
Abstract
Purpose of Review
Clonal hematopoiesis (CH) refers to the expansion of hematopoietic stem cell clones and their cellular progeny due to somatic mutations, mosaic chromosomal alterations (mCAs), or copy number variants which naturally accumulate with age. CH has been linked to increased risk of blood cancers, but CH has also been linked to adverse cardiovascular outcomes.
Recent Findings
A combination of clinical outcome studies and mouse models have offered strong evidence that CH mutations either correlate with or cause atherosclerosis, diabetes mellitus, chronic kidney disease, heart failure, pulmonary hypertension, aortic aneurysm, myocardial infarction, stroke, aortic stenosis, poor outcomes following transcatheter aortic valve replacement (TAVR) or orthotopic heart transplant, death or need of renal replacement therapy secondary to cardiogenic shock, death from cardiovascular causes at large, and enhance anthracycline cardiac toxicity. Mechanistically, some adverse outcomes are caused by macrophage secretion of IL-1β and IL-6, neutrophil invasion of injured myocardium, and T-cell skewing towards inflammatory phenotypes.
Summary
CH mutations lead to harmful inflammation and arterial wall invasion by bone marrow-derived cells resulting in poor cardiovascular health and outcomes. Blockade of IL-1β or JAK2 signaling are potential avenues for preventing CH-caused cardiovascular morbidity and mortality.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Somatic mutations and mosaic chromosomal alterations (mCAs) accumulate in hematopoietic stem cells (HSCs) and their cellular progeny as individuals age. When these genetic lesions lead to clonal expansion of a population in the absence of a malignancy or cytopenia, it is referred to as CH. CH of indeterminate potential (CHIP) is a subset of CH and is generally restricted to somatic mutations in genes associated with hematologic malignancy (DNMT3A, TET2, ASXL1, JAK2, TP53, etc.) with a minimum variant allele frequency (VAF) of 2% [1]. CH has been primarily viewed as a precursor to myeloid malignancies such as acute myeloid leukemia or myelodysplastic syndrome, though it is recognized that some clonal expansions can predispose individuals to lymphoid malignancies as well, leading to the characterization of some mutations and mCAs as either myeloid or lymphoid CH [2].
Beyond increasing the risk of primary or secondary malignancies, clinical outcomes in other disease processes tend to be negatively affected by the presence of CH although there are some identified exceptions. Responses to chimeric antigen receptor T-cell efficacy are likely enhanced by the presence of CH, albeit with increased rates of cytokine release syndrome and neurotoxicity [3,4,5,6]. Moreover, the presence of CH has been associated with improved overall survival (OS) in patients with metastatic colorectal cancer and with a reduction in the risk of developing Alzheimer’s disease [7, 8]. Some CH mutations in the donor of allogeneic hematopoietic stem cell transplants are protective in the recipient and lead to reduced relapse risk and improved OS [9].
These benefits are firmly outweighed in quantity by the known negative health impacts of CH, likely mediated by excessive IL-6 and IL-1β driven inflammation [1]. CH is associated with chronic obstructive pulmonary disease in multiple studies, and mouse models suggest CH mutations can be causative [10, 11]. CH is associated with excessive osteoclast activity and resulting osteoporosis [12]. CH correlates with post-transplant, non-relapse mortality in patients receiving stem cell transplants for multiple myeloma and lymphoma [3, 4]. In a cohort of myeloma patients that had undergone autologous stem cell transplants, the presence of CH was also associated with decreased progression free survival and decreased OS regardless of whether the patients were treated with immune modulating drugs [13]. The immune system’s response to infections also appears impaired by the presence of CH as CH is associated with severe COVID-19 outcomes, as well as with Clostridium Difficile and Enterococcal/Streptococcal infections in solid tumor patients [14]. Diabetes is more common in patients with CH [15]. As will be reviewed here, the evidence implicating CH in poor cardiovascular outcomes is extensive, which is likely a primary driver for the association of CH with decreased OS in the general population [16]. The relationship between CH and coronary artery disease (CAD) was the first non-oncologic outcome to be recognized [15], with further cardiologic studies also linking CH to progression of heart failure [17,18,19,20], aortic aneurysm [21], pulmonary hypertension [22], deterioration from cardiogenic shock [23], aortic stenosis [24], poor outcomes after TAVR [25] or orthotopic heart transplantation [26], and increased sensitivity to cardiotoxic chemotherapy [27]. Here we will review the evidence connecting CH to cardiovascular outcomes, mechanistic explanations, potential practice changes, and future research directions.
A Shot Through the Heart, CH Realized as a Risk Factor for Vascular Disease
Atherosclerosis is the result of inflammation in the blood vessel wall, and myeloid cells such as monocyte-derived macrophages are known to mediate this inflammation [28,29,30]. In CH, lineage restriction can occur leading to “myeloid bias” with mutated myeloid progeny cells that are more prone to causing atherosclerosis [31]. CH was first implicated in cardiovascular disease on a population level by Jaiswal et al. after performing whole-exome sequencing of peripheral blood from 17,182 individuals. Mutations in genes associated with blood cancers were frequently found in DNMT3A, TET2, and ASXL1, and individuals with such mutations were found to be at an increased risk for all-cause mortality (HR 1.4; 1.1 to 1.8 95% CI), CAD (HR 2.0; 1.2 to 3.4 95% CI), and ischemic stroke (HR 2.6; 1.4 to 4.8 95% CI) [15]. A second landmark case–control study using whole-exome sequencing of 4,726 participants with CAD and 3,529 controls found that rates of CH were 1.9 × greater in individuals with CAD [32]. Causality was also demonstrated by modeling CH with atherosclerosis-prone mice and a low-density lipoprotein receptor knockout mutation and engrafting them with bone marrow from Tet2 control or heterozygous or homozygous knockout mice and then feeding them a high-fat, high-cholesterol diet. Loss of Tet2 was associated with higher atherosclerotic burden in the aorta, as well as with macrophage expression of cytokines and chemokines associated with atherosclerosis, including Cxcl1, Cxcl2, Cxcl3, Pf4, Il-1b, and Il-6 [32]. Together, these two studies provide strong evidence not only that CH is associated with atherosclerotic disease, but that CH mutations can cause atherosclerosis likely in part through macrophage recruitment of monocytes and other blood cells to the arterial intima. CH mutations may also contribute to the premature cardiovascular disease in people with HIV, as CH mutations are more common in individuals with HIV and in this patient population CH mutation carriers tend to have greater coronary atherosclerosis burden [33].
CH is also associated with the consequences of atherosclerosis. A prospective cohort study involving 78,752 individuals examined the association of CH with the risk of different types of stroke, and found CH to be associated more with hemorrhagic stroke (HR 1.24; 1.01 to 1.54 95% CI) than with ischemic stroke (HR 1.11; 0.98 to 1.25 95% CI) [34]. TET2 mutations conferred the strongest association with stroke overall and with ischemic stroke among the 74 genes sequenced [34, 35]. CH is also prevalent in patients with ST-segment elevation myocardial infarction (STEMI) and correlates with poor outcomes following STEMI. DNMT3A or TET2 CH mutations have been found in 12.4% of STEMI patients, and patients harboring these mutations have a much higher incidence of death (30.9% vs 15.5%, p = 0.001) or major cardiac events (44.5% vs 21.8%, p < 0.001) over a median 3-year follow-up after STEMI [36]. In patients with cardiogenic shock secondary to myocardial infarction, having a CH mutation is associated with an increased risk of death or requiring renal replacement therapy independent of age or renal function [23]. In summary, CH mutations appear to cause accelerated atherosclerosis and likely lead to the subsequent sequelae of atherosclerotic burden including stroke and myocardial infarction.
Molecular Mechanisms of CH-Mediated Cardiotoxicity
Myeloid CH gene mutations (such as TET2 or DNMT3A) as a class have been strongly associated with all-cause mortality (HR = 1.6, p < 0.001) and an increased risk of CAD (HR = 1.35, p = 0.005) whereas lymphoid CH gene mutations as a class are not associated with either [2]. mCAs have been associated with all-cause mortality but are not associated with increased risk of CAD [2]. Thus, we will emphasize mutations in myeloid CH genes, like TET2. An abundance of literature has been developed around TET2 and has delineated a mechanistic link with adverse cardiovascular outcomes. The TET2 gene is one of three TET genes and resides at 4q24 where it encodes the Ten-eleven translocation 2 protein. TET proteins catalyze the production of cytosine derivatives and may be involved in the demethylation of CpG dinucleotides [37,38,39]. Murine Tet2 knockouts demonstrate that loss or heterozygosity of Tet2 leads to myeloid progenitor cell and multipotent progenitor cell expansion, and that hematopoietic stem cells harboring Tet2 loss can outcompete wildtype hematopoietic stem cells in competitive engraftment experiments [40,41,42,43,44]. TET2 mutations are frequently found in myeloid neoplasms but also in angioimmunoblastic T-cell lymphoma and in peripheral T-cell lymphoma not otherwise specified [39, 45,46,47,48].
TET2 has been firmly implicated in the development of atherosclerosis and subsequent myocardial infarction through the NLRP3 inflammasome-mediated secretion of IL-1β. As mentioned previously, murine data demonstrates that Tet2 loss enhances macrophage cytokine secretion of Il-1β leading to plausible recruitment of monocytes, platelets, and other cell types to the arterial wall [32]. Similarly, Tet2 deficiency in Ldl receptor null mice fed a high fat diet leads to markedly accelerated atherosclerosis that can be rescued by NLRP3 inhibition [49]. Tet2 is also a master regulator of vascular smooth muscle cell plasticity and Tet2 activation also promotes vascular smooth muscle proliferation and intimal hyperplasia, which may further contribute to accelerated atherosclerosis [50,51,52,53,54].
Murine models also suggest a role for myocardial Tet2 in CH-associated heart failure. Tet2 deficiency worsens cardiac remodeling and function after left anterior descending artery ligation or transverse aortic constriction, and increases Il-1β expression in the remodeling heart tissue [55]. Moreover, blocking Il-1β production by inhibiting the NLRP3 inflammasome has an enhanced protective effect following myocardial injury in Tet2 deficient mice [55]. Infusion of Tet2 mutant bone marrow cells induces clonal expansion of Tet2 deficient Ccr2 + macrophages that infiltrate the mouse heart provoking age-related cardiac hypertrophy and fibrosis [56]. Similar results have been found using CRISPR-mediated gene editing of stem cells [57]. Loss of Tet2 also leads to cardiomyocyte hypertrophy through hyperactivation of ERK 1/2 suggesting that the hypertrophic phenotype of Tet2 null mice may arise from both clonal populations of infiltrating cells and cardiomyocyte-autonomous effects [58]. Asxl1 CH mutations have also been shown to have similar negative effects on cardiac remodeling with similar enhancement of macrophage Il-1β production [59].
There is some evidence in humans implicating TET2-regulated IL-1β secretion as pathogenic as well. In the Canakinumab Anti-inflammatory Thrombosis Outcomes Trial (CANTOS), participants with a prior myocardial infarction and an elevated hs-CRP level were randomized to placebo or escalating doses of canakinumab: an anti-IL-1β monoclonal antibody [60•]. Higher doses of canakinumab led to a 15% relative risk reduction in the combined end points of myocardial infarction, stroke, and cardiovascular-related death (major adverse cardiac events, MACE). A follow-up study identified patients as having CH or not having CH mutations using a 74 gene panel, and found that 8.6% of participants in CANTOS had CH mutations, with TET2 gene mutations being the most common variant [60•]. Patients that harbored a TET2 mutation had higher rates of major cardiac events than those that did not, as well as a statically significant response to canakinumab, whereas patients without a TET2 mutation had no significant effect in major myocardial events after treatment with canakinumab [60•]. Together with the data from mouse models, these results strongly implicate TET2 dysfunction as driving the production of IL-1β and causing vascular disease as manifested by higher rates of myocardial infarction, stroke, and cardiovascular-related death.
DNMT3A is another CH gene, which like TET2 regulates the methylation of CpG dinucleotides. In fact, mendelian randomization studies have identified subsets of DNA methylation sites that likely cause the increased risk of CAD associated with these two mutations [61]. In a study of 485 patients with STEMI, those that harbored either a DNMT3A or TET2 mutation had an increased risk of death or MACE and had higher levels of plasma IL-1β and IL-6, but not higher levels of TNFβ or IFNγ [36]. Extensive evidence supports a pathogenic role for IL-6 in numerous forms of cardiovascular disease and the IL-6 antagonist ziltivekimab was associated with favorable atherothrombotic outcomes in a recent phase 2 clinical trial [62, 63]. Patients with CH defined by DNMT3A or TET2 mutations have higher rates of cardiovascular disease and this effect is greater in patients with high VAF (> 10%), but this effect is abrogated in patients with congenital deficiency in IL-6 signaling that carry the IL6R p.Asp358Ala germline mutation [64]. In a macaque model of TET2 CH, macrophages with a TET2 loss-of-function mutation expressed higher levels of IL-6 and IL-1β, and treating the macaques with tocilizumab slowed TET2-mutated clone expansion, suggesting that IL-6 may enable the selective advantage of TET2-mutated clones [65•].
Another commonly mutated CH gene is JAK2, which has now been implicated in the development of pulmonary hypertension: a cause of right heart failure [22]. In a prospective clinical cohort, JAK2V617F-positive CH was more common in patients with pulmonary hypertension than in healthy controls [22]. Extending this finding into mice for mechanistic insight, transgenic mice harboring Jak2V617F had exacerbated pulmonary hypertension and pulmonary arterial remodeling after being exposed to chronic hypoxia. This was accompanied by increased neutrophil invasion of the pulmonary arterial regions and increases in neutrophil-derived elastase activity [22]. Inhibiting Alk1/2, which is upregulated by Jak2V617F, completely stopped the development of pulmonary hypertension in Jak2V617F mice [22]. JAK2V617F may also cause venous thromboembolism and pulmonary embolism further contributing to pulmonary hypertension [66,67,68,69]. One demonstrated mechanism for thromboembolism caused by JAK2 mutations is through enhanced production of neutrophil extracellular traps: large structures that are web-like, pro-thrombotic and made of cytosolic and granule proteins stuck together on a scaffold of decondensed chromatin [69, 70]. Interestingly, because JAK2 can be inhibited by ruxolitinib, this may be a therapy for pulmonary hypertension in the future. TET2 has also been implicated in pulmonary hypertension as germline mutations are disproportionately found in patients with pulmonary hypertension, as circulating TET2 expression is reduced in this disease, and as TET2 knockout mice spontaneously develop pulmonary hypertension which can be reversed by Il-1β blockade [71].
Returning to JAK2, JAK2 mutations have been associated with cardiovascular outcomes other than pulmonary hypertension. Jak2V617F mice are predisposed to developing aortic aneurysms, suggesting vascular effects beyond the pulmonary arterial bed [21]. Jak2V617F bone marrow transplanted into wildtype mice show increased bone marrow-derived hematopoietic cells invading the aorta, and this translocation can be blocked by ruxolitinib [21]. JAK2V617F also accelerates atherosclerosis in part through activation of the AIM2 inflammasome, by promoting neutrophil invasion into early atherosclerotic lesions, and by causing defective macrophage erythrophagocytosis [72•, 73].
Adaptive immune mechanisms have been implicated in poor cardiac outcomes due to CH as well. Patients with TET2 or DNMT3A mutations had significantly reduced OS after undergoing TAVR [25]. No differences in cytokine serum levels were found between patients with and without CH mutations, but patients with TET2 or DNMT3A mutations did have elevated Th17 / Treg ratios on flow cytometry, suggestive of increased systemic T-cell driven inflammation [25].
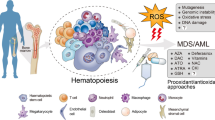
Patients with CH mutations have higher rates of diabetes mellitus at baseline (21.2% vs 33.3%, p = 0.035) [36]. This observation corresponds with evidence from mice showing that Tet2 deficiency modeled through bone marrow transplants enhances insulin resistance in aged mice as well as obese mice while increasing Il-1β production from bone marrow-derived macrophages in adipose tissue [74]. Like in the murine models of heart failure and atherosclerosis, these effects could be rescued through NLRP3 inflammasome inhibition [74]. Large population studies have also found higher rates of diabetes mellitus type 2 in patients with CH [15]. Investigation into the incidence or progression of diabetic kidney disease have not found an association with CH but there is evidence that CH promotes the development of chronic kidney disease at large, especially in the context of myeloid CH mutations and myeloid mCAs [75, 76]. Thus, aggravation of cardiac risk factors like diabetes and chronic kidney disease is an additional mechanism through which CH mutations are able to impact cardiovascular outcomes. Moreover, other established cardiovascular risk factors may cause CH. Smoking may mediate some of its negative cardiovascular effects through promoting the development of CH. Some studies have shown an association between smoke exposure and somatic CH mutations and others have shown an association with mCAs [10, 77, 78]. Unhealthy diets, defined as being low in fruits and vegetables and high in red meat, processed food, and added salt, have also been associated with a higher prevalence of CH [79]. The cardiovascular effects of CH with the corresponding mechanisms are summarized in Fig. 1.

Schematic of clonal hematopoiesis, its cardiovascular effects, and the underlying molecular mechanisms
Saving the Heart or the Blood Within It, CH Enhances Therapy-Related Cardiac Toxicity
Human studies have demonstrated clear evidence that cancer therapy can influence the development of CH and hence influence cardiovascular risk longitudinally [80]. Anthracyclines are chemotherapeutic agents employed against a wide variety of solid and liquid malignancies. Anthracyclines inhibit topoisomerase 2β and in doing so kill tumor cells but also cause cardiomyocyte injury, leading to left ventricular dysfunction [81]. In a mouse model of therapy-related CH in which TP53 heterozygous deficient hematopoietic stem and progenitor cells (HSPCs) were transferred to mice treated with doxorubicin, doxorubicin treatment promoted the expansion of TP53 deficient HSPCs [27]. Furthermore, mice harboring the TP53 heterozygous HSPCs had accelerated doxorubicin cardiac toxicity as measured by decreased capillary density, decreased fractional shortening, decreased left ventricular posterior wall thickness diameter, increased left ventricular end-systolic diameter, and decreased left ventricular mass. While cardiac monocyte and macrophage population sizes were not affected by TP53 deficiency, cardiac neutrophil counts were increased and depleting neutrophils with α-Ly6G antibody rescued the cardiotoxic effects of doxorubicin. These murine results demonstrate that TP53-mediated CH enhances neutrophil invasion and damage of cardiac tissue after treatment with doxorubicin.
While CH is classically driven by the age-related accumulation of somatic mutations, CH can also be caused by the selection of HSPCs and genotoxic stress produced by cancer directed therapy and will be referred to as therapy-related CH (t-CH). Mutations in DNA-damage response pathways are common following chemotherapy or radiotherapy, specifically in PPM1D and in TP53 [82, 83]. PPM1D is another myeloid CHIP gene[2], and mutations in PPM1D are seen in up to 20% of patients with therapy-related myeloid neoplasms [84]. In mice with Ppm1d HSPC mutations introduced by CRISPR-Cas9, Ppm1d mutations caused adverse ventricular remodeling and contractile dysfunction in mice challenged with an angiotensin II infusion [82]. Similar to the studies on Tet2, Ppm1d mutations caused higher levels of Il-1β and the cardiac remodeling effects noted were prevented by NLRP3 inflammasome inhibition suggesting that Ppm1d mutations mediate cardiac toxicity at least in part through the same cytokine pathway as Tet2 mutations [82]. Additionally, Ppm1d-mutated macrophages exhibited impaired DNA damage repair pathways, higher reactive oxygen species levels, and increased Il-18 secretion [82]. Thus, chemotherapy and radiotherapy can cause t-CH via mutations in Ppm1d, which subsequently shifts macrophage polarization towards a pro-inflammatory phenotype (M1), contributing to cardiomyocyte injury that synergizes with the direct cardiotoxicity of prior chemotherapy or radiotherapy. These preclinical findings, that CH mutations enhance anthracycline cardiac toxicity, are corroborated in human studies: within adult lymphoma patients treated with anthracyclines, TET2 mutations markedly increased the risk of developing anthracycline-induced cardiotoxicity (odds ratio: 5.15, 1.10–24.05 95% CI) [85].
Conclusions
-
Available data extensively implicate CH as a cardiovascular risk factor in the settings of atherosclerosis, heart failure, aortic stenosis/TAVR, orthotopic heart transplantation, pulmonary hypertension, aortic aneurysm, and stroke. CH may potentiate cardiac risk via associations with smoking, diabetes mellitus, and chronic kidney disease.
-
CH due to mutations in myeloid genes but not lymphoid genes or mCAs is associated with CAD. Evidence that TET2 mutations are causative is especially strong.
-
TET2, DNMT3A, and ASXL1 mutations cause macrophages to secrete higher levels of cytokines and chemokines, including IL-1β and IL-6, which contribute to accelerated atherosclerosis, heart failure, and diabetes mellitus type 2. Canakinumab, a monoclonal antibody that neutralizes IL-1β as secondary myocardial infarction prophylaxis, has enhanced efficacy in patients that carry a TET2 mutation.
-
CH mutations, specifically in TP53 and TET2, can increase myocardial toxicity from anthracycline chemotherapy in part through enhanced trafficking of neutrophils to the injured myocardium.
-
PPM1D t-CH is common in both patients with solid tumors and myeloid malignancies. Similar to TET2, PPM1D CH promotes non-ischemic heart failure in mice through IL-1β inflammation.
Future Research Directions
-
Generate prospective evidence that canakinumab can lead to improved cardiovascular outcomes in patients harboring myeloid CH mutations. Similarly, evaluate whether inhibiting JAK2V617F with ruxolitinib can treat pulmonary hypertension patients harboring the V617F mutation.
-
More rigorously assess mechanisms alternative to IL-1β production, such as IL-6 or IL-18 production or T-cell driven inflammation for contributions to cardiovascular toxicity of CH mutations.
-
Expand research into uncharacterized CH mutations and mCAs, and into their effects on the cardiovascular system. mCAs as a class have not been implicated in cardiac outcomes, and understanding the effects of individual mCAs remains unknown.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Meier J, Jensen JL, Dittus C, Coombs CC, Rubinstein S. Game of clones: Diverse implications for clonal hematopoiesis in lymphoma and multiple myeloma. Blood Rev. 2022;56:100986. https://doi.org/10.1016/j.blre.2022.100986.
Niroula A, Sekar A, Murakami MA, Trinder M, Agrawal M, Wong WJ, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat Med. 2021;27(11):1921–7.
Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35(14):1598–605.
Husby S, Favero F, Nielsen C, Sørensen BS, Bæch J, Grell K, et al. Clinical impact of clonal hematopoiesis in patients with lymphoma undergoing ASCT: a national population-based cohort study. Leukemia. 2020;34(12):3256–68.
Miller PG, Sperling AS, Brea EJ, Leick MB, Fell GG, Jan M, et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv. 2021;5(15):2982–6.
Saini NY, Swoboda DM, Greenbaum U, Ma J, Patel RD, Devashish K, Das K, Tanner MR, Strati P, Nair R, Fayad L, Ahmed S, Lee HJ, Iyer SP, Steiner R, Jain N, Nastoupil L, Loghavi S, Tang G, Bassett RL, Jain P, Wang M, Westin JR, Green MR, Sallman DA, Padron E, Davila ML, Locke FL, Champlin RE, Garcia-Manero G, Shpall EJ, Kebriaei P, Flowers CR, Jain MD, Wang F, Futreal AP, Gillis N, Neelapu SS, Takahashi K. Clonal hematopoiesis is associated with increased risk of severe neurotoxicity in axicabtagene ciloleucel therapy of large B-cell lymphoma. Blood Cancer Discov. 2022;3(5):385–93. https://doi.org/10.1158/2643-3230.BCD-21-0177.
Bouzid H, Belk J, Jan M, Qi Y, Sarnowski C, Wirth S, et al. Clonal hematopoiesis is associated with reduced risk of Alzheimer’s disease. Blood. 2021;138:5.
Arends CM, Dimitriou S, Stahler A, Hablesreiter R, Strzelecka PM, Stein CM, Tilgner M, Saiki R, Ogawa S, Bullinger L, Modest DP, Stintzing S, Heinemann V, Damm F. Clonal hematopoiesis is associated with improved survival in patients with metastatic colorectal cancer from the FIRE-3 trial. Blood. 2022;139(10):1593–97. https://doi.org/10.1182/blood.2021014108.
Gibson CJ, Kim HT, Zhao L, Murdock HM, Hambley B, Ogata A, et al. Donor clonal hematopoiesis and recipient outcomes after transplantation. J Clin Oncol. 2022;40(2):189–201.
Miller PG, Qiao D, Rojas-Quintero J, Honigberg MC, Sperling AS, Gibson CJ, et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood. 2022;139(3):357–68.
van Zeventer IA, Salzbrunn JB, de Graaf AO, van der Reijden BA, Boezen HM, Vonk JM, et al. Prevalence, predictors, and outcomes of clonal hematopoiesis in individuals aged ≥80 years. Blood Adv. 2021;5(8):2115–22.
Kim PG, Niroula A, Shkolnik V, McConkey M, Lin AE, Słabicki M, Kemp JP, Bick A, Gibson CJ, Griffin G, Sekar A, Brooks DJ, Wong WJ, Cohen DN, Uddin MM, Shin WJ, Pirruccello J, Tsai JM, Agrawal M, Kiel DP, Bouxsein ML, Richards JB, Evans DM, Wein MN, Charles JF, Jaiswal S, Natarajan P, Ebert BL. Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. J Exp Med. 2021;218(12):e20211872. https://doi.org/10.1084/jem.20211872.
Mouhieddine TH, Sperling AS, Redd R, Park J, Leventhal M, Gibson CJ, et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat Commun. 2020;11(1):2996.
Bolton KL, Koh Y, Foote MB, Im H, Jee J, Sun CH, et al. Clonal hematopoiesis is associated with risk of severe Covid-19. Nat Commun. 2021;12(1):5975.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98.
Nowakowska MK, Kim T, Thompson MT, Bolton KL, Deswal A, Lin SH, et al. Association of clonal hematopoiesis mutations with clinical outcomes: a systematic review and meta-analysis. Am J Hematol. 2022;97(4):411–20.
Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4(1):25–33.
Yu B, Roberts MB, Raffield LM, Zekavat SM, Nguyen NQH, Biggs ML, et al. Association of clonal hematopoiesis with incident heart failure. J Am Coll Cardiol. 2021;78(1):42–52.
Pascual-Figal DA, Bayes-Genis A, Díez-Díez M, Hernández-Vicente Á, Vázquez-Andrés D, de la Barrera J, et al. Clonal hematopoiesis and risk of progression of heart failure with reduced left ventricular ejection fraction. J Am Coll Cardiol. 2021;77(14):1747–59.
Cremer S, Kirschbaum K, Berkowitsch A, John D, Kiefer K, Dorsheimer L, et al. Multiple somatic mutations for clonal hematopoiesis are associated with increased mortality in patients with chronic heart failure. Circ: Genomic Precis Med. 2020;13(4):e003003.
Yokokawa T, Misaka T, Kimishima Y, Wada K, Minakawa K, Sugimoto K, et al. Crucial role of hematopoietic JAK2 V617F in the development of aortic aneurysms. Haematologica. 2021;106(7):1910–22.
Kimishima Y, Misaka T, Yokokawa T, Wada K, Ueda K, Sugimoto K, et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nat Commun. 2021;12(1):6177.
Böhme M, Desch S, Rosolowski M, Scholz M, Krohn K, Büttner P, et al. Impact of clonal hematopoiesis in patients with cardiogenic shock complicating acute myocardial infarction. J Am Coll Cardiol. 2022;80(16):1545–56.
Raddatz MA, Silver AJ, Farber-Eger E, Xu Y, Wells QS, Savona MR, et al. Abstract 9334: Clonal hematopoiesis is associated with incident severe aortic stenosis. Circulation. 2021;144(Suppl_1):A9334–A9334.
Mas-Peiro S, Hoffmann J, Fichtlscherer S, Dorsheimer L, Rieger MA, Dimmeler S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J. 2019;41(8):933–9.
Scolari FL, Brahmbhatt DH, Abelson S, Medeiros JJF, Anker MS, Fung NL, Otsuki M, Calvillo-Argüelles O, Lawler PR, Ross HJ, Luk AC, Anker S, Dick JE, Billia F. Clonal hematopoiesis confers an increased mortality risk in orthotopic heart transplant recipients. Am J Transplant. 2022;(12):3078–86. https://doi.org/10.1111/ajt.17172.
Sano S, Wang Y, Ogawa H, Horitani K, Sano M, Polizio AH, et al. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight. 2021;6(13):e146076.
Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(9):2045–51.
Pardali E, Waltenberger J. Monocyte function and trafficking in cardiovascular disease. Thromb Haemost. 2012;108(11):804–11.
Golia E, Limongelli G, Natale F, Fimiani F, Maddaloni V, Pariggiano I, et al. Inflammation and cardiovascular disease: from pathogenesis to therapeutic target. Curr Atheroscler Rep. 2014;16(9):435.
Buscarlet M, Provost S, Zada YF, Bourgoin V, Mollica L, Dubé M-P, et al. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood. 2018;132(3):277–80.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–21.
Wang S, Pasca S, Post WS, Langan S, Pallavajjala A, Haley L, et al. Clonal hematopoiesis in men living with HIV and association with subclinical atherosclerosis. AIDS. 2022;36(11):1521–31.
Bhattacharya R, Zekavat SM, Haessler J, Fornage M, Raffield L, Uddin MM, et al. Clonal hematopoiesis is associated with higher risk of stroke. Stroke. 2022;29(2):788–97.
Murphy AJ, Dragoljevic D, Natarajan P, Wang N. Hematopoiesis of indeterminate potential and atherothrombotic risk. Thromb Haemost. 2022;122(09):1435–42.
Wang S, Hu S, Luo X, Bao X, Li J, Liu M, et al. Prevalence and prognostic significance of DNMT3A- and TET2- clonal haematopoiesis-driver mutations in patients presenting with ST-segment elevation myocardial infarction. eBioMed. 2022;78:103964.
Weber AR, Krawczyk C, Robertson AB, Kuśnierczyk A, Vågbø CB, Schuermann D, et al. Biochemical reconstitution of TET1–TDG–BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat Commun. 2016;7(1):10806.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–3.
Nakajima H, Kunimoto H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci. 2014;105(9):1093–9.
Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25–38.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24.
Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011;108(35):14566–71.
Kunimoto H, Fukuchi Y, Sakurai M, Sadahira K, Ikeda Y, Okamoto S, et al. Tet2 disruption leads to enhanced self-renewal and altered differentiation of fetal liver hematopoietic stem cells. Sci Rep. 2012;2:273.
Shide K, Kameda T, Shimoda H, Yamaji T, Abe H, Kamiunten A, et al. TET2 is essential for survival and hematopoietic stem cell homeostasis. Leukemia. 2012;26(10):2216–23.
Cheng S, Zhang W, Inghirami G, Tam W. Mutation analysis links angioimmunoblastic T cell lymphoma to clonal hematopoiesis and smoking. Elife. 2021;10:e66395. https://doi.org/10.7554/eLife.66395.
Lewis NE, Petrova-Drus K, Huet S, Epstein-Peterson ZD, Gao Q, Sigler AE, et al. Clonal hematopoiesis in angioimmunoblastic T-cell lymphoma with divergent evolution to myeloid neoplasms. Blood Adv. 2020;4(10):2261–71.
Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123(9):1293–6.
Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26(5):934–42.
Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842–7.
Chakraborty R, Ostriker AC, Xie Y, Dave JM, Gamez-Mendez A, Chatterjee P, et al. Histone acetyltransferases p300 and CBP coordinate distinct chromatin remodeling programs in vascular smooth muscle plasticity. Circulation. 2022;145(23):1720–37.
Ostriker AC, Xie Y, Chakraborty R, Sizer AJ, Bai Y, Ding M, et al. TET2 protects against vascular smooth muscle cell apoptosis and intimal thickening in transplant vasculopathy. Circulation. 2021;144(6):455–70.
Zeng Z, Xia L, Fan S, Zheng J, Qin J, Fan X, et al. Circular RNA CircMAP3K5 acts as a MicroRNA-22-3p sponge to promote resolution of intimal hyperplasia via TET2-mediated smooth muscle cell differentiation. Circulation. 2021;143(4):354–71.
Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013;128(18):2047–57.
Prosdocimo DA, Jain R, Jain MK. UnTEThering (smooth muscle) cell plasticity. Circulation. 2013;128(18):2002–4.
Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1B/NLRP3 inflammasome. J Am Coll Cardiol. 2018;71(8):875–86.
Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, Ogawa H, Horitani K, Min KD, Miura-Yura E, Kour A, Evans MA, Zuriaga MA, Hirschi KK, Fuster JJ, Pietras EM, Walsh K. Tet2 mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight. 2020;5(6):e135204. https://doi.org/10.1172/jci.insight.135204.
Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res. 2018;123(3):335–41.
Tao H, Xu W, Qu W, Gao H, Zhang J, Cheng X, et al. Loss of ten-eleven translocation 2 induces cardiac hypertrophy and fibrosis through modulating ERK signaling pathway. Hum Mol Genet. 2021;30(10):865–79.
Min KD, Polizio AH, Kour A, Thel MC, Walsh K. Experimental ASXL1-mediated clonal hematopoiesis promotes inflammation and accelerates heart failure. J Am Heart Assoc. 2022;11(19):e026154.
• Svensson EC, Madar A, Campbell CD, He Y, Sultan M, Healey ML, et al. TET2-driven clonal hematopoiesis and response to canakinumab: an exploratory analysis of the CANTOS randomized clinical trial. JAMA Cardiol. 2022;7(5):521–8. “TET2-driven clonal hematopoiesis and response to canakinumab: an exploratory analysis of the CANTOS randomized clinical trial”: TET2 mutations are associated with improved responses to the IL-1β neutralizing antibody, canakinumab. Studies like this may develop a clinical role for assessing CH in patients with cardiovascular disease in guiding therapy choices.
Uddin MDM, Nguyen NQH, Yu B, Brody JA, Pampana A, Nakao T, Fornage M, Bressler J, Sotoodehnia N, Weinstock JS, Honigberg MC, Nachun D, Bhattacharya R, Griffin GK, Chander V, Gibbs RA, Rotter JI, Liu C, Baccarelli AA, Chasman DI, Whitsel EA, Kiel DP, Murabito JM, Boerwinkle E, Ebert BL, Jaiswal S, Floyd JS, Bick AG, Ballantyne CM, Psaty BM, Natarajan P, Conneely KN. Clonal hematopoiesis of indeterminate potential, DNA methylation, and risk for coronary artery disease. Nat Commun. 2022;13(1):5350. https://doi.org/10.1038/s41467-022-33093-3.
Ridker PM, Rane M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ Res. 2021;128(11):1728–46.
Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2021;397(10289):2060–9.
Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation. 2020;141(2):124–31.
• Shin TH, Zhou Y, Chen S, Cordes S, Grice MZ, Fan X, et al. A macaque clonal hematopoiesis model demonstrates expansion of TET2-disrupted clones and utility for testing interventions. Blood. 2022;140(16):1774–89. “A macaque clonal hematopoiesis model demonstrates expansion of TET2-disrupted clones and utility for testing interventions”: In this primate study, blocking IL-6 with tocilizumab prevented the expansion of TET2-disrupted clones, showing that clonal expansion can be prevented.
Edelmann B, Gupta N, Schnoeder TM, Oelschlegel AM, Shahzad K, Goldschmidt J, et al. JAK2-V617F promotes venous thrombosis through β 1/β 2 integrin activation. J Clin Investig. 2018;128(10):4359–71.
Dikilitas O, Saadatagah S, Satterfield B, Kullo IJ. Abstract 12959: clonal hematopoiesis of indeterminate potential predicts incident venous thromboembolism in the UK biobank cohort. Circulation. 2021;144(Suppl_1):A12959–A12959.
Soudet S, Jedraszak G, Evrard O, Marolleau JP, Garcon L, Pietri MAS. Is hematopoietic clonality of indetermined potential a risk factor for pulmonary embolism? TH Open. 2021;05(03):e338–42.
Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10(436):eaan8292.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134–47.
Potus F, Pauciulo MW, Cook EK, Zhu N, Hsieh A, Welch CL, et al. Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation. 2020;141(24):1986–2000.
• Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592(7853):296–301. “The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis”: In this Jak2VF model of CH, CH was associated with increased proliferation and glycolytic metabolism in macrophages leading to DNA stress and subsequent activation of the AIM2 inflammasome and exacerbation of atherosclerosis. Aberrant cytokine secretion is not the only mechanism through which clonal myeloid cells worsen cardiovascular health outcomes.
Wang W, Liu W, Fidler T, Wang Y, Tang Y, Woods B, et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in <i>Jak2</i><sup><i>V617F</i></sup> mice. Circ Res. 2018;123(11):e35–47.
Fuster JJ, Zuriaga MA, Zorita V, MacLauchlan S, Polackal MN, Viana-Huete V, et al. TET2-loss-of-function-driven clonal hematopoiesis exacerbates experimental insulin resistance in aging and obesity. Cell Rep. 2020;33(4):108326.
Denicolò S, Vogi V, Keller F, Thöni S, Eder S, Heerspink HJL, et al. Clonal hematopoiesis of indeterminate potential and diabetic kidney disease: a nested case-control study. Kidney Int Reports. 2022;7(4):876–88.
Dawoud AAZ, Gilbert RD, Tapper WJ, Cross NCP. Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia. 2022;36(2):507–15.
Levin MG, Nakao T, Zekavat SM, Koyama S, Bick AG, Niroula A, et al. Genetics of smoking and risk of clonal hematopoiesis. Sci Rep. 2022;12(1):7248.
Dawoud AA, Tapper WJ, Cross NC. Clonal myelopoiesis in the UK Biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia. 2020;34(10):2660–72.
Bhattacharya R, Zekavat SM, Uddin MM, Pirruccello J, Niroula A, Gibson C, et al. Association of diet quality with prevalence of clonal hematopoiesis and adverse cardiovascular events. JAMA Cardiol. 2021;6(9):1069–77.
Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52(11):1219–26.
Henriksen PA. Anthracycline cardiotoxicity: an update on mechanisms, monitoring and prevention. Heart. 2018;104(12):971–7.
Yura Y, Miura-Yura E, Katanasaka Y, Min K-D, Chavkin N, Polizio AH, et al. The cancer therapy-related clonal hematopoiesis driver gene Ppm1d promotes inflammation and non-ischemic heart failure in mice. Circ Res. 2021;129(6):684–98.
Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21(3):374-82.e4.
Hsu JI, Dayaram T, Tovy A, De Braekeleer E, Jeong M, Wang F, et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell. 2018;23(5):700-13.e6.
Hatakeyama K, Hieda M, Semba Y, Moriyama S, Wang Y, Maeda T, et al. TET2 clonal hematopoiesis is associated with anthracycline-induced cardiotoxicity in patients with lymphoma. JACC: CardioOncol. 2022;4(1):141–3.
Acknowledgements
Figure 1 is created with BioRender.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jeffrey L. Jensen is an AbbVie and Pfizer shareholder. Saumya Easaw declares no conflicts of interest. Travis Anderson declares no conflicts of interest. Yash Varma declares no conflicts of interest. Jiandong Zhang declares no conflicts of interest. Brian C Jensen has served as a consultant for AstraZeneca. Catherine C. Coombs has served as a consultant for AbbVie, has served on steering committees for AbbVie and Loxo Oncology, has served on independent review committees for AbbVie and Octapharma, has served on Speaker’s Bureau for AbbVie, has received honoraria from AbbVie, AstraZeneca, Beigene, Genentech, Loxo Oncology, MEI Pharma, Novartis, TG Therapeutics, is a CTI Biopharma and Bluebird bio shareholder, and has received research funding (paid to the institution) from AbbVie and Loxo Oncology.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jeffrey L. Jensen and Brian C. Jensen have no familial relationship
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jensen, J.L., Easaw, S., Anderson, T. et al. Clonal Hematopoiesis and the Heart: a Toxic Relationship. Curr Oncol Rep 25, 455–463 (2023). https://doi.org/10.1007/s11912-023-01398-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-023-01398-1