
Abstract
Glioblastoma multiforme (GBM) is a fatal brain tumor in adults with a bleak diagnosis. Expansion of immunosuppressive and malignant CD4 + FoxP3 + GITR + regulatory T cells is one of the hallmarks of GBM. Importantly, most of the patients with GBM expresses the tryptophan-degrading enzyme indoleamine 2,3-dioxygenase (IDO). While IDO1 is generally not expressed at appreciable levels in the adult central nervous system, it is rapidly stimulated and highly expressed in response to ongoing immune surveillance in cancer. Increased levels of immune surveillance in cancer are thus related to higher intratumoral IDO expression levels and, as a result, a worse OS in GBM patients. Conversion of the important amino acid tryptophan into downstream catabolite known as kynurenines is the major function of IDO. Decreasing tryptophan and increasing the concentration of immunomodulatory tryptophan metabolites has been shown to induce T-cell apoptosis, increase immunosuppressive programming, and death of tumor antigen-presenting dendritic cells. This observation supported the immunotherapeutic strategy, and the targeted molecular therapy that suppresses IDO1 activity. We review the current understanding of the role of IDO1 in tumor immunological escape in brain tumors, the immunomodulatory effects of its primary catabolites, preclinical research targeting this enzymatic pathway, and various issues that need to be overcome to increase the prospective immunotherapeutic relevance in the treatment of GBM malignancy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is the most common and deadly malignant brain tumor in adults and children, accounting for more than 45.2 percent of all primary brain and CNS malignancies. Despite multimodal conventional treatment techniques such as surgical resection, radiation, and systemic therapy with adjuvant temozolomide, GBM remains an incurable malignancy with an average survival of less than 2 years [1, 2]. These dismal reports provide a strong incentive for the development of more effective therapies for GBM patients. Immunotherapy, which uses the patient's immune system to direct immune cells to the tumor while causing high rates of side effects, has attracted much attention. Immunotherapy primarily addresses peptide vaccines, dendritic cell vaccines, CAR-T and CAR-NK cell therapy, and immune checkpoint inhibition [3]. Patients with GBM are among the cancer types that do not respond to these approaches and generally have poor outcomes, so they have not benefited from these approaches to date. Recent research suggests that indoleamine 2,3-dioxygenase (IDO), a rate-limiting enzyme of tryptophan (Trp) metabolism that catalyzes the oxidation of this important amino acid, may be involved in such immunological escape via local Trp depletion and the formation of the lethal Trp catabolites [4, 5]. Higher expression of IDO is seen in 90% of glioma cells and has been associated with tumor development and poor survival rates. In addition to GBM cancer, the majority of studies indicated increased IDO expression in malignancies such as malignant melanoma, liver cancer, prostate, colon, and pancreatic cancer [6, 7]. The central dogma underlying the cancer-induced immunosuppressive role of the enzyme IDO is related to the conversion of tryptophan to downstream metabolites known as kynurenines (Kyn), which are associated with inhibition of both adaptive and innate immune responses [7, 8]. Expression of IDO in tumor cells can impede an effective immune response against glioma cells by modulating the T-cell response not only through increased apoptosis of cytotoxic T lymphocytes (CTL) but also by converting naïve T cells into inducible immunosuppressive regulatory T cells (Tregs; CD4 + CD25 + FoxP3 +) [9, 10]. Although GBM cells typically do not express IDO, it is activated when GBM cells are identified by tumor-infiltrating T cells or natural killer cells and exposed to important anti-cancer cytokines such as IFN-γ and TNF-α [11]. In this scenario, increasing levels of GBM-infiltrating T and NK cells are associated with poorer overall survival of GBM patients, due to increased intratumoral expression of the enzyme IDO [8, 11]. This sequence of events suggests that pharmacological IDO enzyme or pathway inhibitors may be a promising therapeutic target for improving cancer patient survival.
IDO in tumor immunological escape from cell immunity
IDO is an enzyme with a prosthetic hemegroup (Fe2+) present in some tumors and tumor-infiltrating cells (macrophages, dendritic cells (DCs), and B cells) that catalyzes the first step of tryptophan catabolism. IDO converting L-tryptophan to N-formylkynurenine (NFK), which is then rapidly converted to Kyn by formamidase [12, 13]. IDO competent DCs (IDO+ DCs) are a type of human DCs that induces immunological tolerance through a number of mechanisms and are crucial in the establishment and maintenance of a tumor-suppressing microenvironment. DCs are not directly affected by Trp deficiency and kynurenine accumulation, but Tregs may limit their physiological function [14, 15]. However, recent studies have shown that due to tryptophan deprivation and the accumulation of immunomodulatory tryptophan metabolites, IDO+ DCs from tumor-draining lymph nodes (TDLNs) are dominant activators of quiescent CD4 + CD25 + Foxp3 + Tregs as well as, direct suppressors of effector T cells (112, 113). Nevertheless, DCs from normal lymph nodes and spleen do not express IDO [16]. Studies have shown that IDO activation or increased kynurenine metabolites are correlated with poor clinical outcomes and increased tumor grade in patients with various malignancies, including malignant gliomas [17, 18]. Higher expression of IDO in gliomas, leading to an influx of immunosuppressive cells such as Treg cells. The accumulation of Treg cells can worsen the prognosis of gliomas by enhancing angiogenic remodeling of the tumor microenvironment, suppressing tumor antigen-specific immune responses, and leading to the death of effector T cells or possibly tumor antigen-presenting DCs [9, 19]. In contrast, downregulation of IDO reduces Treg cell accumulation while increasing T-cell-mediated anticancer effects. In addition to Treg accumulation, molecules such as IL-10, TGF-b, IL-35, and other factors have the potential to be extremely hostile to immunological CD8+ effector T cells and render fully armed specific killer CD8+ T cells impotent. Normally IDO expression may cause T cells to develop antigen-specific tolerance [20, 21]. When tumors promote aberrant IDO expression, this physiological effect may become pathogenic [21]. In most patient studies, higher IDO activity correlates not only with poor prognosis but also with accelerated DNA repair and resistance to treatments such as immune checkpoint inhibitors (anti-PD-1), ipilimumab (a monoclonal antibody against CTLA-4), NAD+ inhibitors, and chemotherapeutic agents such as cisplatin and paclitaxel [22, 23]. Moreover, treatment with anti-PD -1 increases tryptophan degradation via activation of IDO, which follows the release of interferon γ (IFN-γ) as well as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and other cytokines by tumor-infiltrating lymphocytes [24]. Although these factors have an antitumor effect, they also play an important role in cancer development, recurrence, and metastasis through the kynurenine pathway. Increased production of inflammatory cytokines by infiltrating lymphocytes is critical for GBM progression and treatment resistance [25]. This can be interpreted as a major cause of disappointing outcomes in patients with GBM when PD-1 is used as monotherapy. Therefore, there is now an additional molecular rationale for combining PD-(L)1 blockade with IDO inhibition in GBM immunotherapies [24, 26]. Interestingly, Han et al. demonstrated that inhibition of IDO alone or in combination with the conventional treatment such as chemotherapy (temozolomide), had no appreciable effect on overall survival in models with glioma. In contrast, the combined effect of inhibition of CTLA-4, PD-L1, and IDO in established glioma models indicated a substantial decrease in Treg production that was associated with improved survival [27, 28]. However, the response of glioma cells to IDO inhibitors and their adverse effects on other glioma treatments have not been thoroughly investigated. Recently, TDO-2 was found to exert immunoregulatory effects comparable to IDO -1 [29, 30]. Similar to IDO, TDO catalyzes an analogous reaction, namely the oxidation of tryptophan to N-formyl kynurenine. Although, unlike IDO, which is normally expressed in extrahepatic tissues such as the brain and lymph nodes, TDO is mainly expressed in the liver and has a much lower expression level than IDO1, limiting their functions [31]. Moreover, compared with IDO enzymes, TDO enzymes remain unaffected by immune system responses and exclusively use L-tryptophan as a substrate [29]. The key distinguishing features of TDO and IDO were shown in Table 1.
Increased IDO1 and TDO2 expression within the tumor is associated with worse overall survival (OS) in glioma patients [9]. Activation of IDO and the action of enzymes in the kynurenine pathway, the major pathway for Trp degradation in primates, have counterregulatory effects by maintaining homeostasis, suppression of cytotoxic T cells, expansion of Tregs, inhibition of NK cells, and differentiation of immature DCs to a Treg-inducing or tolerogenic status [24, 32]. The primary dogma linking IDO1 and IDO2 (the two major types of IDO genes in mammals) to immune cell suppression is their individual and/or collective activity in local Trp depletion [33]. Moreover, the formation of tryptophan metabolites such as quinolinic acid (QUIN) is one of the processes that may be responsible for the numerous effects observed after IDO stimulation, including Tregs proliferation and worse survival [33, 34]. TRP depletion following activation of IDO has recently become a focus of interest because of its immunosuppressive effects on innate and adaptive immune responses. This dogma assumes that TRP depletion enhances activation of General Control Non-Depressible 2 (GCN2) kinases, which are triggered when Trp deprivation raises uncharged tRNA levels, leading to T-cell dysfunction in vivo via GCN2-activated plasmacytoid DCs that would block an efficient immunological attack on glioma cells [35]. Deprivation of Trp inhibits the molecular target of rapamycin (mTOR) and protein kinase C (PKC) in tumor cells and promotes T-cell autophagy and T-lymphocyte anergy, according to Metz and colleagues [36, 37]. In addition, Lee et al. investigated the biological effects of TRP on T lymphocytes by culturing them in trp-free media and found that naive T lymphocytes activated in trp-free media did not differentiate into cytotoxic effector T cells and cell cycle arrest them in the mid-G1 phase, as well as showing an increased tendency to die by apoptosis [38, 39]. The increased apoptosis or cell cycle arrest of effector T cells in the mid-G1 phase and the greater differentiation of naïve T cells to CD4+ T cells may be due to accumulation of uncharged Trp-tRNA, leading to activation of GCN2 and subsequent phosphorylation and inactivation of eIF2α. Activation of GCN2 and inactivation of eIF2α reduce RNA transcription and protein translation [40]. All these elements come together to create an immunosuppressive tumor environment that supports immune evasion of tumor cells. In this case, IDO inhibition could limit the immune escape of glioma cells from T cells and would be a suitable complementary treatment to combat malignant gliomas. Besides the trp depletion, Trp breakdown may potentially inhibit immune cell activity by forming Kyn and other subsequent metabolites. KYN and its derivatives are physiologically active substances and are effective in tumor progression and recurrence (Fig. 1). KYN is the potent trigger of the KYN pathway (KP), which splits into two sub-branches. The three primary immunomodulatory catabolites formed in KP include quinolinic acid (QUIN), kynurenic acid (KYNA), and picolinic acid (PIC), which are collectively referred to as kynurenines and have received more attention in modulating both innate and adaptive immune responses[41, 42]. Each of these kynurenines could potentially entail different changes in the immunological milieu that could result in immune effector cells becoming more or less active through different processes. QUIN, an N-methyl-D-aspartate (NMDA) receptor exciter, has a neurotoxic effect and causes cell membrane rupture due to oxidative stress. It also stimulates neurons to produce nitric acid synthase, which increases free radical production and leads to neurotoxicity in complex processes, especially in the striatum [43, 44]. KYNA and PIC have various neuroprotective, immunosuppressive, and antiproliferative effects on tumor cells. They act as a competitive antagonist of the NMDA receptor and stimulator of macrophages to synthesize MIP1 α and 1β (macrophage inflammatory protein) respectively [45]. KYNA has also been demonstrated to have a role in a variety of biological activities, including a potent scavenger of reactive oxygen species, serving as a ligand for GPR35 (G-protein coupled receptor 35), and as an agonist for HCAR3 (hydroxycarboxylic acid receptor 3) [46, 47].

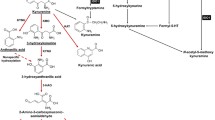
IDO1/TDO-mediated tryptophan catabolism. A;The amino acid tryptophan is converted to the immunosuppressive catabolite kynurenine via IDO /TDO and formidase. Higher overexpression of IDO /TDO in cancer cells leads to accumulation of kynurenine and its metabolites and to a decrease in the amount of the amino acid tryptophan in cancer cells. The accumulation of kynurenine and Trp deficiency lead to the production of Tregs and immunosuppressive molecules such as interleukins and TGF-b, which has many destructive effects on the function of cells of the immune system, such as death of effector CD8 + T cells, inhibition of NK cells, and differentiation of naive DCs toward tolerogenic status. B; Trp deprivation increases the level of uncharged tRNA, which activates GCN2 kinases by phosphorylation, inhibits mTOR, PKC, and STAT3 in tumor cells, promotes T-cell autophagy associated with T-lymphocyte anergy, and induces the formation of Tregs. IDO indoleamine 2, 3-dioxygenase, Tregs regulatory T cells, TGF-b transforming growth factor beta, DCs dendritic cells, GCN2 general control nonderepressible 2, mTOR mechanistic target of rapamycin kinase, PKC protein kinase C, STAT3 signal transducer and activator of transcription 3
T-cell apoptosis by quinolinic acid (QUIN)
Among the other kynurenines, QUIN is a potential excitotoxin in the CNS that can cause selective neuronal lesions by necrosis and/or apoptosis at considerably high micromolar doses, especially in the more sensitive brain regions such as the hippocampus and striatum [48, 49]. However, the neurotoxicity associated with tryptophan deficiency cannot be explained just by NMDA receptor overactivation induced by QUIN accumulation; other processes are likely involved. A high concentration of tryptophan metabolites such as 3-hydroxyanthranilic acid and quinolinic acid in the kynurenine pathway may interfere with immune surveillance of cancers by affecting T-cell responses [49]. In mouse thymocytes in vitro, QUIN modulated T-cell homeostasis by inducing programmed apoptosis of Th1 but not Th2 cells [49]. This process does not require death ligands or Fas interaction and was associated with changes in mitochondria and endoplasmic reticulum. Activation of a family of cysteine proteases known as caspases, particularly caspase-8 and -9, has been shown to play a critical role in triggering the apoptotic cascade. These caspases are the two major upstream caspases in the extrinsic and intrinsic signaling pathways of cell death [50]. Activation of the caspases and subsequent responses are triggered by the release of cytochrome c from mitochondria. This type of apoptosis is characterized by an increase in Bax and a decrease in Bcl-2 protein levels, resulting in fewer Bax: Bcl-2 heterodimers. In addition to apoptosis triggering activity of QUIN, other physiopathological mechanisms may occur in a number of neurodegenerative diseases with high QUIN levels [50, 51].
QUIN stimulates tumor cell cycling and supports the development of concurrent resistance to multiple structurally and mechanistically distinct drugs in chemotherapy regimens [52, 53]. Long-term exposure with QUIN also increases tau phosphorylation (a neuronal microtubule-associated protein whose major biological function is to stabilize already formed microtubules) at multiple sites in human neurons, as well as phosphorylation of the low-molecular-weight neurofilament subunit (NFL) in the head domain, which disrupts neurofilament assembly in vivo in neurons [53, 54]. Therefore, Cytoskeletal homeostasis is disrupted by prolonged exposure to QUIN, which dysregulates intermediate filament accumulation in both glial and neuronal cells; this process is supported by Ca2+ influx through NMDA channels and L-type voltage-gated Ca2+ channels (L-VDCC) [53].
However, the concept that the accumulation of QUIN contributes to tumorigenicity in GBM is neither proven nor disproven. Recent studies have shown that the production of QUIN in GBM patients is significantly lower than in control patients, the damaging burden on brain cells may be negligible [55]. This unexpected finding is thought to indicate that QUIN is degraded more rapidly in GBM patients than in control subjects, resulting in higher NAD+ levels [55]. Adams et al. found that glioma cells have greater expression of quinolinate phosphoribosyltransferase (QPRT) as the major enzyme that catalyzes the degradation of QUIN to NAD+ than do control subjects. This could favor tumor cell survival and proliferation and increase resistance to oxidative stress-induced by radiochemotherapy, leading to a worse prognosis [55,56,57]. The high synthesis rates of NAD+ improve the availability of substrate for DNA repair enzymes, which help in DNA replication and repair in tumor cells and support the maintenance of high energy metabolism [56, 58]. These results shed light on treatments that induce intracellular NAD+ deprivation, such as alkylating compounds or direct NAD+ synthesis blockers and point to QPRT as a potential therapeutic candidate in malignant gliomas [58].
Immunomodulatory properties of KYNA
Kynurenic acid (KYNA) is an important neuroprotective metabolite in the breakdown of L-tryptophan (TRP) via the kynurenine pathway. The effect of KYNA on the immune response and its mechanisms in neurophysiological and neuropathological processes has received much attention in recent years [59]. KYNA is an endogenous antagonist of a variety of receptors, including the NMDA and AMPA receptors (a-amino-3-hydroxy-5-methyl-4-isoxazolepropionate) and the kainic acid receptors (KARs) via an allosteric glycine site [55, 60]. KYNA is also thought to be an endogenous ligand of the aryl hydrocarbon receptor (AHR) [61]. AhR activation increases Kyn production in a manner dependent on IDO-induction in which AhR binds to dendritic cell-responsive elements and increases Kyn production [62, 63]. Increased AhR is another element of the ubiquitin/proteasome system that leads to regulated proteolytic cleavage of IDO1 as well as other enzymes [63]. As a result, even at low concentrations, KYNA has a modulatory effect on neuronal activity in the brain. Fluctuations in KYNA levels in brain tissue have been associated with a number of neurological disorders. KYNA levels have been found to be decreased in neurological diseases such as Huntington's disease (HD) and Alzheimer's disease (AD) [64, 65]. On the other hand, KYNA is increased in diseases such as schizophrenia and various cancers such as lung cancer, glioblastoma, and non-small cell lung cancer (NSCLC) [66]. However, the potential interaction of KYNA with glioma cells has not yet been elucidated. According to the current state of research, KYNA overexpression was detected in all analyzed samples of collected adult glioblastoma tissue. Di Serio et al. observed that the growth rate of N11 (a mouse microglial cell line) and human glioma cells was dramatically increased in the presence of KYNA. When QUIN was added to the culture, the proliferation rate increased as well [67]. In contrast, another study by Walczak et al. showed contradictory results [68]. They showed that KYNA suppressed the growth of T98G (a human glioblastoma cell line) at various doses and also enhanced the anticancer effects of glutamate receptor antagonists while decreasing DNA synthesis [68].
Despite significant breakthroughs in therapy, GB remains the leading cause of death in people with brain tumors, with the only difference being the duration of survival [69]. One treatment option for GB may be the use of glutamate receptor antagonists. Glutamate plays a supporting role in the human CNS and influences both neuronal cells and cancer cells to promote efficient proliferation, migration, and survival; therefore, inhibition of glutamate receptors will limit the proliferation and migration of cancer cells [70, 71]. Rzeski et al. investigated the antitumor effect of glutamate antagonists on tumor cell properties such as division, migration, and invasion. They observed decreased division, migration, increased death, and altered appearance of cancer cells in a variety of malignancies, including colon and thyroid carcinomas and astrocytomas [72, 73]. They have also shown that the antiproliferative effect of glutamate antagonists in combination with routinely used antitumor drugs is greater in cancer treatment than glutamate antagonists or therapy alone [72].
Another compelling reason for glutamate receptor inhibition is that malignant gliomas have a limited amount of space in the skull as they grow. They stimulate the death of neighboring neurons to make room for tumor growth, which is driven by excessive glutamate release into the surrounding area. Excessive stimulation causes neurons to spasm and eventually die by apoptosis or necrosis, making room for tumor cell development [74]. Other studies have shown that synthetic AMPA antagonists such as GYKI and CFM -2 block ERK1/2 signaling in cancer cells, an internal pathway triggered by growth factors that derive the proliferation of lung cancer cells. These results shed light on the antitumor functions of AMPA antagonists, specifically on KYNA, implying that this class of agents might be effective for the treatment of human malignancies [75, 76]. According to studies, KYNA and the KYNA/KYN ratio are decreased in the plasma of GBM patients. The significantly lower KYNA synthesis and KYNA/KYN ratio in plasma of GBM patients compared with controls may indicate that KYNA catabolism is mainly directed to the neurotoxic QUIN pathway and eventually NAD+ [55]. These conflicting findings provide the basis and motivation for further research into the biological effects of KYNA in GBM.
Immunosuppressive functions of PIC
Picolinic Acid (PA) is a terminal metabolite of L-Trp metabolism, whose primary physiological role is not yet clear. The activity of a crucial enzyme called ACMS decarboxylase (ACMSD) regulates whether metabolites in the kynurenine pathway (KP) are converted to QUIN, a necessary substrate for the formation of NAD+, or PIC formation as a neuroprotective metabolite [77, 78]. Interestingly, ACMSD activity is negatively related to the amount of NAD+ formed from tryptophan (64). ACMSD, as a rate-limiting enzyme for PIC synthesis, requires a substrate for PIC synthesis, which depends mainly on IDO activity. Therefore, the pharmacological intervention of IDO will have an impact on PIC production. PIC and its physiological function may be studied to prevent the occurrence of probable PIC-related adverse effects after IDO inhibition [79, 80].
Previous research has shown that PA, a natural metal chelator molecule, inhibits the growth of cultured normal or transformed mammalian cells, as do iron chelators such as desferrioxamine, and is being studied for its anti-tumor activity when cultured together with tumor cells. TESTA et al. have shown that PA both acutely blocks iron uptake into cells in a dose-dependent manner and dramatically reduces intracellular ferritin levels. Thus, PA is thought to suppress cell growth by reducing the amount of iron accessible to cells [81]. Iron chelation has been associated with several biological effects of PA, such as transcriptional stimulation of iNOS production and subsequent synthesis of nitric oxide (NO) [82]. In addition to chelating iron, Suzuki et al. demonstrated that PA possesses strong chelation capabilities for a variety of divalent metals, including Ni, Zn, Cd, and Pb [83].
PA reduces QUIN and kainic acid neurotoxicity, which is thought to be dependent on glutamate uptake to exert its neurotoxic effects. The antiexcitotoxicity function of PA is unique in that this tryptophan metabolite appears to affect excitotoxicity without altering neuronal activity (Robinson et al.) [84]. PA may protect against the effects of some excitotoxins (QUIN and kainic acid) by interacting with and regulating glutamatergic release at the presynaptic level. Thus, glutamate release by the same terminals may be inhibited. However, the main mechanism by which PA inhibits the synaptic release of glutamate is unknown [85]. Based on previous findings, numerous hypotheses have been developed. PA has been shown to attenuate the calcium-dependent component of glutamate release but not the calcium-independent component. The greatest PA impact was very similar in magnitude to the inhibitory effect caused by a calcium deficiency in the media. As a result, PA limits glutamate release by interfering with the translocation of calcium ions to the release mechanism [85,86,87].
Picolinic acid has also been associated with immunological functions. There is evidence that PA is physiologically active on macrophages and can activate them to tumoricidal and antiviral activity [88]. In mice, this metabolite has previously been shown to stimulate the lytic activities of a peritoneal macrophage when administered in vivo. Varesio et al. have shown that PA can theoretically and specifically increase mRNA transcription of macrophage inflammatory proteins-Ia (MIP-Ia) and Iβ (MIP-I β) [89]. MIP-1a and MIP-1β are macrophage-secreted chemotactic CC chemokines with a common affinity for CCR5. These chemokines are essential mediators of inflammatory responses as well as chemoattractants for certain types of leukocyte groups that are necessary for the formation of effector immune responses [90, 91]. In addition, these chemokines have been shown to promote IFN-γ secretion through activation of the CCR5 receptor [89]. IFN-γ is an important stimulator of the inflammatory response that can induce macrophages to produce chemokines. Picolinic acid also interacts with IFN-γ to increase iNOS production by macrophages, resulting in potent cytotoxic/cytostatic effects [88, 92]. MIP-1a and MIP-1b are effective chemoattractants for specific T-cell subsets and therefore play an important role in recruiting T cells and directing them toward migration into sites of inflammation. T lymphocytes are divided into two types: CD4 (helper T cells) and CD8 (cytotoxic T cells). These cells have been shown to produce a large amount of Th-1 and Th-2 cytokines, respectively [93]. Th-1 cytokines are potent pro-inflammatory cytokines that are necessary for host defense against internal viral and bacterial infections. Th2-type cytokines include interleukin-4 (IL -4) and IL -10 (IL-10), which have a more anti-inflammatory effect and therefore attenuate the inflammation triggered by Th1-mediated microbicidal effects [94]. In general, MIP-1b recruits mainly activated CD4+ T lymphocytes (helpers), whereas MIP-1a selectively induces chemotaxis of predominantly CD8+ T cells. Therefore, PA and subsequent chemokine production play a crucial role in determining the direction of the immune system [93, 95]. In vivo studies in mice infected with MBL-2 tumor cells showed that treatment with PIC (100 mg/Kg) resulted in a 46% longer life expectancy compared with the control group. As mentioned earlier, these effects may be related to macrophage activation and subsequent induction of cytotoxicity in mouse peritoneal macrophages by PIC (Fig. 2) [96].

The main physiological activity of PIC in the human CNS. PIC is a terminal metabolite of L-tryptophan metabolism via the kynurenine pathway. The exact role of PIC is not known, but it has been reported to be involved in neuroprotective, immunological, and anti-proliferative functions. PIC acts as an iron chelator, causing iron to be unavailable for normal cell development. PIC also limits presynaptic glutamate release from nerve terminals, selectively modulating QUIN excitotoxicity, which depends on glutamate input. PIC has a dose-dependent negative regulatory function on CD4+ T-cell proliferation and the ability to generate profound anergy and suppress CD4+ T-cell polarization. IDO competent DCs generate an immunosuppressive milieu that converts local T-cell activity from immunogenic to tolerogenic. DCs expressing IDO secrete immunosuppressive cytokines that in turn block effector CD8+ T cells while inducing Tregs. IDO indoleamine 2, 3-dioxygenase, PIC picolinic acid, DCs dendritic cells, CNS central nervous system, QUIN quinolinic acid, Tregs regulatory T cells
A previous study has shown that ACMSD is downregulated in glioma cells compared with controls, which may redirect KP metabolism from the generation of PA to the production of QUIN and consequently to the production of NAD+, which enhances glioma cell survival and proliferation [55, 58]. This decrease in PIC levels in GBM patients would imply that KP is directed toward neurotoxicity rather than neuroprotection. PA may suppress the proliferation of effector T cells in addition to its immunoprotective properties. Recently, it was revealed that the harmony between the immunoprotective and immunosuppressive effects of Trp metabolites is strictly controlled by factors in the tumor microenvironment. These factors include IL-6, IL-2, CD40, IFN-γ, CTLA4, IL-21, IL-10, and TGF-b [97, 98] (Fig. 3).

IDO1 as a prognostic clinicopathological marker for monitoring invasiveness and progression of cancer. Overexpression of IDO1 leads to a decrease in tryptophan and an accumulation of kynurenine in cells and in the microenvironment, which is subsequently taken up into the bloodstream through the highly permeable BBB and increases the ratio of kynurenine to tryptophan ([Kyn]/[Trp]) in plasma. In addition, DCs in peripheral blood have been shown to contribute to higher Kyn/Trp serum levels by converting Trp to Kyn via the enzymatic activity of IDO. Several in vivo studies have associated an elevated systemic Kyn/Trp ratio with poor outcome and limited survival in GBM patients. Several techniques are used to determine the Kyn/Trp ratio in human serum. HPLC, GC, LC, MS, and HVPE are the best known methods for measuring the Kyn/Trp ratio. IDO indoleamine 2, 3-dioxygenase, BBB blood–brain barrier, HPLC high-performance liquid chromatography, GC gas chromatography, LC liquid chromatography, MS mass spectrometry, HVPE high-voltage paper electrophoresis
IDO1 in cancer diagnostics and therapy
Numerous studies have shown that the higher relative concentration of Kyn compared with Trp, and thus the Kyn/Trp ratio, is a promising surveillance biomarker for predicting the clinicopathologic status of tumors to monitor cancer invasion and progression [97, 99]. Considering the recent findings of enhanced Trp degradation, and the active transport of Trp products such as Kyn across the blood–brain barrier (BBB), as well as the immunomodulatory effects of IDO1-mediated Trp degradation, analysis of the Kyn/Trp ratio in the patient's peripheral blood has been considered as a potential marker of cancer progression and to optimize risk stratification and therapy for GBM patients [100, 101]. Moreover, the activity of IDO in non-tumor cells, such as DCs in peripheral blood, has been shown to contribute to higher serum Kyn/Trp levels[102]. Increased IDO1 mRNA and protein expression and a high Kyn/Trp ratio have been associated with poor prognosis and lower survival in patients with many cancers, including prostate, cervical, lung, and glioblastoma [11, 99]. According to various research findings, the K/T ratio could potentially be used as a surrogate prognostic marker in some cancers. The blood Kyn/Trp ratio increases in patients with advanced melanoma and renal cell carcinoma treated with nivolumab (an antibody against PD1), indicating an adaptive resistance mechanism associated with poorer survival [103].
Patients with GBM have a low survival rate of only 14.6 months, prompting the search for new effective therapies, including immunotherapy. However, identifying parameters that actively categorize GBM patients with the potential for treatment response has been challenging. Interestingly, the Kyn/Trp index in the serum of GBM patients appears to be an accurate predictor of enrollment in immunotherapeutic treatments [101]. In GBM patients treated with surgery and immunotherapy, both prognostic and predictive values of the serum Kyn/Trp ratio have been proposed [100, 101]. To date, several techniques have been developed to quantify Trp and seven of its KP metabolites (KYN, 3-HK, 3-HAA, KYNA, XANA, QUIN, and PA) in biological samples, including high-performance liquid chromatography (HPLC), gas chromatography (GC), liquid chromatography (LC), mass spectrometry (MS), and high-voltage paper electrophoresis (HVPE) [104]. Increasing data indicate that the LC–MS-based approach is the analytical gold standard for the detection of and determination of KP metabolites in body fluids and tissues [105].
IDO1 protein expression is high in a variety of cancer tissues, allowing them to evade immune surveillance. As a result, IDO1 may be a suitable therapeutic target for reducing immunosuppression and is thus a key element in cancer development. This notion has motivated the development of several IDO1 inhibitors through pharmacological, genetic, and immunological approaches, some of which are currently in clinical trials. In addition, numerous small-molecule agents and peptide vaccines targeting IDO1 have entered into clinical trials to evaluate their efficacy and safety in cancer treatment [37, 106]. To date, a considerable number of IDO1 inhibitors, including Epacadostat, KHK2455, BGB-5777, BMS-986205, indoximod, PF -06840003, have been tested in clinical trials for cancer therapy [106]. Single-agent blockade of IDO as monotherapy has not shown any effect on overall survival in patients with advanced GBM, suggesting that the strategy for efficient implementation of immunomodulatory therapies in GBM patients, such as combined immunotherapies, needs to be reevaluated [16, 17]. The agents of combined immunotherapies target different immune checkpoints, as has been done in preclinical research targeting CTLA-4 and PD-L1 in mice with malignant gliomas [16]. For example, Ladomersky et al. observed that the combination of irradiation with concomitant administration of an anti-PD -1 treatment followed by an IDO1 enzyme inhibitor increased T-cell recruitment and/or effector T-cell (CD8+) activity in GBM, resulting in effective immune-mediated anti-GBM activity [107, 108]. The combination of these agents, BGB-5777 (a novel IDO1 inhibitor but not TDO) to inhibit IDO1-mediated metabolism, irradiation, and PD -1 blockade dramatically increased survival in mice and resulted in long-term tumor control in 30–40% of mice with advanced GBM [108]. However, neither monotherapy nor dual therapies, such as radiation in combination with PD -1 mAb and IDO1 inhibitors, failed to increase the overall survival of animals with advanced GBM in a long-term way. In the following Table 2, we summarized the current clinical trials using IDO inhibitors as a component of clinical immunotherapy.
Table 2 of 2.
1-Methyl-D, L-tryptophan is another widely used IDO1 inhibitor that has a variety of complicated inhibitory effects. The D-isomer of 1-Methyl-D, L-tryptophan (indoximod) binds with and mostly inhibits IDO2, whereas the L-isomer interacts with and inhibits IDO1 [107]. Indoximod has received the most attention in clinical research because of its relatively potent antitumor properties and unique mechanisms of action. In contrast to direct IDO1 enzyme inhibitors, indoximod acts as a tryptophan mimic by reducing the inhibitory effects of IDO /TDO-mediated tryptophan deprivation on mTORC1 kinase [107, 109]. Downregulation of mTORC1 in T cells promotes autophagy/tolerance while reducing T-cell antitumor efficacy, which is reversed by indoximod as a tryptophan substitute. Recently, researchers revealed that employing IDO inhibitors as monotherapy to improve OS in cancer patients yielded poor results. Nevertheless, their full efficacy could be achieved in combination with other chemotherapeutic agents, including cyclophosphamide, doxorubicin, paclitaxel, cisplatin, and gemcitabine [110, 111]. In contrast to Indoximod, epacadostat binds directly to IDO1 and inhibits Trp degradation by cells in the tumor microenvironment [112]. Consistent with these findings, epacadostat not only promotes the growth of effector immune cells such as DCs (dendritic cells), NK cells, and T cells, but it also inhibits the development of T regulatory cells (Tregs) [113, 114]. Epacadostat generates a potent immune response by altering the abnormal signaling pathways of cancer cells and reducing kynurenine levels by 90% in both plasma and tumor tissue in mouse models [114]. In mouse models, studies showed that epacadostat in combination with other immune checkpoint inhibitors has greater efficacy in improving overall survival. Thus, prompting the initiation of several clinical trials, including NCT03196232, and NCT02327078, to evaluate the combination of epacadostat (INCB024360) with other immune checkpoint inhibitors such as pembrolizumab (mk-3475), and nivolumab [115]. NCT03196232, evaluated the efficacy, safety, and tolerability of epacadostat in combination with pembrolizumab in the treatment of patients with gastric cancer. It is assumed that a combination of epacadostat and anti-PD-1 antibodies (pembrolizumab or nivolumab) may be more effective in treating patients with gastric or melanoma cancer. It is worth noting that this combination improved response rate (RR) and progression-free survival (> 6 months) in patients with gastric cancer [106, 116]. Other IDO1 inhibitors, such as KHK2455, BMS-986205, and PF -06840003, are currently under investigation in preclinical and clinical trials for various tumor types [117].
IDO vaccination
In addition to the previously described IDO inhibitors, several peptide-based vaccines targeting IDO1 for cancer treatment have entered clinical trials to evaluate their efficacy and safety. Peptides" used in these vaccines are 20–30 amino acid sequences. They aim to induce a protective immune response in a naïve host by introducing immunodominant epitopes derived from tumor-specific antigens. IDO vaccines differ from IDO inhibitors in many ways. Interestingly, these vaccines have shown promising antitumor responses in patients with various malignancies by supporting the immune system, which response inefficiently to tumor antigens [118, 119]. Therapeutic cancer vaccines attempt to target antitumor immunity by targeting highly expressed cancer-related antigens [118, 120]. Recently, IDO has emerged as a potential vaccine target because it is expressed in a variety of cell types, including cancer cells, antigen-presenting cells (APCs), stomatal cells [121]. Compared with typical IDO inhibitors, IDO vaccines can be effective in tumors even when the cancer cells do not express the target antigen and have low HLA expression [122,123,124]. Recent research has revealed that vaccination with an IDO-derived HLA-A2 epitope might boost the antitumor response against IDO+cancer cells and increase their identification by CTLs [125]. All cancer vaccines, such as the IDO vaccine, attempt to increase the presentation of tumor-associated antigens by APCs to induce lengthy T-cell protection against MHC class I-antigen complexes on tumor cell surfaces. CD8+ T lymphocytes are the major players in cancer cell recognition and eradication. In vitro, these IDO-reactive T cells were able to recognize and destroy IDO-expressing cells, including tumor cells, as well as IDO-expressing regulatory DCs, which are among the most common immunosuppressive cell types [121]. In vitro, these IDO-reactive T cells were able to detect and destroy IDO-expressing cells including tumor cells, as well as IDO-expressing regulatory DCs, which are one of the most common immune-suppressive cell types [126]. In addition to suppressing IDO immunoregulatory functions, IDO-specific T cells also disrupt other pathways of immunosuppression driven by IDO+ target cells. As a result, IDO -specific T cells can alter the tumor microenvironment from favorable to hostile to tumor cells by enhancing the growth and activation of various immune cells such as DCs, NK cells, and effector T cells [126, 127]. Recently, it has been hypothesized that this IDO1-targeted therapeutic vaccine strategy may act synergistically when combined with other types of immunotherapy such as immune checkpoint inhibitors and other immunomodulatory agents [128]. Several clinical trials have been conducted based on this hypothesis, and numerous patients are currently being recruited to evaluate the efficacy and safety of this combinational therapy. For instance, BJOERN et al. investigated the efficacy and safety of a combined therapy consisting of a peptide vaccine (21aa) derived from IDO with routine ipilimumab ( ipi) therapy in patients with stage III or IV malignant melanoma [129]. The peptide has several distinct HLA class I epitopes that are expected to bind many of the common HLA alleles, making tissue-type-based patient screening unnecessary [130]. They showed that the administration of the IDO peptide vaccine with ipi therapy resulted in low toxicity in the majority of patients, and the vaccine was generally safe and well-tolerated [129]. Vaccination elicited IDO T-cell responses observed ex vivo. However, administration of ipilimumab alone had no significant effect on clinical activity compared with combined administration in this cohort. Ipilimumab is a fully human immunoglobulin G1 antibody that targets CTLA-4 and inhibits its interaction with ligands such as CD80 and CD86. Ipilimumab has been shown to prolong overall survival in patients with high-risk stage melanoma III [131].
Interestingly, surgical resection of gliomas significantly improves the outcomes of immunologic interventions. In particular, vaccine treatment would be most efficient if administered in the early postoperative period or when tumor excision is nearly complete surgically [128]. Similarly, vaccination as adjuvant therapy after surgical excision in stage II and III melanoma patients inhibited tumor recurrence in a manner similar to how vaccines are used to prevent infection [132]. Several factors influence the overall response in cancer vaccine recipients, including host genetics, time to diagnosis, grading, and lifespan [133]. Some clinical responses may be observed within a few weeks, whereas other responses to vaccination may take longer, sometimes up to 6–12 months [132]. In addition to the peptide vaccines based on IDO, other peptide vaccines targeting specific antigens in GBM have also been developed. Tumor-specific antigens used in peptide vaccine development include EGFRvIII and heat shock proteins [134]. Despite promising results from the phase I/ II clinical trials, no successful phase III clinical trials of glioblastoma immunotherapies, including glioblastoma vaccines, have been achieved in recent years, and further research is needed to improve the efficacy of peptide-based vaccines in GBM immunotherapy.
Conclusion
Expression of IDO has been shown to be associated with suppression of adaptive T-cell responses, not only by enhancing CTL death but also by converting naïve T cells into immunosuppressive regulatory T cells, which correlates with tumorigenesis. In contrast, IDO insufficiency reduces Treg recruitment and increases T-cell-mediated tumor defense. IDO may serve as a broadly used target for cancer immunotherapies whose function and expression patterns are fundamentally different from previously identified antigens, and such IDO -based immunotherapy may function effectively in conjunction with other forms of immunotherapy. The fact that IDO-vaccines elicit CD4+ and CD8+ T cells as well as pro-inflammatory T cells in the tumor microenvironment that are active against IDO -expressing cancer cells underscores the efficacy of this novel vaccine-based approach in improving patient response rates to other conventional therapies. IDO1-Inhibitor treatment improves anti-tumor immune function and can be used in conjunction with other types of immunotherapy, including anti-tumor vaccines. Nevertheless, due to the immunosuppressive nature of the GBM microenvironment and the concept of IDO1's versatility to function as either an enzyme or a signal transducer, IDO1 is a difficult molecule to target. Therefore, future therapeutic approaches should consider such a pleiotropic effect and explore the possibility of combining it with other immunotherapeutic modalities. Interestingly, due to the increasing importance of biotechnology and the basis of research in a variety of sectors, there is a promising future for IDO1 vaccination and IDO inhibitors to reach patients with the goal that these vaccine-induced cytotoxic T cells remove malignant cells.
Data availability
Not applicable.
Abbreviations
- Trp:
-
Tryptophan
- CAR:
-
Chimeric antigen receptor
- IDO:
-
Indoleamine 2, 3-dioxygenase
- Kyn:
-
Kynurenine
- CTL:
-
Cytotoxic T lymphocyte
- Tregs:
-
T regulatory cells
- DCs:
-
Dendritic cells
- NFK:
-
N-formylkynurenine
- IFN-γ:
-
Interferon γ
- TNF-α:
-
Tumor necrosis factor-alpha
- OS:
-
Overall survival
- QUIN:
-
Quinolinic acid
- GCN2:
-
General control non-depressible 2
- mTOR:
-
Molecular target of rapamycin
- PKC:
-
Protein kinase C
- KYNA:
-
Kynurenic acid
- PIC:
-
Picolinic acid
- NMDA:
-
N-Methyl-d-aspartate
- MIP1:
-
Macrophage inflammatory protein
- GPR35:
-
G-protein coupled receptor 35
- HCAR3:
-
Hydroxycarboxylic acid receptor 3
- L-VDCC:
-
L-type voltage-dependent Ca2+ channels
- QPRT:
-
Quinolinate phosphoribosyltransferase
- KARs:
-
Kainic acid receptors
- AHR:
-
Aryl hydrocarbon receptor
- HD:
-
Huntington's disease
- AD:
-
Alzheimer's disease
- NSCLC:
-
Non-small cell lung cancer
- ACMSD:
-
ACMS decarboxylase
- TDLNs:
-
Tumor-draining lymph nodes
References
Thakkar JP, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomark Prev. 2014;23(10):1985–96.
Davis ME. Glioblastoma: overview of disease and treatment. Clin J Oncol Nurs. 2016;20(5 Suppl):S2–8.
Yu MW, Quail DF. Immunotherapy for glioblastoma: current progress and challenges. Front Immunol 2021;12(1637).
Muller AJ, et al. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312–9.
Uyttenhove C, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–74.
Zhai L, et al. Tumor cell IDO enhances immune suppression and decreases survival independent of tryptophan metabolism in glioblastoma. Clin Cancer Res. 2021;27(23):6514.
Söderlund J, Erhardt S, Kast RE. Acyclovir inhibition of IDO to decrease Tregs as a glioblastoma treatment adjunct. J Neuroinflamm. 2010;7(1):44.
Zhai L, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol. 2018;15(5):447–57.
Wainwright DA, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–21.
Mezrich JD, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190.
Zhai L, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res. 2017;23(21):6650–60.
Mellor AL, Munn DH. Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4(10):762–74.
Ball HJ, et al. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int J Biochem Cell Biol. 2009;41(3):467–71.
Dhainaut M, Moser M. Chapter six—mechanisms of surveillance of dendritic cells by regulatory T lymphocytes. In: Liston A, editor. Progress in molecular biology and translational science. New York: Academic Press; 2015. p. 131–54.
Munn DH, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297(5588):1867–70.
Bai R-Y, et al. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro Oncol. 2011;13(9):974–82.
Brandacher G, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. 2006;12(4):1144–51.
Prendergast GC. Cancer: why tumours eat tryptophan. Nature. 2011;478(7368):192–4.
Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Can Res. 2012;72(9):2162–71.
Balachandran VP, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–100.
Vinay DS, Kwon BS. CD11c+CD8+ T cells: two-faced adaptive immune regulators. Cell Immunol. 2010;264(1):18–22.
Meireson A, Devos M, Brochez L. IDO expression in cancer: different compartment, different functionality? Front Immunol 2020;11(2340).
Botticelli A, et al. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J Transl Med. 2018;16(1):219.
Gomes B, et al. Characterization of the selective indoleamine 2,3-dioxygenase-1 (IDO1) catalytic inhibitor EOS200271/PF-06840003 supports IDO1 as a critical resistance mechanism to PD-(L)1 blockade therapy. Mol Cancer Ther. 2018;17(12):2530.
Campbell B, et al. Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci. 2014;8:12.
Kim M, Tomek P. Tryptophan: a rheostat of cancer immune escape mediated by immunosuppressive enzymes IDO1 and TDO. Front Immunol 2021;12(21).
Wainwright DA, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20(20):5290–301.
Reardon DA, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic. Immunocompetent Model Cancer Immunol Res. 2016;4(2):124–35.
Schmidt SK, et al. Antimicrobial and immunoregulatory properties of human tryptophan 2,3-dioxygenase. Eur J Immunol. 2009;39(10):2755–64.
Bilir C, Sarisozen C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J Oncol Sci. 2017;3(2):52–6.
Ye Z, et al. Role of IDO and TDO in cancers and related diseases and the therapeutic implications. J Cancer. 2019;10(12):2771–82.
Mándi Y, Vécsei L. The kynurenine system and immunoregulation. J Neural Transm (Vienna). 2012;119(2):197–209.
Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Can Res. 2012;72(21):5435.
Munn DH, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–42.
Zhai L, et al. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res. 2015;21(24):5427.
Metz R, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1(9):1460–8.
Liu M, et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. 2018;11(1):100.
Tomek P, et al. Discovery and evaluation of inhibitors to the immunosuppressive enzyme indoleamine 2,3-dioxygenase 1 (IDO1): probing the active site-inhibitor interactions. Eur J Med Chem. 2017;126:983–96.
Crosignani S, et al. Discovery of a novel and selective indoleamine 2,3-dioxygenase (IDO-1) inhibitor 3-(5-fluoro-1H-indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and its characterization as a potential clinical candidate. J Med Chem. 2017;60(23):9617–29.
Koromilas AE. Roles of the translation initiation factor eIF2α serine 51 phosphorylation in cancer formation and treatment. Biochim Biophys Acta. 2015;1849(7):871–80.
Tanaka M, et al. Immune influencers in action: metabolites and enzymes of the tryptophan-kynurenine metabolic pathway. Biomedicines. 2021;9(7):734.
Botticelli A, et al, Tryptophan catabolism as immune mechanism of primary resistance to anti-PD-1. Front Immunol 2020:11.
Ribeiro CA, et al. Evidence that quinolinic acid severely impairs energy metabolism through activation of NMDA receptors in striatum from developing rats. J Neurochem. 2006;99(6):1531–42.
Zinger A, et al. The involvement of neuroinflammation and kynurenine pathway in Parkinson's disease. Parkinson’s Dis 2011;2011.
Bosco MC, et al. The tryptophan catabolite picolinic acid selectively induces the chemokines macrophage inflammatory protein-1 alpha and -1 beta in macrophages. J Immunol. 2000;164(6):3283–91.
Badawy AAB. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res. 2017;10:1178646917691938–1178646917691938.
Lugo-Huitrón R, et al. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol Teratol. 2011;33(5):538–47.
Chiarugi A, Meli E, Moroni F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J Neurochem. 2001;77(5):1310–8.
Fallarino F, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9(10):1069–77.
Stone TW, Perkins MN. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol. 1981;72(4):411–2.
Lugo-Huitrón R, et al. Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxid Med Cell Longev. 2013;2013:104024–104024.
Heng B, et al. Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget. 2016;7(6):6506–20.
Pierozan P, et al. Acute intrastriatal administration of quinolinic acid provokes hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons of rats. Exp Neurol. 2010;224(1):188–96.
Rahman A, et al. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE. 2009;4(7):e6344.
Adams S, et al. Involvement of the kynurenine pathway in human glioma pathophysiology. PLoS ONE. 2014;9(11):e112945–e112945.
Ying W. NAD+ and NADH in brain functions, brain diseases and brain aging. Front Biosci. 2007;12:1863–88.
Braidy N, et al. Changes in kynurenine pathway metabolism in the brain, liver and kidney of aged female Wistar rats. FEBS J. 2011;278(22):4425–34.
Sahm F, et al. The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer Res. 2013;73(11):3225–34.
Moroni F, et al. Kynurenic acid: a metabolite with multiple actions and multiple targets in brain and periphery. J Neural Transm. 2012;119(2):133–9.
Prescott C, et al. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci Lett. 2006;402:108–12.
Stephens GL, et al. Kynurenine 3-monooxygenase mediates inhibition of Th17 differentiation via catabolism of endogenous aryl hydrocarbon receptor ligands. Eur J Immunol. 2013;43(7):1727–34.
Vogel CFA, et al. Aryl hydrocarbon receptor signaling regulates NF-κB RelB activation during dendritic-cell differentiation. Immunol Cell Biol. 2013;91(9):568–75.
Pallotta MT, et al. AhR-mediated, non-genomic modulation of IDO1 function. Front Immunol. 2014;5:497.
Braidy N, Grant R. Kynurenine pathway metabolism and neuroinflammatory disease. Neural Regen Res. 2017;12(1):39–42.
Beal MF, et al. Kynurenine pathway measurements in huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem. 1990;55(4):1327–39.
Schwarcz R, et al. Increased cortical kynurenate content in schizophrenia. Biol Psychiat. 2001;50(7):521–30.
Serio CD, et al. Kynurenic acid inhibits the release of the neurotrophic fibroblast growth factor (FGF)-1 and enhances proliferation of glia cells, in vitro. Cell Mol Neurobiol. 2005;25(6):981–93.
Walczak K, et al. Kynurenic acid inhibits proliferation and migration of human glioblastoma T98G cells. Pharmacol Rep. 2014;66(1):130–6.
Nabi S, et al. Predictors of venous thromboembolism in patients with glioblastoma multiforme. J Clin Oncol. 2015;33(15_Suppl):e13022–e13022.
Cavalheiro EA, Olney JW. Glutamate antagonists: deadly liaisons with cancer. Proc Natl Acad Sci USA. 2001;98(11):5947–8.
Corsi L, Mescola A, Alessandrini A. Glutamate receptors and glioblastoma multiforme: an old “route” for new perspectives. Int J Mol Sci. 2019;20(7):1796.
Rzeski W, Turski L, Ikonomidou C. Glutamate antagonists limit tumor growth. Proc Natl Acad Sci USA. 2001;98(11):6372–7.
Pereira MSL, et al. Metabotropic glutamate receptors as a new therapeutic target for malignant gliomas. Oncotarget. 2017;8(13):22279–98.
Lyons SA, et al. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67(19):9463–71.
Stepulak A, et al. AMPA antagonists inhibit the extracellular signal regulated kinase pathway and suppress lung cancer growth. Cancer Biol Ther. 2007;6(12):1908–15.
Stepulak A, et al. NMDA antagonist inhibits the extracellular signal-regulated kinase pathway and suppresses cancer growth. Proc Natl Acad Sci USA. 2005;102(43):15605–10.
Pucci L, et al. Tissue expression and biochemical characterization of human 2-amino 3-carboxymuconate 6-semialdehyde decarboxylase, a key enzyme in tryptophan catabolism. FEBS J. 2007;274(3):827–40.
Salter M, Knowles RG, Pogson CI. Quantification of the importance of individual steps in the control of aromatic amino acid metabolism. Biochem J. 1986;234(3):635–47.
Grant RS, Coggan SE, Smythe GA. The physiological action of picolinic acid in the human brain. Int J Tryptophan Res 2009;2:IJTR.S2469.
Ikeda M, et al. Studies on the biosynthesis of nicotinamide adenine dinucleotide. II. A role of picolinic carboxylase in the biosynthesis of nicotinamide adenine dinucleotide from tryptophan in mammals. J Biol Chem 1965;240:1395–401.
Testa U, et al. The iron-chelating agent picolinic acid enhances transferrin receptors expression in human erythroleukaemic cell lines. Br J Haematol. 1985;60(3):491–502.
Melillo G, et al. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J Exp Med. 1995;182(6):1683–93.
Suzuki K, Yasuda M, Yamasaki K. Stability constants of picolinic and quinaldic acid chelates of bivalent metals. J Phys Chem. 1957;61(2):229–31.
Robinson MB, et al. Structure-function relationships for kynurenic acid analogues at excitatory pathways in the rat hippocampal slice. Brain Res. 1985;361(1–2):19–24.
Guillemin GJ, et al. Characterization of the kynurenine pathway in human neurons. J Neurosci. 2007;27(47):12884–92.
Vrooman L, et al. Picolinic acid modulates kainic acid-evoked glutamate release from the striatum in vitro. Brain Res. 1993;627(2):193–8.
Young AM, Crowder JM, Bradford HF. Potentiation by kainate of excitatory amino acid release in striatum: complementary in vivo and in vitro experiments. J Neurochem. 1988;50(2):337–45.
Bosco MC, et al. Macrophage activating properties of the tryptophan catabolite picolinic acid. Adv Exp Med Biol. 2003;527:55–65.
Bosco MC, et al. The tryptophan catabolite picolinic acid selectively induces the chemokines macrophage inflammatory protein-1α and -1β in macrophages. J Immunol. 2000;164(6):3283–91.
Ben-Baruch A, Michiel DF, Oppenheim JJ. Signals and receptors involved in recruitment of inflammatory cells. J Biol Chem. 1995;270(20):11703–6.
Lillard JW, et al. MIP-1α and MIP-1β differentially mediate mucosal and systemic adaptive immunity. Blood. 2003;101(3):807–14.
Varesio L, et al. Picolinic acid, a catabolite of tryptophan, as the second signal in the activation of IFN-gamma-primed macrophages. J Immunol. 1990;145(12):4265–71.
Taub DD, et al. Preferential migration of activated CD4+ and CD8+ T cells in response to MIP-1 alpha and MIP-1 beta. Science. 1993;260(5106):355–8.
Bhavsar I, Miller CS, Al-Sabbagh M. Macrophage inflammatory protein-1 alpha (MIP-1 alpha)/CCL3: as a biomarker. General Methods Biomark Res Appl 2015:223–249.
Cook DN, et al. CD8+ T cells are a biologically relevant source of macrophage inflammatory protein-1 alpha in vivo. J Immunol. 1999;162(9):5423–8.
Ruffmann R, et al. Antiproliferative activity of picolinic acid due to macrophage activation. Drugs Exp Clin Res. 1987;13(10):607–14.
Hornyák L, et al. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol 2018;9(151).
Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–43.
Hascitha J, et al. Analysis of Kynurenine/Tryptophan ratio and expression of IDO1 and 2 mRNA in tumour tissue of cervical cancer patients. Clin Biochem. 2016;49(12):919–24.
Mandarano M, et al. Kynurenine/tryptophan ratio as a potential blood-based biomarker in non-small cell lung cancer. Int J Mol Sci. 2021;22(9):4403.
Zhai L, et al. The kynurenine to tryptophan ratio as a prognostic tool for glioblastoma patients enrolling in immunotherapy. J Clin Neurosci. 2015;22(12):1964–8.
Meireson A, Devos M, Brochez L. IDO expression in cancer: different compartment, different functionality? Front Immunol 2020;11.
Li H, et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun. 2019;10(1):4346.
Möller M, Du Preez JL, Harvey BH. Development and validation of a single analytical method for the determination of tryptophan, and its kynurenine metabolites in rat plasma. J Chromatogr B. 2012;898:121–9.
Gulaj E, et al. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv Med Sci. 2010;55(2):204–11.
Tang K, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol. 2021;14(1):68.
Fox E, et al. Indoximod: an immunometabolic adjuvant that empowers T cell activity in cancer. Front Oncol 2018;8(370).
Ladomersky E, et al. IDO1 inhibition synergizes with radiation and PD-1 blockade to durably increase survival against advanced glioblastoma. Clin Cancer Res. 2018;24(11):2559–73.
Ahlstedt J, et al. Increased effect of two-fraction radiotherapy in conjunction with IDO1 inhibition in experimental glioblastoma. PLoS ONE. 2020;15(5):e0233617.
Khalil DN, et al. Chapter one—the new era of cancer immunotherapy: manipulating T-cell activity to overcome malignancy. In: Wang X-Y, Fisher PB, editors., et al., Advances in cancer research. New York: Academic Press; 2015. p. 1–68.
Banerjee T, et al. A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene. 2008;27(20):2851–7.
Dhiman V, et al. Determination of epacadostat, a novel IDO1 inhibitor in mouse plasma by LC-MS/MS and its application to a pharmacokinetic study in mice. Biomed Chromatogr 2017;31(2).
Jochems C, et al. The IDO1 selective inhibitor epacadostat enhances dendritic cell immunogenicity and lytic ability of tumor antigen-specific T cells. Oncotarget. 2016;7(25):37762–72.
Prendergast GC, et al. Chapter four - indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. In: Galluzzi L, editor., et al., International review of cell and molecular biology. New York: Academic Press; 2018. p. 175–203.
Kötzner L, et al. Chapter one - small molecules—giant leaps for immuno-oncology. In: Witty DR, Cox B, editors., et al., Progress in medicinal chemistry. New York: Elsevier; 2020. p. 1–62.
Yue EW, et al. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med Chem Lett. 2017;8(5):486–91.
Prendergast GC, et al. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017;77(24):6795–811.
Dey S, et al. Peptide vaccination directed against IDO1-expressing immune cells elicits CD8(+) and CD4(+) T-cell-mediated antitumor immunity and enhanced anti-PD1 responses. J Immunother Cancer 2020:8(2).
Miyazaki T, et al. Therapeutic strategies for overcoming immunotherapy resistance mediated by immunosuppressive factors of the glioblastoma microenvironment. Cancers. 2020;12(7):1960.
Keilholz U, et al. Immunologic monitoring of cancer vaccine therapy: results of a workshop sponsored by the society for biological therapy. J Immunother 2002;25(2).
Sørensen RB, et al. The immune system strikes back: cellular immune responses against indoleamine 2,3-dioxygenase. PLoS ONE. 2009;4(9):e6910.
Dey S, et al. Peptide vaccination directed against IDO1-expressing immune cells elicits CD8+ and CD4+ T-cell-mediated antitumor immunity and enhanced anti-PD1 responses. J Immunother Cancer. 2020;8(2):e000605.
Sharma MD, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Investig. 2007;117(9):2570–82.
Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Investig. 2007;117(5):1147–54.
Munir S, et al. Natural CD4+ T-cell responses against indoleamine 2,3-dioxygenase. PLoS ONE. 2012;7(4):e34568.
Andersen MH. The specific targeting of immune regulation: T-cell responses against Indoleamine 2,3-dioxygenase. Cancer Immunol Immunother. 2012;61(8):1289–97.
Dey S, et al. Peptide vaccination directed against IDO1-expressing immune cells elicits CD8(+) and CD4(+) T-cell-mediated antitumor immunity and enhanced anti-PD1 responses. J Immunother Cancer. 2020;8(2):e000605.
Winograd EK, Ciesielski MJ, Fenstermaker RA. Novel vaccines for glioblastoma: clinical update and perspective. Immunotherapy. 2016;8(11):1293–308.
Bjoern J, et al. Safety, immune and clinical responses in metastatic melanoma patients vaccinated with a long peptide derived from indoleamine 2,3-dioxygenase in combination with ipilimumab. Cytotherapy. 2016;18(8):1043–55.
Hjortsø MD, et al. Tryptophan 2,3-dioxygenase (TDO)-reactive T cells differ in their functional characteristics in health and cancer. OncoImmunology. 2015;4(1):e968480.
Fellner C. Ipilimumab (yervoy) prolongs survival in advanced melanoma: serious side effects and a hefty price tag may limit its use. P & T. 2012;37(9):503–30.
Coventry BJ. Therapeutic vaccination immunomodulation: forming the basis of all cancer immunotherapy. Ther Adv Vaccin Immunother. 2019;7:2515135519862234–2515135519862234.
Ryan SO, et al. Tumor antigen epitopes interpreted by the immune system as self or abnormal-self differentially affect cancer vaccine responses. Can Res. 2010;70(14):5788.
Kong Z, Wang Y, Ma W. Vaccination in the immunotherapy of glioblastoma. Hum Vaccin Immunother. 2018;14(2):255–68.
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this artice.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hosseinalizadeh, H., Mahmoodpour, M., Samadani, A.A. et al. The immunosuppressive role of indoleamine 2, 3-dioxygenase in glioblastoma: mechanism of action and immunotherapeutic strategies. Med Oncol 39, 130 (2022). https://doi.org/10.1007/s12032-022-01724-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-022-01724-w