 14: 5-17.
Vojdani A, Choppa PC, Tagle C, Andrin R, Samimi B, Lapp CW: Detection of mycoplasma genus and mycoplasma fermentans by PCR in patients with chronic fatigue syndrome. FEMS Immunol Med Microbiol. 1998, 22: 355-365.
Seishima M, Mizutani Y, Shibuya Y, Arakawa C: Chronic fatigue syndrome after human parvovirus B19 infection without persistent viremia. Dermatology. 2008, 216: 341-346.
Chia JK, Chia AY: Chronic fatigue syndrome is associated with chronic enterovirus infection of the stomach. J Clin Pathol. 2008, 61: 43-48.
Goudsmit EM, Howes S: Pacing to manage chronic fatigue syndrome. Pacing: an additional strategy to manage fatigue in chronic fatigue syndrome. http://freespace.virgin.net/david.axford/pacing.htm.
LaManca JJ, Peckerman A, Sisto SA, DeLuca J, Cook S, Natelson BH: Cardiovascular responses of women with chronic fatigue syndrome to stressful cognitive testing before and after strenuous exercise. Psychosom Med. 2001, 63: 756-764.
Kerr JR, Mattey DL: Preexisting psychological stress predicts acute and chronic fatigue and arthritis following symptomatic parvovirus B19 infection. Clin Infect Dis. 2008, 46: e83-e87.
Ebers GC: Environmental factors and multiple sclerosis. Lancet Neurol. 2008, 7: 268-277.
Simpson S, Blizzard L, Otahal P, Van der Mei I, Taylor B: Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry. 2011, 82: 1132-1141.
Buchwald D, Umali P, Umali J, Kith P, Pearlman T, Komaroff AL: Chronic fatigue and the chronic fatigue syndrome: prevalence in a Pacific Northwest health care system. Ann Intern Med. 1995, 123: 81-88.
Acheson D: The clinical syndrome variously called benign myalgic encephalomyelitis, Iceland disease and epidemic neuromyasthenia. Am J Med. 1959, 26: 569-595.
Holmes GP, Kaplan JE, Gantz NM, Komaroff AL, Schonberger LB, Straus SE, Jones JF, Dubois RE, Cunningham-Rundles C, Pahwa S, Tosato G, Zegans LS, Purtilo DT, Brown N, Schooley RT, Brus I: Chronic fatigue syndrome: a working case definition. Ann Intern Med. 1988, 108: 387-389.
Jason LA, King CP, Frankenberry EL, Jordan KM, Tryon WW, Rademaker F, Huang CF: Chronic fatigue syndrome: assessing symptoms and activity level. J Clin Psychol. 1999, 55: 411-424.
Reyes M, Nisenbaum R, Hoaglin DC, Unger ER, Emmons C, Randall B, Stewart JA, Abbey S, Jones JF, Gantz N, Minden S, Reeves WC: Prevalence and incidence of chronic fatigue syndrome in Wichita. Kansas. Arch Intern Med. 2003, 2003: 1530-1536.
Harbo HF, Gold R, Tintore M: Sex and gender issues in multiple sclerosis. Ther Adv Neurol Disord. 2013, 6: 237-248.
Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, Esterbauer H, Binder CJ, Witztum JL, Lassmann H: Oxidative damage in multiple sclerosis lesions. Brain. 2011, 134: 1914-1924.
Gonsette RE: Review: Oxidative stress and excitotoxicity: a therapeutic issue in multiple sclerosis?. Mult Scler. 2008, 14: 22-34.
Toshniwal PK, Zarling EJ: Evidence for increased lipid peroxidation in multiple sclerosis. Neurochem Res. 1992, 17: 205-207.
van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, van der Valk P, de Vries HE: Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol Med. 2008, 45: 1729-1737.
Smith KJ, Kapoor R, Felts A: Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999, 9: 69-92.
Hill KE, Zollinger LV, Watt HE, Carlson NG, Rose JW: Inducible nitric oxide synthase in chronic active multiple sclerosis plaques: distribution, cellular expression and association with myelin damage. J Neuroimmunol. 2004, 151: 171-179.
Koch M, Mostert J, Arutjunyan A, Stepanov M, Teelken A, Heersema D, De Keyser J: Peripheral blood leukocyte NO production and oxidative stress in multiple sclerosis. Mult Scler. 2008, 14: 159-165.
Basu S: Isoprostanes: novel bioactive products of lipid peroxidation. Free Radic Res. 2004, 38: 105-122.
Greco A, Minghetti L, Levi G: Isoprostanes, novel markers of oxidative injury, help understanding the pathogenesis of neurodegenerative diseases. Neurochem Res. 2000, 25: 1357-1364.
Morrow JD: The isoprostanes - unique products of arachidonate peroxidation: their role as mediators of oxidant stress. Curr Pharm. 2006, 12: 895-902.
Mattsson N, Haghighi S, Andersen O, Yao Y, Rosengren L, Blennow K, Pratico D, Zetterberg H: Elevated cerebrospinal fluid F2- isoprostane levels indicating oxidative stress in healthy siblings of multiple sclerosis patients. Neurosci Lett. 2007, 414: 233-236.
Maes M, Mihaylova I, Leunis JC: Chronic fatigue syndrome is accompanied by an IgM-related immune response directed against neopitopes formed by oxidative or nitrosative damage to lipids and proteins. Neuro Endocrinol Lett. 2006, 27: 615-621.
Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E: Increased 8-hydroxy-deoxyguanosine, a marker of oxidative damage to DNA, in major depression and myalgic encephalomyelitis/chronic fatigue syndrome. Neuro Endocrinol Lett. 2009, 30: 715-722.
Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E: Coenzyme Q10 deficiency in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is related to fatigue, autonomic and neurocognitive symptoms and is another risk factor explaining the early mortality in ME/CFS due to cardiovascular disorder. Neuro Endocrinol Lett. 2009, 30: 470-476.
Maes M, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E: Increased plasma peroxides as a marker of oxidative stress in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med Sci Monit. 2011, 17: SC11-SC15.
Manuel y Keenoy B, Moorkens G, Vertommen J, Noe M, Neve J, De Leeuw I: Magnesium status and parameters of the oxidant-antioxidant balance in patients with chronic fatigue: effects of supplementation with magnesium. J Am Coll Nutr. 2000, 19: 374-382.
Manuel-y-Keenoy B, Moorkens G, Vertommen J, De Leeuw I: Antioxidant status and lipoprotein peroxidation in chronic fatigue syndrome. Life Sci. 2001, 68: 2037-2049.
Vecchiet J, Cipollone F, Falasca K, Mezzetti A, Pizzigallo E, Bucciarelli T, De Laurentis S, Affaitati G, De Cesare D, Giamberardino MA: Relationship between musculoskeletal symptoms and blood markers of oxidative stress in patients with chronic fatigue syndrome. Neurosci Letters. 2003, 335: 151-154.
Miwa K, Fujita M: Fluctuation of serum vitamin E (alpha-tocopherol) concentrations during exacerbation and remission phases in patients with chronic fatigue syndrome. Heart Vessels. 2010, 25: 319-323.
Kennedy G, Spence VA, McLaren M, Hill A, Underwood C, Belch JJ: Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic Biol Med. 2005, 39: 584-589.
Maes M, Mihaylova I, Kubera M, Bosmans E: Not in the mind but in the cell: increased production of cyclo-oxygenase-2 and inducible NO synthase in chronic fatigue syndrome. Neuro Endocrinol Lett. 2007, 28: 463-469.
Jammes Y, Steinberg JG, Delliaux S: Chronic fatigue syndrome: acute infection and history of physical activity affect resting levels and response to exercise of plasma oxidant/antioxidant status and heat shock proteins. J Intern Med. 2011, 272: 74-84.
Jammes Y, Steinberg JG, Mambrini O, Bregeon F, Delliaux S: Chronic fatigue syndrome: assessment of increased oxidative stress and altered muscle excitability in response to incremental exercise. J Intern Med. 2005, 257: 299-310.
Fulle S, Pietrangelo T, Mancinelli R, Saggini R, Fano G: Specific correlations between muscle oxidative stress and chronic fatigue syndrome: a working hypothesis. J Muscle Res Cell Motil. 2007, 28: 355-362.
Richards RS, Roberts TK, McGregor NR, Dunstan RH, Butt HL: Blood parameters indicative of oxidative stress are associated with symptom expression in chronic fatigue syndrome. Redox Rep. 2000, 5: 35-41.
Shungu DC, Weiduschat N, Murrough JW, Mao X, Pillemer S, Dyke JP, Medow MS, Natelson BH, Stewart JM, Mathew SJ: Increased ventricular lactate in chronic fatigue syndrome. III. Relationships to cortical glutathione and clinical symptoms implicate oxidative stress in disorder pathophysiology. NMR Biomed. 2012, 25: 1073-1087.
Syburra C, Passi S: Oxidative stress in patients with multiple sclerosis. Ukr Biokhim Zh. 1999, 71: 112-115.
de Bustos F, Jiménez-Jiménez FJ, Molina JA, Gómez-Escalonilla C, de Andrés C, del Hoyo P, Zurdo M, Tallón-Barranco A, Berbel A, Porta-Etessam J, Parrilla G, Arenas J: Serum levels of coenzyme Q10 in patients with multiple sclerosis. Acta Neurol Scand. 2000, 101: 209-211.
Choi IY, Lee SP, Denney DR, Lynch SG: Lower levels of glutathione in the brains of secondary progressive multiple sclerosis patients measured by 1H magnetic resonance chemical shift imaging at 3 T. Mult Scler. 2011, 17: 289-296.
Ghazavi A, Kianbakht S, Ghasami K, Mosayebi G: High copper and low zinc serum levels in Iranian patients with multiple sclerosis: a case control study. Clin Lab. 2012, 58: 161-164.
Maes M, Mihaylova I, Leunis JC: In chronic fatigue syndrome, the decreased levels of omega-3 poly-unsaturated fatty acids are related to lowered serum zinc and defects in T cell activation. Neuro Endocrinol Lett. 2005, 26: 745-751.
Dujmovic I, Mangano K, Pekmezovic T, Quattrocchi C, Mesaros S, Stojsavljevic N, Nicoletti F, Drulovic J: The analysis of IL-1 beta and its naturally occurring inhibitors in multiple sclerosis: the elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J Neuroimmunol. 2009, 207: 101-106.
Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, Raine CS, Brosnan CF, John GR: IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol. 2006, 177: 5574-5584.
Hauser SL, Doolittle TH, Lincoln R, Brown RH, Dinarello CA: Cytokine accumulations in CSF of multiple sclerosis patients: frequent detection of interleukin-1 and tumor necrosis factor but not interleukin-6. Neurology. 1990, 40: 1735-1739.
Sharief MK, Hentges R: Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. 1991, 325: 467-472.
Al-Omaishi J, Bashir R, Gendelman HE: The cellular immunology of multiple sclerosis. J Leukoc Biol. 1999, 65: 444-452.
Frei K, Fredrikson S, Fontana A, Link H: Interleukin-6 is elevated in plasmain multiple sclerosis. J Neuroimmunol. 1991, 31: 147-153.
Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O: Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis. A correlation with clinical activity. Immunopharmacol Immunotoxicol. 1998, 20: 373-382.
Gajewski TF, Schell SR, Nau G, Fitch FW: Regulation of T-cell activation: differences among T-cell subsets. Immunol Rev. 1989, 111: 79-110.
Murphy KM, Reiner SL: The lineage decisions of helper T cells. Nat Rev Immunol. 2002, 2002: 933-944.
Imam SA, Guyton MK, Haque A, Vandenbark A, Tyor WR, Ray SK, Banik NL: Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J Neuroimmunol. 2007, 190: 139-145.
Koguchi K, Anderson DE, Yang L, O’Connor KC, Kuchroo VK, Hafler DA: Dysregulated T cell expression of TIM3 in multiple sclerosis. J Exp Med. 2006, 203: 1413-1418.
Cannella B, Raine CS: The adhesion molecule and cytokine profile of MS lesions. Ann Neurol. 1995, 37: 424-435.
Navikas V, Link H: Cytokines and the pathogenesis of multiple sclerosis. J Neurosci Res. 1996, 45: 322-333.
Rodriguez-Sainz Mdel C, Sanchez-Ramon S, de Andres C, Rodriguez-Mahou M, Munoz-Fernandez MA: Th1/Th2 cytokine balance and nitric oxide in cerebrospinal fluid and serum from patients with multiple sclerosis. Eur Cytokine Netw. 2002, 13: 110-114.
Maes M, Twisk FNM, Kubera M, Ringel K: Evidence for inflammation and activation of cell-mediated immunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): increased interleukin-1, tumor necrosis factor-α, PMN-elastase, lysozyme and neopteri. J Affect Disord. 2012, 136: 933-939.
Morris G, Maes M: Increased nuclear factor-κB and loss of p53 are key mechanisms in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med Hypotheses. 2012, 79: 607-613.
Maher KJ, Klimas NG, Fletcher MA: Chronic fatigue syndrome is associated with diminished intracellular perforin. Clin Exp Immunol. 2005, 142: 505-511.
Broderick G, Fuite J, Kreitz A, Vernon SD, Klimas N, Fletcher MA: A formal analysis of cytokine networks in chronic fatigue syndrome. Brain Behav Immun. 2010, 24: 1209-1217.
Rose JW, Hill KE, Watt HE, Carlson NG: Inflammatory cell expression of cyclooxygenase-2 in the multiple sclerosis lesion. J Neuroimmunol. 2004, 149: 40-49.
Carlson NG, Rojas MA, Redd JW, Tang P, Wood B, Hill KE, Rose JW: Cyclooxygenase-2 expression in oligodendrocytes increases sensitivity to excitotoxic death. J Neuroinflammation. 2010, 7: 25.
Gveric D, Kaltschmidt C, Cuzner ML, Newcombe J: Transcription factor NF-κB and inhibitor IκBα are localized in macrophages in active multiplesclerosis lesions. J Neuropathol Exp Neurol. 1998, 57: 168-178.
Bonetti B, Stegagno C, Cannella B, Rizzuto N, Moretto G, Raine CS: Activation of NF-kappaB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am J Pathol. 1999, 155: 1433-1438.
Mattson MP, Camandola S: NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001, 107: 247-254.
Link H, Fredrikson S: HLA-DR expression and neopterin levels as activity markers in multiple sclerosis. Riv Neurol. 1987, 57: 154-158.
Khorami H, Neyestani TR, Kadkhodaee M, Lotfi J: Increased urinary neopterin: creatinine ratio as a marker of activation of cell- mediated immunity and oxidative stress in the Iranian patients with multiple sclerosis. Iran J Allergy Asthma Immunol. 2003, 2: 155-158.
Giovannoni G, Lai M, Kidd D, Thorpe JW, Miller DH, Thompson AJ, Keir G, Feldmann M, Thompson EJ: Daily urinary neopterin excretion as an immunological marker of disease activity in multiple sclerosis. Brain. 1997, 120: 1-13.
Rejdak K, Leary SM, Petzold A, Thompson AJ, Miller DH, Giovannoni G: Urinary neopterin and nitric oxide metabolites as markers of interferon beta-1a activity in primary progressive multiple sclerosis. Mult Scler. 2010, 16: 1066-1072.
Fredrikson S, Link H, Eneroth P: CSF neopterin as marker of disease activity in multiple sclerosis. Acta Neurol Scand. 1987, 75: 352-355.
Buchwald D, Wener MH, Pearlman T, Kith P: Markers of inflammation and immune activation in chronic fatigue and chronic fatigue syndrome. J Rheumatol. 1997, 24: 372-376.
Matsuda J, Gohchi K, Gotch N: Serum concentrations of 2′,5′-oligoadenylate synthetase, neopterin, and beta-glucan in patients with chronic fatigue syndrome and in patients with major depression. J Neurol Neurosurg Psychiatry. 1994, 57: 1015-1016.
Chao CC, Gallagher M, Phair J, Peterson PK: Serum neopterin and interleukin-6 levels in chronic fatigue syndrome. J Infect Dis. 1990, 162: 1412-1413.
Chao CC, Janoff EN, Hu SX, Thomas K, Gallagher M, Tsang M, Peterson PK: Altered cytokine release in peripheral blood mononuclear cell cultures from patients with the chronic fatigue syndrome. Cytokine. 1991, 3: 292-298.
Fransson ME, Liljenfeldt LS, Fagius J, Totterman TH, Loskog AS: The T-cell pool is anergized in patients with multiple sclerosis in remission. Immunology. 2009, 126: 92-101.
Saresella M, Marventano I, Longhi R, Lissoni F, Trabattoni D, Mendozzi L, Caputo D, Clerici M: CD4 + CD25 + FoxP3 + PD1- regulatory T cells in acute and stable relapsing-remitting multiple sclerosis and their modulation by therapy. FASEB J. 2008, 22: 3500-3508.
Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA: Loss of functional suppression by CD4 + CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004, 199: 971-979.
Ni L, Ma CJ, Zhang Y, Nandakumar S, Zhang CL, Wu XY, Borthwick T, Hamati A, Chen XY, Kumaraguru U, Moorman JP, Yao ZQ: PD-1 modulates regulatory T cells and suppresses T-cell responses in HCV-associated lymphoma. Immunol Cell Biol. 2011, 89: 535-539.
Costantino CM, Baecher-Allan C, Hafler DA: Multiple sclerosis and regulatory T cells. J Clin Immunol. 2008, 28: 697-706.
Zozulya AL, Wiendl H: The role of regulatory T cells in multiple sclerosis. Nat Clin Pract Neurol. 2008, 4: 384-398.
Libera DD, Mitri DD, Bergami A, Centonze D, Gasperini C, Grasso MG, Galgani S, Martinelli V, Comi G, Avolio C, Martino G, Borsellino G, Sallusto F, Battistini L, Furlan R: T regulatory cells are markers of disease activity in multiple sclerosis patients. PLoS ONE. 2011, 6: e21386.
D’Souza M, Fontenot AP, Mack DG, Lozupone C, Dillon S, Meditz A, Wilson CC, Connick E, Palmer BE: Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J Immunol. 2007, 179: 1979-1987.
Song SJ, Feng X, Guo JJ, Iu YN, Lun WH, Wei HS, Liu SA: Relationship of CD4+ CD25hi regulatory T (Treg) cells to disease progression in HIV-infected patients. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2009, 23: 361-363.
Hofstetter HH, Gold R, Hartung HP: Th17 cells in MS and experimental autoimmune encephalomyelitis. Int MS J. 2009, 16: 12-18.
Peelen E, Damoiseaux J, Smolders J, Knippenberg S, Menheere P, Tervaert JW, Hupperts R, Thewissen M: Th17 expansion in MS patients is counterbalanced by an expanded CD39+ regulatory T cell population during remission but not during relapse. J Neuroimmunol. 2011, 240–241: 97-103.
Weissert R: Differential response to treatment of relapsing-remitting multiple sclerosis with IFN-β: is there a dichotomy into T-helper-1 and −17 driven disease?. Future Neurol. 2010, 5: 481-484.
Edwards LJ, Robins RA, Constantinescu CS: Th17/Th1 phenotype in demyelinating disease. Cytokine. 2010, 50: 19-23.
Kostic M: Role of Th1 and Th17 immune responces in pathogenesis of multiple sclerosis. Acta Medica Medianae. 2010, 49: 61-69.
Ziegler SF, Buckner JH: FOXP3 and the regulation of Treg/Th17 differentiation. Microbes Infect. 2009, 11: 594-598.
Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR: TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008, 453: 236-240.
Liu DH, Liu ZD, Li YZ, Zhang HY, Hu CJ, Zhang YB, Whang DX: Expression of lymphocyte subsets and CD25 ~ + regulative T Cells in peripheral blood of patients with chronic fatigue syndrome. Labeled Immunoassays and Clinical Medicine. Abstract 2011, 2011–02. http://en.cnki.com.cn/Article_en/CJFDTOTAL-BJMY201102012.htm.
Gallo P, Piccinno MG, Pagni S, Argentiero V, Giometto B, Bozza F, Tavolato B: Immune activation in multiple sclerosis: study of IL-2, sIL-2R, and gamma-IFN levels in serum and cerebrospinal fluid. J Neurol Sci. 1989, 92: 9-15.
Gallo P, Piccinno MG, Tavolato B, Siden A: A longitudinal study on IL-2, sIL-2R, IL-4 and IFN-gamma in multiple sclerosis CSF and serum. J Neurol Sci. 1991, 101: 227-232.
Gallo P, Pagni S, Piccinno MG, Giometto B, Argentiero V, Chiusole M, Bozza F, Tavolato B: On the role of interleukin-2 (IL-2) in multiple sclerosis (MS). IL-2-mediated endothelial cell activation. Ital J Neurol Sci. 1992, 13: 65-68.
Sharief MK, Hentges R, Ciardi M, Thompson EJ: In vivo relationship of interleukin-2 and soluble IL-2 receptor to blood-brain barrier impairment in patients with active multiple sclerosis. J Neurol. 1993, 240: 46-50.
Cheney PR, Dorman SE, Bell DS: Interleukin-2 and the chronic fatigue syndrome. Ann Intern Med. 1989, 110: 321.
Lombardi V, Hagen KS, Hunter KW, Diamond JW, Smith-Gagen J, Yang W, Mikovits JA: Xenotropic murine leukemia virus-related virus-associated chronic fatigue syndrome reveals a distinct inflammatory signature. In Vivo. 2011, 25: 307-314.
Popmihajlov Z, Smith KA: Negative feedback regulation of T cells via interleukin-2 and FOXP3 reciprocity. PLoS ONE. 2008, 3: e1581.
Klimas N, Koneru AO: Chronic fatigue syndrome: inflammation, immune function, and neuroendocrine interactions. Curr Rheumatol Rep. 2007, 9: 482-487.
Capelli E, Zola R, Lorusso L, Venturini L, Sardi F, Ricevuti G: Chronic fatigue syndrome/myalgic encephalomyelitis: an update. Int J Immunopathol Pharmacol. 2010, 23: 981-989.
Jones JF, Straus SE: Chronic Epstein-Barr virus infection. Annu Rev Med. 1987, 38: 195-209.
Borysiewicz LK, Haworth SJ, Cohen J, Mundin J, Rickinson A, Sissons JG: Epstein-Barr virus - specific immune defects in patients with persistent symptoms following infectious mononucleosis. Q J Med. 1986, 58: 111-121.
Klimas N, Salvato F, Morgan R, Fletcher MA: Immunologic abnormalities in chronic fatigue syndrome. J Clin Microbiol. 1990, 28: 1403-1410.
Behan PO, Behan WHM, Bell EJ: The postviral fatigue syndrome - an analysis of the findings in 50 cases. J Infect. 1985, 10: 211-222.
Tobi M, Morag A, Ravid Z, Chowers I, Feldman-Weiss V, Michaeli Y, Ben-Chetrit E, Shalit M, Knobler H: Prolonged atypical illness associated with serological evidence of persistent Epstein-Barr infection. Lancet. 1982, 1: 61-64.
Morimoto C, Torimoto Y, Levinson G, Rudd CE, Schrieber M, Dang NH, Letvin NL, Schlossman SF: 1 F7, a novel cell surface molecule involved in helper function of CD4 cells. J Immunol. 1989, 143: 3430-3439.
Nakao H, Eguchi K, Kawakami A, Migita K, Otsubo T, Ueki Y, Shimomura C, Tezuka H, Matsunaga M, Maeda K, et al: Increment of Tal positive cells in peripheral blood of patients with rheumatoid arthritis. J Rheumatol. 1989, 16: 904-910.
Eguchi K, Ueki Y, Shimomura C, Otsubo T, Nakao H, Migita K, Kawakami A, Matsunaga M, Tezuka H, Ishikawa N: Increment in the Tal + in the peripheral blood and thyroid tissue of patients with Graves’ disease. J Immunol. 1989, 142: 4233-4240.
Preller V, Gerber A, Togni M, Wrenger S, Schraven B, Rocken C, Marguet D, Ansorge S, Brocke S, Reinhold D: CD26/DP IV in T cell activation and autoimmunity. Adv Exp Med Biol. 2006, 575: 187-193.
Ishii T, Ohnuma K, Murakami A, Takasawa N, Kobayashi S, Dang NH, Schlossman SF, Morimoto C: CD26-mediated signaling for T cell activation occurs in lipid rafts through its association with CD45RO. Proc Natl Acad Sci U S A. 2001, 98: 12138-12143.
Pacheco R, Lluis C, Franco R: Role of CD26-adenosine deaminase interaction in T cell-mediated immunity. Inmunologia. 2005, 24: 235-245.
Bengsch B, Seigel B, Flecken T, Wolanksi J, Blum HE, Thimme R: Human Th17 cells express high levels of enzymatically active dipeptidylpeptidase IV (CD26). J Immunol. 2012, 188: 5438-5447.
Krakauer M, Sorenson PS, Sellebjerg F: CD4(+) memory T cells with high CD26 surface expression are enriched for Th1 markers and correlate with clinical severity of multiple sclerosis. J Neuroimmunol. 2006, 181: 157-164.
Constantinescu CS, Kamoun M, Dotti M, Farber RE, Galetta SL, Rostami A: A longitudinal study of the T cell activation marker CD26 in chronic progressive multiple sclerosis. J Neurol Sci. 1995, 130: 178-182.
Sellebjerg F, Ross C, Koch-Henriksen N, Sorensen PS, Frederiksen JL, Bendtzen K, Sorensen TL: CD26 + CD4 + T cell counts and attack risk in interferon-treated multiple sclerosis. Mult Scler. 2005, 11: 641-645.
Fletcher MA, Zeng XR, Maher K, Levis S, Hurwitz B, Antoni M, Broderick G, Klimas NG: Biomarkers in chronic fatigue syndrome: evaluation of natural killer cell function and dipeptidyl peptidase IV/CD26. PLoS ONE. 2010, 5: e10817.
Martin P, Sanchez-Madrid F: CD69: an unexpected regulator of TH17 cell-driven inflammatory responses. Sci Signal. 2011, 4: pe14.
Perrella O, Carrieri PB, De Mercato R, Buscaino GA: Markers of activated T Lymphocytes and T cell receptor Gamma/Delta + in patients with multiple sclerosis. Eur Neurol. 1993, 33: 152-155.
Huang YM, Hussien Y, Jin YP, Soderstrom M, Link H: Multiple sclerosis: deficient in vitro responses of blood mononuclear cells to IFN-beta. Acta Neurol Scand. 2001, 104: 249-256.
Mihaylova I, DeRuyter M, Rummens JL, Bosmans E, Maes M: Decreased expression of CD69 in chronic fatigue syndrome in relation to inflammatory markers: evidence for a severe disorder in the early activation of T lymphocytes and natural killer cells. Neuro Endocrinol Lett. 2008, 28: 477-483.
Zhang C, Zhang J, Tian Z: The regulatory effect of natural killer Cells: do “NK-reg cells” exist?. Cell Mol Immunol. 2006, 3: 241-254.
Benczur M, Petrynyl GG, Palffy G, Varga M, Talas M, Kotsy B, Foldes I, Hollan SR: Dysfunction of natural killer cells in multiple sclerosis: a possible pathogenetic factor. Clin Exp Immunol. 1980, 39: 657-662.
Jiang W, Chai NR, Maric D, Bielekova B: Unexpected role for granzyme K in CD56 bright NK cell-mediated immunoregulation of multiple sclerosis. J Immunol. 2011, 187: 781-790.
Kastrukoff LF, Lau A, Wee R, Zecchini D, White R, Paty DW: Clinical relapses of multiple sclerosis are associated with ‘novel’ valleys in natural killer cell functional activity. J Neuroimmunol. 2003, 145: 103-114.
Takahashi K, Aranami T, Endoh M, Miyake S, Yamamura T: The regulatory role of natural killer cells in multiple sclerosis. Brain. 2004, 127: 1917-1927.
Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H, Henkart PA, Martin R: Regulatory CD56 bright natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006, 103: 5941-5946.
Caligiuri M, Murray C, Buchwald D, Levine H, Cheney P, Peterson D, Komaroff AL, Ritz J: Phenotypic and functional deficiency of natural killer cells in patients with chronic fatigue syndrome. J Immunol. 1987, 139: 3306-3313.
Whiteside TL, Friberg D: Natural killer cells and natural killer cell activity in chronic fatigue syndrome. Am J Med. 1998, 105: 27S-34S.
Genc K, Dona DL, Reder AT: Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest. 1997, 99: 2664-2671.
Nijs J, McGregor NR, De Becker P, Verhas M, Englebienne P, De Meirleir K: Monitoring a hypothetical channelopathy in chronic fatigue syndrome: preliminary observations. JCFS. 2003, 11: 117-133.
Nijs J, Coomans D, Nicolson GL, De Becker P, Christian D, De Meirleir K: Immunophenotyping predictive of mycoplasma infection in patients with chronic fatigue syndrome. JCFS. 2003, 11: 51-69.
Tirelli U, Marotta G, Improta S, Pinto A: Immunological abnormalities in patients with chronic fatigue syndrome. Scand J Immunol. 1994, 40: 601-608.
Anderton SM, Fillatreau S: Activated B cells in autoimmune diseases: the case for a regulatory role. Nat Clin Pract Rheumatol. 2008, 4: 657-666.
Barned S, Goodman AD, Mattson DH: Frequency of anti-nuclear antibodies in multiple sclerosis. Neurology. 1995, 45: 384-385.
Fukazawa T, Moriwaka F, Mukai M, Hamada T, Koike T, Tashiro K: Anticardiolipin antibodies in Japanese patients with multiple sclerosis. Acta Neurol Scand. 1993, 88: 184-189.
IJdo JW, Conti-Kelly AM, Greco P, Abedi M, Amos M, Provenzale JM, Greco TP: Anti-phospholipid antibodies in patients with multiple sclerosis and MS-like illnesses: MS or APS?. Lupus. 1999, 8: 109-115.
Vyshkina T, Kalman B: Autoantibodies and neurodegeneration in multiple sclerosis. Lab Invest. 2008, 88: 796-807.
Nordal GJ, Vandvik B: Evidence of local synthesis of smooth-muscle antibodies in the central nervous system in isolated cases of multiple sclerosis and chronic lymphocytic meningoencephalitis. Scand J Immunol. 1977, 6: 327-334.
Arnon R, Crisp E, Kelley R, Ellison GW, Myers LW, Tourtellotte WW: Anti-ganglioside antibodies in multiple sclerosis. J Neurol Sci. 1980, 46: 179-186.
Schott K, Schaefer JE, Richartz E, Batra A, Eusterschulte B, Klein R, Berg PA, Bartels M, Mann K, Buchkremer G: Autoantibodies to serotonin in serum of patients with psychiatric disorders. Psychiatr Res. 2003, 121: 51-57.
Hokama Y, Camproa CE, Hara C, Higa N, Siu N, Lau R, Kuribayashi T, Yabusaki K: Acute phase phospholipids related to the cardiolipin of mitochondria in the sera of patients with chronic fatigue syndrome (CFS), chronic Ciguatera fish poisoning (CCFP), and other diseases attributed to chemicals, Gulf War, and marine toxins. J Clin Lab Anal. 2008, 22: 99-105.
Hokama Y, Camproa CE, Hara C, Kuribayashi T, Le Huynh D, Yabusaki K: Anticardiolipin antibodies in the sera of patients with diagnosed chronic fatigue syndrome. J Clin Lab Anal. 2009, 23: 210-212.
Konstantinov K, von Mikecz A, Buckwalk D, Jones J, Gerace L, Tan EM: Autoantibodies to nuclear envelope antigens in chronic fatigue syndrome. J Clin Invest. 1996, 98: 1888-1896.
Buchwald MD, Wener MH, Komaroff AL: Antineuronal antibody levels in chronic fatigue syndrome patients with neurologic abnormalities. Arthritis Rheum. 1991, 34: 1485-1486.
Nishikai M: Antinuclear antibodies in patients with chronic fatigue syndrome. Nippon Rinsho. 2007, 65: 1067-1070.
Bassi N, Amital D, Amital H, Doria A, Shoenfeld Y: Chronic fatigue syndrome: characteristics̃and possible causes for its pathogenesis. Isr Med Assoc J. 2008, 10: 79-82.
Klein R, Berg PA: High incidence of antibodies to 5-hydroxytryptamine, gangliosides and phospholipids in patients with chronic fatigue and fibromyalgia syndrome and their relatives: evidence for a clinical entity of both disorders. Eur J Med Res. 1995, 1: 21-26.
Tanaka S, Kuratsune H, Hidaka Y, Hakariya Y, Tatsumi KI, Takano T, Kanakura Y, Amino N: Autoantibodies against muscarinic cholinergic receptor in chronic fatigue syndrome. Int J Mol Med. 2003, 12: 225-230.
Geffard M, Bodet D, Martinet Y, Dabadie MP: Detection of the specific IgM and IgA circulating in sera of multiple sclerosis patients: interest and perspectives. Immunoanalyse Biologie Specialisee. 2002, 17: 302-310.
Daverat P, Geffard M, Orgogozo JM: Identification and characterization of anti-conjugated azelaic acid antibodies in multiple sclerosis. J Neuroimmunol. 1989, 22: 129-314.
Maneta-Peyret L, Daverat P, Geffard M, Cassagne C, Orgogozo JM: Natural seric anti-fatty acid antibodies in multiple sclerosis. Neurosci Lett. 1987, 80: 235-239.
Boullerne A, Petry KG, Geffard M: Circulating antibodies directed against conjugated fatty acids in sera of patients with multiple sclerosis. J Neuroimmunol. 1996, 65: 75-81.
Naidoo R, Knapp ML: Studies of lipid peroxidation products in cerebrospinal fluid and serum in multiple sclerosis and other conditions. Clin Chem. 1992, 38: 2449-2454.
Maes M, Mihaylova I, Leunis JC: Increased serum IgM antibodies directed against phosphatidyl inositol (Pi) in chronic fatigue syndrome (CFS) and major depression: evidence that an IgM-mediated immune response against Pi is one factor underpinning the comorbidity between both CFS and depression. Neuro Endocrinol Lett. 2007, 28: 861-867.
McFarland HF: The B cell-old player, new position on the team. N Engl J Med. 2008, 2008: 664-665.
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH, HERMES Trial Group: B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. New Engl J Med. 2008, 358: 676-688.
Monson NL, Cravens PD, Frohman EM, Hawker K, Racke MK: Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch Neurol. 2005, 62: 258-264.
Maurer MA, Rakocevic G, Leung CS, Quast I, Lukacisin M, Goebels N, Munz C, Wardemann H, Dalakas M, Lnemann JD: Rituximab induces sustained reduction of pathogenic B cells in patients with peripheral nervous system autoimmunity. J Clin Invest. 2012, 122: 1393-1402.
Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA: Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006, 180: 63-70.
Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, Calabresi PA, Waubant E, Hauser SL, Zhang J, Smith CH: Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS?. Ann Neurol. 2010, 67: 452-461.
Reichardt P, Dornbach B, Rong S, Beissert S, Gueler F, Loser K, Gunzer M: Naive B cells generate regulatory T cells in the presence of a mature immunologic synapse. Blood. 2007, 110: 1519-1529.
Petereit HF, Moeller-Hartmann W, Reske D, Rubbert A: Rituximab in a patient with multiple sclerosis-effect on B cells, plasma cells and intrathecal IgG synthesis. Acta Neurol Scand. 2008, 117: 399-403.
Fluge O, Bruland O, Risa K, Storstein A, Kristoffersen EK, Sapkota D, Næss H, Dahl O, Nyland H, Mella O: Benefit from B-lymphocyte depletion using the anti-CD20 antibody rituximab in chronic fatigue syndrome: a double-blind and placebo-controlled study. PLoS ONE. 2011, 6: e26358.
Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, Racewicz AJ, van Vollenhoven RF, Li NF, Agarwal S, Hessey EW, Shaw TM, DANCER Study Group: The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006, 54: 1390-1400.
Eisenberg R: Update on rituximab. Ann Rheum Dis. 2005, 64: iv55-iv57.
Gurcan HM, Keskin DB, Stern JN, Nitzberg MA, Shekhani H, Ahmed AR: A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. 2009, 9: 10-25.
Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T: Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004, 350: 2572-2581.
Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC, REFLEX Trial Group: Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006, 54: 2793-2806.
Tokunaga M, Saito K, Kawabata D, Imura Y, Fujii T, Nakayamada S, Tsujimura S, Nawata M, Iwata S, Azuma T, Mimori T, Tanaka Y: Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann Rheum Dis. 2007, 66: 470-475.
Devauchelle-Pensec V, Pennec Y, Morvan J, Pers JO, Daridon C, Jousse-Joulin S, Roudaut A, Jamin C, Renaudineau Y, Roue IQ, Cochener B, Youinou P, Saraux A: Improvement of Sjogren’s syndrome after two infusions of rituximab (anti-CD20). Arthritis Rheum. 2007, 57: 310317.
Ireland S, Monson N: Potential impact of B cells on T cell function in multiple sclerosis. Mult Scler Int. 2011, 2011: 423971.
van de Veerdonk FL, Lauwerys B, Marijnissen RJ, Timmermans K, Di Padova F, Koenders MI, Gutierrez-Roelens I, Durez P, Netea MG, van der Meer JW, van den Berg WB, Joosten LA: The anti-CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum. 2011, 63: 1507-1516.
Lederer JA, Liou JS, Kim S, Rice N, Lichtman AH: Regulation of NF-kappa B activation in T helper 1 and T helper 2 cells. J Immunol. 1996, 156: 56-63.
Jazirehi AR, Huerta-Yepez S, Cheng G, Bonavida B: Rituximab (chimericanti-CD20 monoclonal antibody) inhibits the constitutive nuclear factor-{kappa}B signaling pathway in non-Hodgkin’s lymphoma B-cell lines: role in sensitization to chemotherapeutic drug-induced apoptosis. Cancer Res. 2005, 65: 264-276.
Bonavida B: Rituximab-induced inhibition of antiapoptotic cell survival pathways: implications in chemo/immunoresistance, rituximab unresponsiveness, prognostic and novel therapeutic interventions. Oncogene. 2007, 26: 3629-3636.
Vigna-Perez M, Hernandez-Castro B, Paredes-Saharopulos O, Portales-Perez D, Baranda L, Abud-Mendoza C, Gonzalez-Amaro R: Clinical and immunological effects of rituximab in patients with lupus nephritis refractory to conventional therapy: a pilot study. Arthritis Res Ther. 2006, 8: R83.
Vigna-Perez M, Abud-Mendoza C, Cuevas-Orta E, Baranda L, Paredes-Saharopulos O, Moreno R, Gonzalez-Amaro R: In vivo effect of rituximab on regulatory T cells and apoptosis in patients with rheumatoid arthritis. Immunologia. 2006, 25: 167-172.
Tavazzi B, Batocchi AP, Amorini AM, Nociti V, D’Urso S, Longo S, Gullotta S, Picardi M, Lazzarino G: Serum metabolic profile in multiple sclerosis patients. Mult Scler Int. 2011, 2011: 167156.
Amorini AM, Petzold A, Tavazzi B, Eikelenboom J, Keir G, Belli A, Giovannoni G, Di Pietro V, Polman C, D’Urso S, Vagnozzi R, Uitdehaag B, Lazzarino G: Increase of uric acid and purine compounds in biological fluids of multiple sclerosis patients. Clin Biochem. 2009, 42: 1001-1006.
Amorini M, Lazzarino G, Galvano F, Fazzina G, Tavazzi B, Galvano G: Cyanidin-3-O-beta-glucopyranoside protects myocardium and erythrocytes from oxygen radical-mediated damages. Free Radic Res. 2003, 37: 453-460.
Signoretti S, Di Pietro V, Vagnozzi R, Lazzarino G, Amorini AM, Belli A, D’Urso S, Tavazzi B: Transient alterations of creatine, creatine phosphate, N-acetylaspartate and high-energy phosphates after mild traumatic brain injury in the rat. Mol Cell Biochem. 2010, 333: 269-277.
Lazzarino G, Amorini AM, Eikelenboom MJ, Killestein J, Belli A, Di Pietro V, Tavazzi B, Barkhof F, Polman CH, Uitdehaag BM, Petzold A: Cerebrospinal fluid ATP metabolites in multiple sclerosis. Mult Scler. 2010, 16: 549-554.
Lassman H, van Horssen J: The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011, 585: 3715-3723.
Lu F, Selak M, O’Connor J, Croul S, Lorenzana C, Butunoi C, Kalman B: Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J Neurol Sci. 2000, 177: 95-103.
Mahad D, Ziabreva I, Lassmann H, Turnbull D: Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008, 131: 1722-1735.
Mahad D, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, Lassmann H, Turnbull DM: Mitochondrial changes within axons in multiple sclerosis. Brain. 2009, 132: 1161-1174.
Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM, Mahad DJ: Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol. 2011, 69: 481-492.
Higgins GC, Beart PM, Shin YS, Chen MJ, Cheung NS, Nagley P: Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. J Alzheimers Dis. 2010, 20: S453-S473.
Mao P, Reddy H: Is multiple sclerosis a mitochondrial disease?. Biochim Biophys Acta. 2010, 1802: 66-79.
van Horssen J, Witte ME, Schreibelt G, de Vries HE: Radical changes in multiple sclerosis pathogenesis. Biochem Biophys Acta. 2011, 1812: 141-150.
Behan WM, More IA, Behan PO: Mitochondrial abnormalities in the postviral fatigue syndrome. Acta Neuropathol. 1991, 83: 61-65.
Plioplys AV, Plioplys S: Electron-microscopic investigation of muscle mitochondria in chronic fatigue syndrome. Neuropsychobiology. 1995, 32: 175-181.
Lane RJ, Barrett MC, Taylor DJ, Kemp GJ, Lodi R: Heterogeneity in chronic fatigue syndrome: evidence from magnetic resonance spectroscopy of muscle. Neuromuscul Disord. 1998, 8: 204-209.
Myhill S, Booth NE, McLaren-Howard J: Chronic fatigue syndrome and mitochondrial dysfunction. Int J Clin Exp Med. 2009, 2: 1-16.
Vermeulen RCW, Kurt RM, Visser FC, Sluiter W, Scholte HR: Patients with chronic fatigue syndrome performed worse than controls in a controlled repeated exercise study despite a normal oxidative phosphorylation capacity. J Transl Med. 2010, 8: 93.
Arnold DL, Bore PJ, Radda GK, Styles P, Taylor DJ: Excessive intracellular acidosis of skeletal muscle on exercise in a patient with a post-viral exhaustion/fatigue syndrome. Lancet. 1984, 1: 1367-1369.
Lane RJM, Barrett MC, Woodrow D, Moss J, Fletcher R, Archard LC: Muscle fibre characteristics and lactate responses to exercise in chronic fatigue syndrome. J Neurol Neurosurg Psychiatry. 1998, 64: 362-367.
Allen DG, Lamb GD, Westerbland H: Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008, 88: 287-332.
Wong R, Lopaschuk G, Zhu G, Walker D, Catellier D, Burton D, Teo K, Collins-Nakai R, Montague T: Skeletal muscle metabolism in the chronic fatigue syndrome. In vivo assessment by 31P nuclear magnetic resonance spectroscopy. Chest. 1992, 102: 1716-1722.
Mathew SJ, Mao X, Keegan KA, Levine SM, Smith EL, Heier LA, Otcheretko V, Coplan JD, Shungu DC: Ventricular cerebrospinal fluid lactate is increased in chronic fatigue syndrome compared with generalized anxiety disorder: an in vivo 3.0 T (1)H MRS imaging study. NMR Biomed. 2009, 22: 251-258.
Murrough JW, Mao X, Collins KA, Kelly C, Andrade G, Nestadt P, Levine SM, Mathew SJ, Shungu DC: Increased ventricular lactate in chronic fatigue syndrome measured by 1H MRS imaging at 3.0 T. II: comparison with major depressive disorder. NMR Biomed. 2010, 23: 643-650.
Pozzilli C, Passafiume D, Bernardi S, Incoccia C, Bastianello S, Bozzao L, Lenzi GL, Fleschi C: SPECT, MRI and cognitive functions in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1991, 54: 110-115.
Adhya S, Johnson G, Herbert J, Jaggi H, Babb JS, Grossman RI, Inglese M: Pattern of hemodynamic impairment in multiple sclerosis: dynamic susceptibility contrast perfusion MR imaging at 3.0 T. Neuroimage. 2006, 33: 1029-1035.
Brooks DJ, Leenders KL, Head G, Marshall J, Legg NJ, Jones T: Studies on regional cerebral oxygen utilisation and cognitive function in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1984, 47: 1182-1191.
Raschid W, Parkes LM, Ingle GT, Chard DT, Toosy AT, Altmann DR, Symms MR, Tofts PS, Thompson AJ, Miller DH: Abnormalities of cerebral perfusion in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2004, 75: 1288-1293.
Costa DC, Tannock C, Brostoff J: Brainstem perfusion is impaired in chronic fatigue syndrome. QJM. 1995, 88: 767-773.
Fischler B, D’Haenen H, Cluydts R, Michiels V, Demets K, Bossuyt A, Kaufman L, De Meirleir K: Comparison of 99m Tc HMPAO SPECT scan between chronic fatigue syndrome, major depression and healthy controls: an exploratory study of clinical correlates of regional cerebral blood flow. Neuropsychobiol. 1996, 34: 175-183.
Schwartz RB, Garada BM, Komaroff AL, Tice HM, Gleit M, Jolesz FA, Holman BL: Detection of intracranial abnormalities in patients with chronic fatigue syndrome: comparison of MR imaging and SPECT. AJR Am J Roentgenol. 1994, 162: 935-941.
Ichise M, Salit IE, Abbey SE, Chung DG, Gray B, Kirsh JC, Freedman M: Assessment of regional cerebral perfusion by 99Tcm-HMPAO SPECT in chronic fatigue syndrome. Nucl Med Commun. 1992, 13: 767-772.
Tirelli U, Chierichetti F, Tavio M, Simonelli C, Bianchin G, Zanco P, Ferlin G: Brain positron emission tomography (PET) in chronic fatigue syndrome: preliminary data. Am J Med. 1998, 105: 54S-58S.
Il’ves AG, Prakhova LN, Kataeva GV, Rudas MS, Tomolian NA, Skoromets AA, Stoliarov ID: Changes of cerebral glucose metabolism in patients with multiple sclerosis and their role in formation of the clinical picture and progression of the disease. Zh Nevrol Psikhiatr Im SS Korsakova. 2003, 2: 53-60.
Roelcke U, Kappos L, Lechner-Scott J, Brunnschweiler H, Huber S, Ammann W, Plohmann A, Dellas S, Maguire RP, Missimer J, Radu EW, Steck A, Leenders KL: Reduced glucose metabolism in the frontal cortex and basal ganglia of multiple sclerosis patients with fatigue A 18F fluorodeoxyglucose positron emission tomography study. Neurology. 1997, 48: 1566-1571.
Paulasu E, Perani D, Fazio F, Comi G, Pozzilli C, Martinelli V, Filippi M, Bettinardi V, Sirabian G, Passafiume D, Anzini A, Lenzi GL, Canal N, Fieschi C: Functional basis of memory impairment in multiple sclerosis: a [18F]FDG PET study. Neuroimage. 1996, 4: 87-96.
Siessmeier T, Nix W, Hardt J, Schreckenberger M, Egle U, Bartenstein P: Observer independent analysis of cerebral glucose metabolism in patients with chronic fatigue syndrome. J Neurol Neurosurg Psychiatry. 2003, 74: 922-928.
Neema M, Stankiewicz J, Arora A, Guss ZD, Bakshi R: MRI in multiple sclerosis: what’s inside the toolbox?. Neurotherapeutics. 2007, 4: 602-617.
Kohler SJ, Yen Y, Wolber J, Chen AP, Albers MJ, Bok R, Zhang V, Tropp J, Nelson S, Vigneron DB, Kurhanewicz J, Hurd RE: In vivo 13 carbon metabolic imaging at 3T with hyperpolarized 13C-1-pyruvate. Magn Reson Med. 2007, 58: 65-69.
Dousset V, Brochet B, Deloire MS, Lagoarde L, Barroso B, Caille JM, Petry KG: MR imaging of relapsing multiple sclerosis patients using ultra-small-particle iron oxide and compared with gadolinium. Am J Neuroradiol. 2006, 27: 1000-1005.
Chen JW, Querol Sans M, Bogdanov A, Weissleder R: Imaging of myeloperoxidase in mice by using novel amplifiable paramagnetic substrates. Radiology. 2006, 240: 473-481.
Bakshi R, Dmochowski J, Shaikh ZA, Jacobs L: Gray matter T2 hypointensity is related to plaques and atrophy in the brains of multiple sclerosis patients. J Neurol Sci. 2001, 185: 19-26.
Laule C, Vavasour IM, Kolind SH, Traboulsee AL, Moore GR, Li DK, Mackay AL: Long T2 water in multiple sclerosis: what else can we learn from multi-echo T2 relaxation?. J Neurol. 2007, 254: 1579-1587.
Bo L, Geurts JJ, Mork SJ, van der Valk P: Grey matter pathology in multiplesclerosis. Acta Neurol Scand Suppl. 2006, 183: 48-50.
Miller DH, Soon D, Fernando KT, MacManus DG, Barker GJ, Yousry TA, Fisher E, O’Connor PW, Phillips JT, Polman CH, Kappos L, Hutchinson M, Havrdova E, Lublin FD, Giovannoni G, Wajgt A, Rudick R, Lynn F, Panzara MA, Sandrock AW, AFFIRM Investigators: MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology. 2007, 68: 1390-1401.
Horakova D, Kalincik T, Dusankova JB, Dolezal O: Clinical correlates of grey matter pathology in multiple sclerosis. BMC Neurol. 2012, 12: 10.
Prinster A, Quarantelli M, Orefice G, Lanzillo R, Brunetti A, Mollica C, Salvatore E, Morra VB, Coppola G, Vacca G, Alfano B, Salvatore M: Grey matter loss in relapsing-remitting multiple sclerosis: a voxel-based morphometry study. Neuroimage. 2006, 29: 859-867.
Morgen K, Sammer G, Courtney SM, Wolters T, Melchior H, Blecker CR, Oschmann P, Kaps M, Vaitl D: Evidence for a direct association between cortical atrophy and cognitive impairment in relapsing-remitting MS. Neuroimage. 2005, 30: 891-898.
Okada T, Tanak M, Kuratsune H, Watanabe Y, Sadato N: Mechanisms underlying fatigue: a voxel-based morphometric study of chronic fatigue syndrome. BMC Neurol. 2004, 4: 14.
Barnden LR, Crouch B, Kwiatek R, Burnet R, Mernone A, Chryssidis S, Scroop G, Del Fante P: A brain MRI study of chronic fatigue syndrome: evidence of brainstem dysfunction and altered homeostasis. NMR Biomed. 2011, 24: 1302-1312.
Puri BK, Jakeman PM, Agour M, Gunatilake KD, Fernando KA, Gurusinghe AI, Treasaden IH, Waldman AD, Gishen P: Regional grey and white matter volumetric changes in myalgic encephalomyelitis (chronic fatigue syndrome): a voxel-based morphometry 3-T MRI study. Br J Radiol. 2011, 85: e270-e273.
Natelson BH, Cohen JM, Brassloff I, Lee HJ: A controlled study of brain magnetic resonance imaging in patients with the chronic fatigue syndrome. J Neurol Sci. 1993, 120: 213-217.
Lange G, DeLuca J, Maldjian JA, Lee H, Tiersky LA, Natelson BH: Brain MRI abnormalities exist in a subset of patients with chronic fatigue syndrome. J Neurol Sci. 2005, 171: 3-7.
Matthews PM, Francis G, Antel J, Arnold DL: Proton magnetic resonance spectroscopy for metabolic characterization of plaques in multiple sclerosis. Neurology. 1991, 41: 1251-1256.
Butteriss DJA, Ismail A, Ellison DW, Birchall D: Use of serial proton magnetic resonance spectroscopy to differentiate low grade glioma from tumefactive plaque in a patient with multiple sclerosis. BJR. 2003, 76: 662-665.
Hattingen E, Magerkurth J, Pilatus U, Hubers A, Wahl M, Ziemann U: Combined (1)H and (31)P spectroscopy provides new insights into the pathobiochemistry of brain damage in multiple sclerosis. NMR Biomed. 2011, 24: 536-546.
Inglese M, Liu S, Babb JS, Mannon LJ, Grossman RI, Gonen O: Three-dimensional proton spectroscopy of deep gray matter nuclei in relapsing-remitting MS. Neurology. 2004, 63: 170-172.
Gustafsson MC, Dahlqvist O, Jaworski J, Lundberg P, Landtblom AM: Low choline concentrations in normal-appearing white matter of patients with multiple sclerosis and normal MR imaging brain scans. AJNR Am J Neuroradiol. 2007, 28: 1306-1312.
Richards TL: Proton MR spectroscopy in multiple sclerosis: value in establishing diagnosis, monitoring progression, and evaluating therapy. Am J Roentgenol. 1991, 157: 1073-1078.
Davie CA, Hawkins CP, Barker GJ, Brennan A, Tofts PS, Miller DH, McDonald WI: Serial proton magnetic resonance spectroscopy in acute multiple sclerosis lesions. Brain. 1994, 117: 49-58.
Simone IL, Federico F, Trojano M, Tortorella C, Liguori M, Giannini P, Picciola E, Natile G, Livrea P: High resolution proton MR spectroscopy of cerebrospinal fluid in MS patients. Comparison with biochemical changes in demyelinating plaques. J Neurol Sci. 1996, 144: 182-190.
Brooks JC, Roberts N, Whitehouse G, Majeed T: Proton magnetic resonance spectroscopy and morphometry of the hippocampus in chronic fatigue syndrome. Br J Radiol. 2000, 73: 1206-1208.
Chaudhuri A, Condon B, Gow J, Brennan D, Hadley D: Proton magnetic resonance spectroscopy of basal ganglia in chronic fatigue syndrome. Brain Imaging. 2003, 14: 225-228.
Puri BK, Counsell SJ, Zaman R, Main J, Collins AG, Hajnal JV, Davey NJ: Relative increase in choline in the occipital cortex in chronic fatigue syndrome. Acta Psychiatr Scand. 2002, 106: 224-226.
Tomoda A, Miike T, Yamada E, Honda H, Moroi T, Ogawa M, Ohtani Y, Morishita S: Chronic fatigue syndrome in childhood. Brain Dev. 2001, 22: 60-64.
Sharma KR, Kent-Braun J, Mynhier MA, Weiner MW, Miller RG: Evidence of an abnormal intramuscular component of fatigue in multiple sclerosis. Muscle Nerve. 1995, 18: 1403-1411.
Steens A, de Vries A, Hemmen J, Heersema T, Heerings M, Maurits N, Zijdewind I: Fatigue perceived by multiple sclerosis patients is associated with muscle fatigue. Neurorehabil Neural Repair. 2012, 26: 48-57.
Flachenecker P, Bihler I, Weber F, Gottschalk M, Toyka KV, Rieckmann P: Cytokine mRNA expression in patients with multiple sclerosis and fatigue. Mult Scler. 2004, 10: 165-169.
Goebel MU, Baase J, Pithan V, Exton M, Saller B, Schedlowski M, Limmroth V: Acute interferon beta-1b administration alters hypothalamic-pituitary-adrenal axis activity, plasma cytokines and leukocyte distribution in healthy subjects. Psychoneuroendocrinol. 2002, 27: 881-892.
Hautecoeur P, Forzy G, Gallois P, Demirbilek V, Feugas O: Variations of IL2, IL6, TNF alpha plasmatic levels in relapsing remitting multiple sclerosis. Acta Neurol Belg. 1997, 97: 240-243.
Pokryszko-Dragan A, Frydecka I, Kosmaczewska A, Ciszak L, Bilinska M, Gruszka E, Podemski R, Frydecka D: Stimulated peripheral production of interferon-gamma is related to fatigue and depression in multiple sclerosis. Clin Neurol Neurosurg. 2012, 114: 1153-1158.
Heesen C, Nawrath L, Reich C, Bauer N, Schulz KH, Gold SM: Fatigue in multiple sclerosis: an example of cytokine mediated sickness behaviour?. J Neurol Neurosurg Psychiatry. 2006, 77: 34-39.
Sharma KR, Kent-Braun J, Mynhier MA, Weiner MW, Miller RG: Evidence of an abnormal intramuscular coponet of fatigue in multiple sclerosis. Muscle Nerve. 2009, 18: 1403-1411.
Konecny L, Pospisil P, Dufek M, Drlikova L, Anbais FH, Erajhi AA, Dobsak P, Vank P, Siegelova J: Functional impairment in multiple sclerosis. Scripta Medica (BRNO). 2007, 80: 225-232.
Lenman AJ, Tulley FM, Vrbova G, Dimitrijevic MR, Towle JA: Muscle fatigue in some neurological disorders. Muscle Nerve. 1989, 12: 938-942.
Kent-Braun JA, Sharma KR, Miller RG, Weiner MW: Postexercise phosphocreatine resynthesis is slowed in multiple sclerosis. Muscle Nerve. 1994, 17: 835-841.
Kent-Braun JA, Ng AV, Castro M, Weiner MW, Gelinas D, Dudley GA, Miller RG: Strength, skeletal muscle composition, and enzyme activity in multiple sclerosis. J Appl Physiol. 1997, 83: 1998-2004.
Petajan JH, White AT: Motor-evoked potentials in response to fatiguing grip exercise in multiple sclerosis. Clin Neurophysiol. 2000, 111: 2188-2195.
Leocani L, Rovaris M, Boneschi FM, Medaglini S, Rossi P, Martinelli V, Amadio S, Comi G: Multimodal evoked potentials to assess the evolution of multiple sclerosis: a longitudinal study. J Neurol Neurosurg Psychiatry. 2006, 77: 1030-1035.
Di Lazzaro V, Oliviero A, Profice P, Ferrara L, Saturno E, Pilato F, Tonali P: The diagnostic value of motor evoked potentials. Clin Neurophysiol. 1999, 110: 1297-1307.
Colombo B, Martinelli Boneschi F, Rossi P, Rovaris M, Maderna L, Filippi M, Comi G: MRI And motor evoked potential findings in nondisabled multiple sclerosis with and without fatigue. J Neurol. 2000, 247: 506-509.
Zeller D, Aufm Kampe K, Biller A, Stefan K, Gentner R, Schutz A, Bartsch A, Bendszus M, Toyka KV, Rieckmann P, Classen J: Rapid-onset central motor plasticity in multiple sclerosis. Neurology. 2010, 74: 728-735.
Ng AV, Miller RG, Gelinas D, Kent-Braun JA: Functional relationships of central and peripheral muscle alterations in multiple sclerosis. Muscle Nerve. 2004, 29: 843-852.
Kuspinar A, Andersen RE, Teng SY, Asano M, Mayo NE: Predicting exercise capacity through submaximal fitness tests in persons with multiple sclerosis. Arch Phys Med Rehabil. 2010, 91: 1410-1417.
Savci S, Inal-Ince D, Arikan H, Guclu-Gunduz A, Cetisli-Korkmaz N, Armutlu K, Karabudak R: Six-minute walk distance as a measure of functional exercise capacity in multiple sclerosis. Disabil Rehabil. 2005, 27: 1365-1371.
Bakshi R, Miletich RS, Kinkel PR, Emmet ML, Kinkel WR: High-resolution fluorodeoxyglucose positron emission tomography shows both global and regional cerebral hypometabolism in multiple sclerosis. J Neuroimaging. 1998, 8: 228-234.
Bakshi R, Shaikh ZA, Miletich RS, Czarnecki D, Dmochowski J, Henschel K, Janardhan V, Dubey N, Kinkel PR: Fatigue in multiple sclerosis and its relationship to depression and neurologic disability. Mult Scler. 2000, 6: 181-185.
Blinkenberg M, Rune K, Jensen CV, Ravnborg M, Kyllingsbaek S, Holm S, Paulson OB, Sorensen PS: Cortical cerebral metabolism correlates with MRI lesion load and cognitive dysfunction in MS. Neurology. 2000, 54: 558-564.
Murai H, Kiyosawa M, Suzuki Y, Mizoguchi S, Ishii K, Ishikawa K, Akashi T: A case of multiple sclerosis with homonymous hemianopia examined by positron emission tomography. Jpn J Ophthalmol. 2004, 48: 591-593.
De Keyser J, Steen C, Mosert JP, Koch MW: Hypoperfusion of the cerebral white matter in multiple sclerosis: possible mechanisms and pathophysiological significance. J Cereb Blood Flow Metabol. 2008, 28: 1645-1651.
Zamboni P, Menegatti E, Weinstock-Guttman B, Dwyer MG, Schirda CV, Malagoni AM, Hojnacki D, Kennedy C, Carl E, Bergsland N, Magnano C, Bartolomei I, Salvi F, Zivadinov R: Hypoperfusion of brain parenchyma is associated with the severity of chronic cerebrospinal venous insufficiency in patients with multiple sclerosis: a cross-sectional preliminary report. BMC Med. 2011, 9: 22.
Tang PPZ, White AT, Topaz S, Petajan JH: Abstract. Muscle energy metabolism in multiple sclerosis measured by in vivo 31P Mrs 476. Med Sci Sports Exerc. 1997, 29: 83.
Hu J, Xu Y, Jiang Q, Sehgal V, Shen Y, Xuan Y, Xia Y: Spectral pattern of total creatine and trimethyl ammonium in multiple sclerosis. Magn Reson Imaging. 2004, 22: 427-429.
Inglese M, Park SJ, Johnson G, Babb JS, Miles L, Jaggi H, Herbert J, Grossman RI: Deep gray matter perfusion in multiple sclerosis: dynamic susceptibility contrast perfusion magnetic resonance imaging at 3 T. Arch Neurol. 2007, 64: 196-202.
Sacco P, Hope PA, Thickbroom GW, Byrnes ML, Mastaglia FL: Corticomotor excitability and perception of effort during sustained exercise in chronic fatigue syndrome. Clin Neurophysiol. 1999, 110: 1883-1891.
Samii A, Wassermann E, Ikoma K, Mercuri B, George MS, O’Fallon A, Dale JK, Straus SE, Hallett M: Decreased postexercise facilitation of motor evoked potentials with chronic fatigue or depression. Neurology. 1996, 47: 1410-1414.
Samii A, Wassermann EM, Ikoma K, Mercuri B, Hallett M: Characterization of postexercise facilitation and depression of motor evoked potentials to transcranial magnetic stimulation. Neurology. 1996, 46: 1376-1382.
Starr A, Scalise A, Gordon R, Michalewski H, Caramia M: Motor excitability in chronic fatigue syndrome. Clin Neurophysiol. 2000, 111: 2025-2031.
Chaudhuri A, Behan P: Fatigue and basal ganglia. J Neurol Sci. 2000, 179: 34-42.
Chaudhuri A, Behan P: Neurological dysfunction in chronic fatigue syndrome. JCFS. 2000, 6: 51-68.
Schillings ML, Kalkman JS, van der Werf SP, van Engelen BG, Bleijenberg G, Zwartz MJ: Diminished central activation during maximal voluntary contraction in chronic fatigue syndrome. Clin Neurophysiol. 2004, 115: 2518-2524.
Kent-Braun JA, Sharma KR, Weiner MW, Massie B, Miller RG: Central basis of muscle fatigue in chronic fatigue syndrome. Neurology. 1993, 43: 125-131.
Newham DJ, Davies JM, Mayston MJ: Voluntary force generation and activation in the knee muscles of stroke patients with mild spastic hemiparesis. J Physiol. 1995, 483: 128.
Kent-Braun JA, Miller RG: Central fatigue during isometric exercise in amyotrophic lateral sclerosis. Muscle Nerve. 2000, 2000: 909-914.
Davey NJ, Puri BK, Nowicky AV, Main J, Zaman R: Voluntary motor function in patients with chronic fatigue syndrome. J Psychosom Res. 2001, 50: 17-20.
Davey NJ, Puri BK, Catley M, Main J, Nowicky AV, Zaman R: Deficit in motor performance correlates with changed corticospinal excitability in patients with chronic fatigue syndrome. Int J Clin Pract. 2003, 57: 262-264.
Araque A: Astrocyte-neuron signaling in the brain-implications for disease. Curr Opin Investig Drugs. 2006, 7: 619-624.
Chvatal A, Anderov M, Neprasova H, Prajerova I, Benesova J, Butenko O, Verkhratsky A: Pathological potential of astroglia. Physiol Res. 2008, 57: S101-S110.
Yoshiuchi K, Farkas J, Natelson BH: Patients with chronic fatigue syndrome have reduced absolute cortical blood flow. Clin Physiol Funct Imaging. 2006, 26: 83-86.
Unger ER, Miller AH, Jones JF, Drake DF, Tian H, Pagnoni G: Decreased basal ganglia activation in chronic fatigue syndrome subjects is associated with increased fatigue. FASEB J. 2012, 26: 1035.20.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
No specific funding was obtained for this review. The authors declare that they have no competing interests.
Authors’ contributions
GM and MM participated in the design of this review and contributed equally to this paper. All authors read and approved the final version.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Morris, G., Maes, M. Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med 11, 205 (2013). https://doi.org/10.1186/1741-7015-11-205
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-11-205