
Abstract
Bile acids (BA) are amphipathic molecules originating from cholesterol in the liver and from microbiota-driven biotransformation in the colon. In the gut, BA play a key role in fat digestion and absorption and act as potent signaling molecules on the nuclear farnesoid X receptor (FXR) and membrane-associated G protein-coupled BA receptor-1 (GPBAR-1). BA are, therefore, involved in the maintenance of gut barrier integrity, gene expression, metabolic homeostasis, and microbiota profile and function. Disturbed BA homeostasis can activate pro-inflammatory pathways in the gut, while inflammatory bowel diseases (IBD) can induce gut dysbiosis and qualitative and/or quantitative changes of the BA pool. These factors contribute to impaired repair capacity of the mucosal barrier, due to chronic inflammation. A better understanding of BA-dependent mechanisms paves the way to innovative therapeutic tools by administering hydrophilic BA and FXR agonists and manipulating gut microbiota with probiotics and prebiotics. We discuss the translational value of pathophysiological and therapeutic evidence linking BA homeostasis to gut inflammation in IBD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bile acids (BA) are amphipathic lipid components of the human bile with non-esterified cholesterol and phospholipids. The BA pool is composed of primary BA synthesized from cholesterol in the liver and secondary BA from microbiota-driven biotransformation in the colon. In the gut, BA are involved in the emulsification and absorption of dietary fat and fat-soluble vitamins [1], have regulatory functions on epithelial cell proliferation [2,3,4] and gut barrier [4], affect expression of several genes involved in metabolic homeostasis [1, 5,6,7], stimulate epigenetic profiles [8, 9], modulate gut microbiota [6, 10], and have antimicrobial properties [10]. Substantial changes of the BA pool also affect the integrity of the intestinal barrier [4, 11], promote immune-modulatory effects [12,13,14], and modulate inflammatory pathways through signaling mechanisms that involve the nuclear receptor farnesoid X receptor (FXR) [15] and the membrane-associated G-protein-coupled BA receptor-1 (GPBAR1).
Evidence points to a close link between BA homeostasis and gut integrity in health and disease. Inflammatory bowel disease (IBD) is associated with disturbances in the gut microbiota and immune imbalance, which, in parallel with the influence of environmental factors, can greatly affect the integrity of the gut barrier [16]. In addition, IBD patients display a consistent shift of the BA pool, e.g., increased fecal concentrations of primary and conjugated BA [17].
In this review, we discuss the bidirectional intersection of BA homeostasis and chronic intestinal inflammation considering novel therapeutic approaches. In recent reviews, we focused on specific aspects of BA homeostasis, enterohepatic circulation, and function as signaling molecules [7, 15].
BA synthesis secretion and absorption
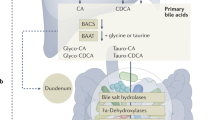
Primary BA (cholic acid [CA] and chenodeoxycholic acid [CDCA]) are synthetized as catabolic products of cholesterol in the pericentral hepatocyte and undergo subsequent conjugation with taurine (2-aminoethanesulfonic acid) and the amino acid glycine (ratio 3:1) through N-acyl amidation at carbon 24 of the aliphatic side chain [18]. This step increases BA solubility in bile (an aqueous solutions) and decreases BA toxicity. BA are actively secreted mainly by the bile salt export pump (BSEP; ABCB11/Abcb11) into the canaliculi [19] and then appear in bile, stored and concentrated in the gallbladder and periodically delivered to the intestine during fasting and mainly during the fat-cholecystokinin-dependent stimulation of the gallbladder in the postprandial period [1].
Reabsorption of about 95% of BA occurs in terminal ileum with uptake by the apical sodium-dependent bile salt transporter (ASBT; SLC10A2/Slc10a2) [20] and binding and transport across the enterocyte by the ileal BA-binding protein (IBABP) [21, 22]. The basolateral BA efflux into the portal circulation requires a third transporter, the organic solute transporters (OSTα and OSTβ heterodimer) [23]. The hepatic reuptake of BA occurs at the basolateral (sinusoidal) membrane, and requires the sodium taurocholate co-transporting polypeptide (NTCP; SLC10A1/Slc10a1) [24]. The sodium-independent basolateral BA uptake into hepatocytes accounts for only 25% of the uptake of mainly unconjugated BA, and is mediated by organic anion transporting polypeptides (OATPs) [1, 19, 25]
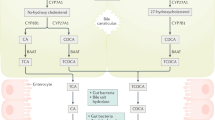
A small amount of primary BA escapes ileal re-absorption and enters the colon, where the resident microbiota promotes the deconjugation, dehydrogenation, and dihydroxylation of primary BA to secondary BA, mainly deoxycholic acid (DCA), small amount of lithocholic acid (LCA), and the “tertiary” ursodeoxycholic acid (UDCA). This additional pool of colonic unconjugated BA undergoes passive diffusion, i.e., ~ 50% DCA, minimal LCA (both mainly insoluble) and UDCA and is transported back to the liver through the portal circulation where both secondary and tertiary BA are conjugated again with taurine or glycine in the liver and re-secreted [26]. This amount of colonic BA which is passively reabsorbed contributes to the enterohepatic circulation of BA with 95% re-absorption at every cycle [27]. The remaining DCA, and a small amount of LCA and UDCA are lost in the feces, accounting for ~ 5% of the total BA pool at every cycle [28]. In health, this BA fecal loss is a fraction of the total amount lost daily according to the number of enterohepatic cycles, and must be compensated by the daily de novo synthesis in the liver [29, 30] (Fig. 1). In general, if the pool cycles 2–3 times per meal, according to the meal frequency, size, and composition, i.e., 4–12 times/day, this increases the BA pool to a “dynamic” size (3 g x − 12 cycles = 12–36 g/day), and a capacity to reabsorb 10–30 g of BA per day [1].

The enterohepatic circulation of bile acids (BA) and qualitative/quantitative composition of the BA pool. Events linked to the synthesis, conjugation, secretion, biotransformation, re-absorption, and excretion of primary, secondary, and tertiary bile acids (BA) in humans at every cycle of the enterohepatic circulation. “Primary” BA, synthetized in the liver starting from cholesterol are the trihydroxy cholic acid (CA) hydroxylated at the 3α,7α,12α positions and the dihydroxy chenodeoxycholic acid (CDCA) hydroxylated at the 3α,7α positions. The two biosynthetic pathways are the classical pathway involving the 7a-hydroxylase which stimulates the 7a-hydroxylation of cholesterol. This major enzyme contributes to more than 75% of total production of primary BA. The alternative pathway is initiated by the sterol-27-hydroxylase which produces mainly CA. BA are actively re-absorbed at the terminal ileum. About 5% of primary BA escape ileal absorption and enter the colon where the resident microbiota initiate BA deconjugation from taurine and glycine, dehydrogenation, dehydroxylation, and epimerization to produce «secondary» BA: the dihydroxy deoxycholic acid (DCA) hydroxylated at the 3α,12α positions and the monohydroxy lithocholic acid (LCA) hydroxylated at the 3α position. The 7α-dehydrogenation of CDCA form the dihydroxy 7α-oxo (keto)-LCA which is metabolized to the “tertiary’ 7β-epimer, the dihydroxy hydrophilic ursodeoxycholic acid (UDCA) hydroxylated at the 3α,7β positions in the colon and to CDCA again in the liver. The 7α-dehydroxylation of the primary BA is the essential reaction to produce DCA and LCA from CA and CDCA, respectively. Both LCA, 7-oxo (keto)-LCA, and UDCA are mainly excreted in feces while about 50% DCA is passively reabsorbed from the colon into the portal tract [27] by ionic more than nonionic diffusion (the remaining part being excreted with feces). Their fate depends on specific physicochemical properties and BA binding to luminal contents. In the liver, a small amount of LCA is quickly transformed in the sulphonated “tertiary” (S-LCA). Altogether, the BA pool at every cycle undergoes re-conjugation with taurine and glycine and new secretion in bile. Fecal loss is minimal (< 5% at every cycle). As an example, when a CA or CDCA pool of 1 g cycles 6 times a day, the daily loss is 5% × 6 cycles = 30% and 300 mg must be resynthesized in the liver [25]. B Relative composition of hepatic and gallbladder bile in health as main solutes (left) and individual bile acids (right). Altogether, the glyco-, tauro-conjugated CA, CDCA and DCA represent more than 90% of the total pool of BA. Abbreviations: G/T glycine, taurine, CA cholic acid, CDCA chenodeoxycholic acid, DCA deoxycholic acid, LCA lithocholic acid, UDCA ursodeoxycholic acid
Deranged BA homeostasis
IBD patients can develop changes of BA synthesis and enterohepatic circulation, both predisposing factors to deranged BA homeostasis. Table 1 depicts the main pathogenic mechanisms able to derange BA homeostasis in IBD patients [17]. In general, mechanisms include changes of BA absorption, microbiota biosynthesis, altered signaling mechanisms, and or deranged BA metabolism.
Early findings documented increased levels of unconjugated BA in subjects with ulcerative colitis (UC) or Crohn’s disease (CD), as compared with healthy subjects. Changes included a decreased BA pool size in CD, but not in UC patients [31]. Nihlin et al. [32] used tauroselcholic [(selenium-75) acid] to assess BA malabsorption and BA pool loss. The authors found BA malabsorption in CD patients and this finding can explain, at least in part, the chronic diarrhea.
Zhen-Huan Yang et al. [33] investigated the relationships between BA, gut microbiota, and gut inflammation in patients with UC. The study found gut dysbiosis with decreased population of Firmicutes, Butyricicoccus, Clostridium XlVa, Faecalibacterium, and Roseburia, and increased pathogens such as E. Coli, Proteobacteria, Klebsiella, and Streptococcus. This deranged microbiota profile was associated in feces with decreased amounts of secondary BA concentration (LCA, DCA, glyco-conjugated GDCA, GLCA, and tauro-conjugated TLCA), and with increased concentrations of primary BA (TCA, CA, TCDA, and GCDA).
In patients with active IBD, another study showed increased rates of conjugated BA and decreased rates of secondary BA profile, as compared with controls [34]. Changes of BA profiles were associated with altered fecal microbiota, i.e., decreased ratio between Faecalibacterium prausnitzii and Escherichia coli, and with significantly decreased bacterial activities of deconjugation, transformation and de-sulphation of BA. The evidence suggests that the presence of gut dysbiosis reduces the anti-inflammatory effects promoted by secondary BA, due to their increased sulphation [34].
Decreased serum levels of BA have been documented in patients with CD, depending on altered intestinal re-absorption of BA at the level of terminal ileum. In UC patients, the level of deoxy-BA such as DCA, LCA, and conjugates was decreased in comparison to healthy and CD subjects, depending on altered colonic microbiota and, in turn, on a decreased deoxidation capacity (7α- dihydroxylation) responsible for the biotransformation of unconjugated to secondary BA [35].
A cross-sectional study measuring the plasma concentrations of 12 BA in patients with CD found decreased GCDCA, TCA, and LCA, and increased GDCA and GCA in patients, as compared with controls [36].
A recent and comprehensive review described, in IBD patients, increased fecal concentrations of CA, CDCA, conjugated BA, sulphated BA, and decreased DCA and secondary BA, as compared with healthy controls [17]. In serum, few studies reported increased GCA concentrations, and reduced LCA, GCDCA, TCDCA, TCA levels in IBD [17]. These findings can be affected by different disease type such as CD or UC, clinical status such as active/inactive disease, and location of inflammatory changes [17, 37]. Recently, however, significantly increased excretion of fecal BA was reported in patients with UC and pan-colonic disease, in a significant proportion of patients with CD affecting ileum or colon, and even in UC or CD patients with quiescent inflammation [38].
During colitis, the activation of hepatic CYP8B1, the cytochrome synthetizing CA, promotes the accumulation of intestinal CA. Consequently, CA inhibits peroxisome proliferator-activated receptor alpha (PPARα) with a decrease in fatty acid oxidation, and markedly affects the renewal of Lgr5 + intestinal stem cells. This pathway ultimately impairs the repairing ability of the gut mucosal barrier, promoting more severe colitis [39].
A longitudinal study of 1 year assessing the gut microbiome in 132 subjects found that gut dysbiosis was associated with IBD. Microbial changes included increased facultative anaerobes, decreased obligate anaerobes, and this profile occurred with decreased rates of secondary BA, i.e., LCA and DCA, and molecular disruptions in microbial transcription and metabolite pools such as short-chain fatty acids [40].
Both T helper 17 cells (Th17) expressing interleukin-17A, and regulatory T cells (Treg) play a critical role in IBD and become sensitive to changes of BA homeostasis. An imbalance between Th17 which promotes tissue inflammation and Treg which suppresses autoimmunity contribute to the onset and progression of IBD. Both gut microbiota and BA [13] can influence the production and maintenance of these immunological cells [41]. The LCA metabolite 3-oxo-LCA inhibits the differentiation of TH17 cells, while the other LCA derivative isoallo-LCA increases the differentiation of Treg cells [13]. Notably, IBD patients display a marked reduction of 3-oxoLCA, iso-LCA and the 3α-hydroxysteroid dehydrogenase (3α-HSDH) genes required for their biosynthesis. The reduced expression of TH17 cell-associated genes depends on the increased levels of these BA, which can strongly influence the onset and progression of IBD [14].
Deranged BA signaling
BA are well-known signaling molecules interacting with nuclear and membrane-associated receptors [7, 15].
FXR is the main sensor of BA in the intestine and the liver and regulates BA synthesis by negative feedback mechanisms which also involve the intestinal secretion of the fibroblast growth factor 19 in humans [7, 15, 42, 43]. Feedbacks are deeply connected with the enterohepatic circulation of BA [44] and with the profile of gut microbiota in health and disease [1, 45]. The signaling role of BA additional receptors include the GPBAR-1 [2, 46], and the sphingosine-1-phosphate receptor 2 (S1PR2) [47, 48] in the intestine, in the liver, in the muscle and in the brown adipose tissue [7, 49], and the retinoid X receptor (RXR), the small heterodimer partner (SHP), the liver receptor homologous-1 (LRH-1), and liver X receptor (LXR) in the liver [49].
As a consequence of these inter-related pathways, altered signaling secondary to disrupted BA homeostasis may lead to multi-level dysfunction in the liver, i.e., intrahepatic cholestasis [50], liver steatosis, fibrosis, and hepatocellular carcinoma [1, 51]. At the extrahepatic level, derangement of BA homeostasis can contribute to extraintestinal cancer [51] and may affect energy expenditure [52, 53], glucose homeostasis [54], lipid homeostasis [55,56,57,58], inflammatory and immune responses [59, 60].
In the liver, FXR plays an anti-inflammatory role by reducing the availability of toxic BA especially during cholestasis [61, 62]. FXR activation inhibits monocytes/macrophages migration and tissue infiltration promoted by the chemokine monocyte chemoattractant protein-1 (MCP-1/CCL2). This step contributes to reduce liver fibrosis [62]. The use of FXR agonists leads to a reduced hepatic inflammation and fibrosis secondary to a concentration-dependent suppression of NF-κB-mediated inflammation [61]. FXR activation also stimulates anti-fibrotic gene expression in hepatic stellate cells (HSCs) through activation of FXR, induction of SHP, increased expression of peroxisomal proliferator-activated receptor γ (PPARγ) [63, 64]. Finally, in the liver, BA can have pro-inflammatory effects mediated by the intracellular assembly of the inflammasome. In this context, FXR is able to interact with the NACHT LRR and PYD domains-containing protein 3 (NLRP3) protein machinery, generating anti-inflammatory effects [65].
In the intestine, FXR has anti-inflammatory effects mainly preserving the integrity of the gut barrier [4, 7], modulating immune and inflammatory pathways by a regulation of cells involved in innate immunity [66, 67], and modulating the composition of gut microbiota [68].
In a context of deranged BA signaling, FXR- and GPBAR1-mediated immune effects can play a role through the modulatory effector functions in cells of innate immunity. In these cells, FXR activation generates a tolerogenic phenotype either at hepatic and intestinal level, with marked anti-inflammatory and anti-fibrogenic effects. However, the translational value of results from animal studies showing a role of BA as effective immune modulators in humans is still poorly documented [69].
As observed in the liver, the relationships between BA, FXR, and inflammatory pathways involving NF-κB are also active at intestinal level. DCA levels in feces can increase in response to a high-fat diet and this step is associated with increased rate of Gram-positive bacteria [70]. In the intestine, increased DCA concentration has been linked with gut inflammation and carcinogenesis. DCA-treated APC (min/ +) mice showed altered gut barrier, low-grade gut inflammation, and tumor progression [71]. DCA is able to promote colonic pro-inflammatory macrophage infiltration, pro-inflammatory cytokine production, and macrophage polarization through NF-κB/ERK/JNK signaling downstream of toll-like receptor 2 (TLR2), driving colonic inflammation [70].
FXR activation inhibits NF-κB at the intestinal level, with local anti-inflammatory effects. In animal models, FXR target gene expression (but nor mRNA expression) is decreased by inflammatory stimuli through NF-κB [66]. In addition, FXR activation decreases epithelial permeability and modulates the expression of genes involved in gut inflammation [66].
Besides FXR, the NF-κB-mediated inflammatory pathway in the intestine can be suppressed by the pregnane X receptor (PXR) [72], another nuclear receptor involved in IBD pathogenesis [66, 67, 72]. In the animal model, the administration of a PXR agonist protected wild type but not PXR-null mice from colitis induced by dextran sulphate sodium, decreasing mRNA expression of several NF-κB target genes [72]
The anti-inflammatory role of FXR is evident in Fxr − / − mice. These animals show a marked pro-inflammatory cytokine mRNA expression in the colon. Of note, the administration of the FXR ligand 6-ECDCA inhibits the expression of pro-inflammatory molecules in wild type but not in Fxr − / − animals [67].
FXR modulates the expression of several genes involved in gut permeability and inflammation, two factors involved in intestinal bacterial overgrowth [66,67,68]. FXR can inhibit bacterial overgrowth and mucosal injury in the ileum following bile duct ligation. This FXR-mediated effect protects the distal ileum from bacterial invasion and epithelial damage [68]. The beneficial role of FXR activation on intestinal inflammation seems to depend on FXR interaction with genes promoting antibacterial effects, i.e., genes encoding angiogenin, carbonic anhydrase 12, and inducible nitric oxide synthase, and on induction of IL-18 [68].
A study exploring the relationships between plasma BA profile and FXR/PXR activation in patients with CD found a reduced activation of target genes secondary to the deranged BA composition and, in turn, to the altered BA signaling [36]. Notably, the reduced FXR/PXR agonism can negatively affect the progression of IBD [66, 67, 72, 73].
Finally, a critical role is emerging for GPBAR1, the cell surface BA-activated receptor highly expressed in the ileum and colon [7]. The susceptibility to develop a severe colitis is significantly increased in GPBAR1(-/-) mice, due to marked alterations in the intestinal barrier [74]. On the other hand, in animal models, GPBAR1 agonists prevent gut inflammation [75]. A recent study in patients with CD demonstrated that GPBAR1 can modulate, in the colon, the expression of ACE2 [76], a receptor involved in intestinal inflammatory processes [77] and able to attenuate intestinal inflammation [76].
Deranged BA–microbiota axis
The gut barrier is an anatomical and functional structure at the border between external environment, i.e., the gut lumen and the host body [4]. The integrity of the barrier depends on the dynamic interaction between several factors: gut microbiota, luminal content of nutrients, mucin, gastrointestinal motility, and secretions, i.e., gastric acid, bile, pancreatic juice, intestinal cells, i.e., enterocytes, Paneth cells, Goblet cells with their tight junctions. Essential components of the gut barrier include also immune-modulating components such as antimicrobial peptides, i.e., microbial- [MAMPs] and pathogen-[PAMPs] associated molecular patterns, toll-like receptors [TLRs], B/T lymphocytes, and cells composing the gut-vascular barrier, i.e., endothelium associated with pericytes and enteric glial cells with specific tight junction and adherens junctions.
As part of the gut barrier machinery, the microbiota and BA have a critical role in maintaining the integrity of the intestinal barrier due to the close bidirectional crosstalk [49, 78,79,80,81] and potential influence on the onset and progression of chronic intestinal inflammation [82]. Of note, a dysfunction of the gut barrier can precede and predict the development of IBD by years [83, 84]. Table 2 lists the main mechanisms linking gut dysbiosis with the pathogenesis of IBD, all pointing to a critical involvement in both local inflammation and altered intestinal barrier.
As compared with healthy individuals, IBD patients show reduced bacterial abundance and diversity [82, 85], with a decrease of Firmicutes and Bacteroidetes, and increased Proteobacteria and Enterobacteriaceae [33, 86,87,88,89]. Reduced bacterial diversity has been described in both inflamed and non-inflamed colon sites in patients with IBD, although inflamed sites seem enriched with specific bacterial species i.e., Cloacibacterium and Tissierellaceae, as compared with non-inflamed tissues [90]. The relative abundance of gut microbes also changes with the activity of IBD, and a lower abundances of Clostridium coccoides, Clostridium leptum, F. prausnitzii, and Bifidobacterium has been linked with periods of disease remission [91]. Despite the association between gut dysbiosis and IBD has been well documented, the causal role of altered gut microbiota in the determination of chronic intestinal inflammation is still under debate. The shift of microbiome in IBD patients may represent a microbial response secondary to local inflammatory changes, rather than having a causal role [85, 92]. Nevertheless, several gut inflammatory pathways can be activated by unbalance between harmful and beneficial gut microbes [93, 94].This condition occurs during upregulation of pathogenic bacteria species, i.e., Enterobacteriaceae [95, 96], Clostridium difficile [97], and decreased abundances of beneficial bacteria species, i.e., Clostridium clusters IV and XIVa, Faecalibacterium prausnitzii, Eubacterium [98, 99]. This unbalance may also lead to increased production of pro-inflammatory lipopolysaccharides (LPSs) and their filtration across the altered gut barrier unable to maintain a selective normal permeability [95]. In line with this evidence, specific bacterial species, (i.e., Lactobacillus, and Faecalibacterium within Firmicutes; Bifidobacterium within Actinobacteria) [92] can have a beneficial role in IBD patients.
Of note, the reduced microbial abundance in IBD patients involves bacteria like Bacteroides, Clostridium, Lactobacillus, Bifidobacterium, and Listeria carrying bile salt hydrolase (BSH), the enzyme involved in the biotransformation of conjugated into unconjugated BA [100, 101], and microbes (mainly Bacteroides, Clostridium, Eubacterium, and Lactobacillus) responsible for the 7α-dehydroxylation of unconjugated BA and, therefore, for their bio-transformation to secondary BA [102].
As shown in an animal models, the cecal concentrations of UDCA and LCA, its primary metabolite, were protective against the disruption of epithelial permeability and colonic inflammation, inhibiting colonic epithelial caspase-3 cleavage and epithelial apoptosis [103].
In a group of patients with UC, the reduced diversity of gut microbiota as compared with healthy controls was in line with decreased microbes such as Firmicutes, Clostridium IV, Butyricicoccus, Clostridium XlVa, Faecalibacterium, and Roseburia, and enrichment in Proteobacteria, Escherichia, Enterococcus, Klebsiella, and Streptococcus. These changes caused a significant decrease of secondary BA, with increased primary BA, altered GPBAR1 expression, and increased production of pro-inflammatory cytokines [33].
As previously mentioned, the link between gut dysbiosis and altered profile of gut BA can reduce the FXR/PXR agonism, while promoting the IBD progression through altered BA signaling functions [66, 67, 72, 73]. The altered intestinal profile of BA secondary to dysbiosis can affect the intestinal permeability, together with the dysregulation of bacterial metabolites usually contributing to the maintenance of the integrity of gut barrier, as short-chain fatty acids (SCFA) like butyrate, acetate, and propionate [104, 105]. In a mouse model of autism spectrum disorders, a reduction in the relative abundance of Bifidobacterium and Blautia, bile-metabolizing species, was linked in the intestine with deficient BA and tryptophan metabolism and with increased intestinal macromolecular permeability [106]. Cytotoxic effects of elevated concentration of BA on the intestinal epithelium have been observed in cells, animals, and humans, and are able to affect the integrity of the gut barrier [107,108,109]. These effects are mediated by different inflammatory and apoptotic molecules as phospholipase A2 (PLA2)- cyclooxygenase (COX)-protein kinase C (PKC), extracellular signal-regulated kinase 1 (ERK1), p38 mitogen-activated protein kinase (p38 MAPK), and phosphatidylinositol 3-kinase (PI3K), which can be activated by altered intestinal BA profile [110,111,112,113,114,115,116,117].
In vitro models of gut barrier based on monolayers of human intestinal Caco-2 cells contributed to document the negative, cytotoxic effects of hydrophobic BA (mainly unconjugated BA), possibly leading to increased gut permeability and inflammation. In this model, CA decreases the transepithelial electrical resistance (TEER) and increases intracellular ROS generation. These effects seem to be mediated by the activation of signaling pathway involving PLA2, COX, PKC ERK1/2, PI3K, p38 MAPK, MLCK, NADH dehydrogenase, and XO (xanthine oxidase) [118]. In the same cellular model, CA, DCA, and CDCA, but not UDCA, decreases TEER and increase paracellular permeability [119]. Furthermore, CDCA or DCA promoted a ligand-independent activation of the epithelial growth factor receptor (EGFR), which correlates with increased paracellular permeability via occludin dephosphorylation and cytoskeletal rearrangement at the tight junctions [119].
In animal models (mice) of colitis, increased intestinal permeability at the level of the colon was linked with decreased proportion of UDCA, increased DCA, and increased jejunal FXR expression [120, 121]. Furthermore, mice with colitis induced by dextran sodium sulphate (DSS) show increased fecal BA hydrophobicity. Notably, the severity of symptoms correlated positively with fecal BA hydrophobicity and fecal DCA concentration [122].
Mice fed a choline-deficient, l-amino acid- deficient, high-fat diet showed reduced concentrations of conjugated BA, which was paralleled by increased gut permeability. In vitro, conjugated BA protected gut epithelial monolayers from the damage induced by unconjugated BA through micelle formation [123].
In mice with DSS-induced colitis, gut inflammation worsened after administration of a ketogenic diet, which induced an upregulation of serum and colon inflammatory cytokines and chemokines (IL-1α, IL-6, TNF-α, IL-17, GM-CSF and IL-10), increased gut permeability, and decreased the expression of intestinal-epithelial-barrier-associated genes. These changes were linked with significant variations in bacterial abundance, i.e., increased pathogenic taxa as Proteobacteria, Enterobacteriaceae, Helicobacter, Escherichia-Shigella; reduced beneficial taxa as Erysipelotrichaceae, and with altered concentration of microbial metabolites, including BA (i.e., increased TCDCA, CA, GCA) [124].
Impaired BA homeostasis can significantly affect the modulatory role of BA on the proliferation of epithelial cells [2, 3], gene expression [5, 6], and epigenetic mechanisms [8, 9], including the interactions between microbial and host genes [125] and the gut metabolome, the molecular interface between host and microbiota [126]. In IBD patients, variations in the relative abundance of mucosa-adherent microorganisms are able to modulate the expression of several host genes [40, 127, 128], and an altered BA homeostasis seems to have a critical role in this process [129].
A study on colonic biopsies from patients with primary sclerosing cholangitis (PSC), who frequently have colitis, UC patients and healthy controls reported different microbiota profiles and significantly different colonic transcriptome, with 939 genes sharing differential gene expression in patients (both UC and PSC), as compared with controls. In patients, imputed pathways were linked with upregulation of immune response and microbial defense, and BA signaling pathways were upregulated in PSC-IBD, as compared with UC [129].
Finally, a study on endoscopic mucosal biopsies (ileum and colon) from IBD patients documented a deficient microbial gene pathway involved in the biosynthesis of secondary BA in inflamed terminal ileum. In samples from non-inflamed colon, the relative abundance of BA-inducible microbial genes directly correlated with the expression, in the host, of Angiopoietin-like 4 (Angptl4) [125], a gene able to attenuate colonic inflammation in animal models [130]. The correlation between BA-inducible microbial genes and Angptl4 gene expression disappeared with inflammation [125].
Potential therapeutic implications
The available evidence suggests that there is a link between IBD and BA homeostasis, and that there is a room for potential therapeutic approaches that can modify the clinical course of disease. Most relevant approaches include BA therapy, gut microbiota modulation, and use of potent FXR agonists.
Therapy with BA
Therapeutic approaches for liver diseases have used hydrophilic BA, i.e., the “tertiary” UDCA acid, the conjugated tauro-UDCA, and, more recently, nor-UDCA [131]. This strategy decreases the hydrophobicity of the BA pool and the cytotoxic effect which occurs at the level of enterocytes [132].
In an animal model of CD, the administration of UDCA was beneficial through positive effects on the intestinal barrier and by reducing the oxidative stress [133]. In the animal models of IBD, the intraperitoneal administration of UDCA and LCA had protective effects against increased epithelial permeability and colonic inflammation. The mechanism included the inhibition of epithelial apoptosis [103] and cytoprotective and anti-inflammatory effects [134].
The beneficial effects of tertiary BA also depend, at least in part, on changes in gut microbiota secondary to the mutated intraluminal BA concentration. In mice UDCA, TUDCA, GUDCA restored the Firmicutes to Bacteroidetes ratio after a colitis-induced dysbiosis, prevented the loss of Clostridium cluster XIVa, and increased the abundance of protective species (in particular Akkermansia muciniphila) [135].
Looking at the effect of BA therapy in IBD, available results in humans are scarce and need further confirmation. Preliminary evidence in UC patients found better therapeutic effects, i.e., reduced Mayo and IBDQ scores when UDCA 200 mg b.i.d. was added to mesalamine. Of note, the combined treatment was also able to modulate the gut microbiota by increased Firmicutes and reduced Proteobacteria, as compared with subjects on mesalamine alone [136].
To counteract the altered BA balance documented in IBD patients, a displacement therapy should be aimed to inhibit the synthesis of primary BA or to increase the fecal elimination of toxic BA through BA binders, as cholestyramine. In an animal model of IBD, cholestyramine attenuates intestinal ulceration [137]. In subjects with collagenous colitis, adding cholestyramine (4 g/day) to mesalamine increases the rate of beneficial therapeutic response (100%, as compared with 73% in mesalamine alone) [138]. The use of cholestyramine is indicated to counteract chronic diarrhea linked with BA malabsorption in CD [32]. In patients with IBD linked with primary sclerosing cholangitis and receiving optimized anti-TNF therapy for IBD, the use of cholestyramine induced a rapid and sustained drop in fecal calprotectin levels [139].
Therapy with probiotics and prebiotics
According to WHO and FAO, probiotics are “live microorganisms when administered in adequate amounts confer a health benefit on the host”. The administration of probiotics (mainly Lactobacillus [140], Bifidobacterium [141, 142], S. boulardii [143, 144], L. rhamnosus GG [145,146,147,148], L. johnsonii LA1 [149, 150], E. faecium [146], VSL#3 [151, 152], E. Nissle 1917 [153,154,155]) can have beneficial effects in IBD patients by acting on the microbiota/BA axis. The therapeutic effects of probiotics likely involve improved gut barrier function and the recovery of physiological gut microbiota involved in the bio-transformation and homeostasis of BA, and ultimately modulating the profile of the luminal pool of BA [156].
In animal models and in humans, additional therapeutic effects of probiotics (mainly Lactobacillus plantarum CCFM8661, Lactobacillus reuteri NCIMB 30242, VSL#3) involve the activation of the fibroblast growth factor (FGF)19 and 15 [157,158,159] and, in turn, enhanced synthesis and excretion of BA [15].
Results of controlled trials using probiotics, however, are controversial with few studies reporting no effects on relieving relapse [143, 150, 160] and uncertain beneficial effects [153, 161]. A meta-analysis exploring ten randomized controlled trials found that probiotics can induce remission during the active period of UC, but have no significant effects in maintaining CD and UC remission [162]. Another recent systematic review on the use of probiotics in IBD patients reported no clear beneficial effects in CD patients, but positive effects in inducing remission in patients with active UC [163].
In a recent study in IBD patients, the probiotic strain Bacillus clausii UBBC-07 positively modulated the gut microbiota and cytokine secretion, and was associated with a significant decrease of symptoms [164].
Akkermansia muciniphila represents 1–4% of gut microbiota in healthy humans [165]. IBD patients show decreased rates of A. muciniphila [165, 166] and, in the mice models of colitis, the administration of A. muciniphila improves intestinal permeability [167], decreases colon inflammation and the expression of pro-inflammatory cytokines (TNF-α, IFN-γ) [168]. In mice, the administration of protein components of the outer membrane protein from A. muciniphila protects from the development of colitis [169]. A. muciniphila can also play a role in the modulation of immune responses mediated by the Toll-like receptor 4 (TLR4), a sensor of gut microbiota alterations sensible to the intestinal concentration of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [170, 171]. A recent study in TLR4-/- mice reported a protective role of TLR4 against the development of intestinal inflammation, linked with the relative abundance of A. muciniphila and the proportion of suppressive RORγt + Treg cells [172]. The close crosstalk between microbiota and BA was disclosed by an experimental model of dextran sodium sulfate-induced colitis in mice, since the administration of UDCA decreased the inflammatory changes and increased the abundance of A. muciniphila [135]. In another animal model of early obesity and non-alcoholic fatty liver disease, the administration of A. muciniphila was associated with increased plasma levels of unconjugated, hydrophilic BA and with increased expression of hepatic genes involved in BA synthesis and transport, pointing to a critical role of A. muciniphila in the modulation of BA signaling [173].
Prebiotics are substrates selectively employed by gut microbes, providing beneficial effects as the development of probiotics (including Ruminococcaceae, Lachnospiraceae, and Bifidobacterium) and the formation of metabolites as SCFAs and BA [174,175,176,177].
The most common prebiotics employed in IBD are lactulose [178], fructo-oligosaccharide (FOS) [179], germinated barley foodstuff [180, 181], ispaghula husk [182], Plantago ovata seeds [183], and inulin [184, 185]. However, the effects of chronic supplementation with these prebiotics on BA homeostasis in IBD patients are still scarcely explored and lead to uncertain results.
In humans, chronic ingestion of lactulose seems to be able to increase Bifidobacteria but not to significantly change fecal BA [186, 187]. Nevertheless, in a previous study, 12 weeks of lactulose 60 g/day decreased secondary BA absorption, decreasing the DCA pool size, with a rise in primary BA [188].
In healthy subjects, it has been reported that long-term FOS administration is able to decrease fecal DCA [189]. A previous evidence, however, was unable to demonstrate significant changes in fecal BA concentration [190].
In an experimental model of colitis, mice receiving germinated barley foodstuff showed a reduced epithelial inflammatory response, paralleled by increased butyrate production and lower BA concentration, as compared with control animals [191]. This dietary fiber is able, in vitro, to strongly adsorb hydrophobic bile salts [192]. However, studies exploring the effects of germinated barley foodstuff on BA homeostasis in humans are still lacking.
The effects of long-term (8 weeks) supplementation with ispaghula husk on the fecal output of BA have been explored in healthy adult subjects, showing a significant decrease of fecal LCA and iso-LCA and the weighted ratio of LCA to DCA, pointing to a reduction of the hydrophobicity of the BA pool [193].
Plantago ovata seeds had no effect on fecal BA excretion in a small group of normal subjects [194]. In guinea pigs, however, the husks from Plantago ovata significantly increased fecal BA, affecting BA absorption [195].
Finally, in animals, inulin increases the fecal concentration of DCA and LCA [177] and changes the composition of gut microbiota and the levels of related metabolites, as BA [185]. The effects of inulin decrease with deletion of FXR, and modulate the pathogenic mechanisms involved in chronic gut inflammation [185]. In patients with an ileal pouch-anal anastomosis, the administration of 24 g of inulin during 3 weeks decreased the numbers of Bacteroides fragilis and reduced the fecal concentrations of secondary BA, with beneficial effects on the mucosal inflammation in the ileal reservoir[196].
Agonists of BA receptors
Studies in animal models documented beneficial effects from FXR activation by specific agonists as INT-747 [11], fexaramine [197], and GW4064 [198] documented by prevention of colitis, anti-inflammatory effects, restored BA homeostasis, and gut microbiota modulation. In a mouse model of colitis, INT-747 alleviated colon inflammation downregulating pro-inflammatory cytokines and preserving gut barrier function [11, 67]. In mice with DCA-induced intestinal damage, the administration of fexaramine decreased the injury, increased the abundance of SCFA-producing bacteria, and normalized BA homeostasis through beneficial effects of the FXR/FGF15 axis [197]. The administration of GW4064 generated favorable effects in an animal model of ileum injury induced by lipopolysaccharides decreasing tight junction dysfunction, macrophage infiltration, inflammatory pathways, and mitochondrial dysfunction with FXR-dependent mechanisms [198]. However, results from another in vitro study on colonic epithelial restitution and wound healing in T84 cell monolayers documented an harmful inhibition of wound closure by GW4064, with a downregulation of CFTR gene expression [199].
Conclusion and future perspectives
A dynamic crosstalk exists between BA homeostasis which includes signaling effects on nuclear and membrane receptors, gut microbiota, and maintenance of gut barrier integrity. These critical factors can become actors in the onset and progression of chronic intestinal inflammatory diseases (Fig. 2). More studies must identify key aspects lacking the full translational value. In particular:
-
1.
The potential links between gut dysbiosis, BA homeostasis, and IBD pathogenesis, point to novel therapeutic strategies. The translational value of available animal and experimental studies, however, must be confirmed in clinical trials considering the role of confounders such as age, dietary habits, lifestyle, ethnicity, drugs, possible chronic ingestion of toxic chemicals with diet, altered metabolic homeostasis, and comorbidities. In humans, the combination of these factors limits the ultimate identification of the causal role of gut dysbiosis in the onset and progression of chronic intestinal inflammation. Well-designed, accurate, and prospective studies are needed with respect to gene–environment interactions, and epigenetic mechanisms. oth artificial intelligence and multiomics can provide additional information in this respect [200, 201]. Starting from machine learning models [202, 203], these techniques will likely contribute to the advancement in the knowledge of the pathogenic mechanisms linking BA, gut microbiota, and gut inflammation. Results will facilitate disease management and can pave the way to primary prevention measures.
-
2.
Animal studies reveal beneficial interplays between gut microbiota variations and anti-inflammatory effects of secondary and tertiary BA. However, the effective value of microbiota transplantation and/or of BA therapy in the management of humans with IBD is still uncertain. Although promising results derive from experimental studies with hydrophilic BA, large randomized, controlled trials targeting the role of BA therapy are still needed. Further studies are also expected to verify the possible convenience, in humans, of BA displacement therapy using BA binders as cholestyramine.
-
3.
BA are signaling molecules for FXR and GPBAR-1. The interactions between BA and membrane/nuclear receptors can generate anti-inflammatory and immune-modulating effects at the intestinal level, mainly acting on cells involved in innate immunity. Preclinical studies indicate that external manipulation of the BA receptors (mainly FXR) with specific agonists can have positive effects in terms of both clinical remission during active periods and maintenance of remissions. To date, however, results in humans are scarce and need further confirmation, also in terms of combination with standard treatments. Although a number of clinical trials are on the way using FXR agonists in chronic liver diseases and in several metabolic disorders [15], no evidence exists on the use of these agents in humans with IBD. We need caution when considering the potential negative effects on healing of the inflamed colon and on expression of genes involved in the maintenance of gut barrier [199].
-
4.
The precise therapeutical efficacy of prebiotics and/or probiotics in patients with IBD requires additional validation and well-designed randomized controlled trials. Promising results derive from the supplementation with some probiotics (mainly Lactobacilli, Bifidobacteria, and Akkermansia muciniphila) and prebiotics (mainly germinated barley foodstuff and inulin). Long-term effects of such therapeutic approaches are also uncertain.
-
5.
We need to clarify if combined multifaceted approaches (including lifestyle changes, environmental exposures, and innovative drugs) aimed at restoring BA homeostasis and gut dysbiosis, do have additional value in the short and in the long term as compared with conventional drug treatment.

Summary of major mechanisms involved in the interaction of bile acids with gut inflammation in the pathogenesis of inflammatory bowel disease. Disturbed BA homeostasis can activate pro-inflammatory pathways in the gut, and inflammatory bowel disease (IBD) can, in turn, induce qualitative and/or quantitative changes in the BA pool in response to gut dysbiosis, leading to impaired mucosal barrier repair due to chronic inflammation. Legend: BA bile acids, FXR farnesoid X receptor, GPBAR1 G protein -coupled bile acid receptor-1
Data availability
Not applicable.
References
Di Ciaula A, Garruti G, Lunardi Baccetto R, Molina-Molina E, Bonfrate L, Wang DQH et al (2017) Bile Acid Physiology. Ann Hepatol 16:S4–S14
Sorrentino G, Perino A, Yildiz E, El Alam G, Bou Sleiman M, Gioiello A et al (2020) Bile Acids Signal via TGR5 to Activate Intestinal Stem Cells and Epithelial Regeneration. Gastroenterology 159(956–68):e8
Guillot A, Guerri L, Feng D, Kim SJ, Ahmed YA, Paloczi J et al (2021) Bile acid-activated macrophages promote biliary epithelial cell proliferation through integrin alphavbeta6 upregulation following liver injury. J Clin Invest 131(9):e132305
Portincasa P, Bonfrate L, Khalil M, Angelis M, Calabrese FM, D'Amato M et al (2021) Intestinal barrier and permeability in health, obesity and NAFLD. Biomed 10(1):83
Hylemon PB, Takabe K, Dozmorov M, Nagahashi M, Zhou H (2017) Bile acids as global regulators of hepatic nutrient metabolism. Liver Res 1:10–16
Jiao N, Baker SS, Chapa-Rodriguez A, Liu W, Nugent CA, Tsompana M et al (2018) Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 67:1881–1891
Portincasa P, Di Ciaula A, Garruti G, Vacca M, De Angelis M, Wang DQ (2020) Bile Acids and GPBAR-1: dynamic interaction involving genes. Environ Gut Microb Nutr 12:3709
Pavlovic N, Stanimirov B, Mikov M (2017) Bile acids as novel pharmacological agents: the interplay between gene polymorphisms, epigenetic factors and drug response. Curr Pharm Des 23:187–215
Kim YC, Jung H, Seok S, Zhang Y, Ma J, Li T et al (2020) MicroRNA-210 Promotes Bile Acid-Induced Cholestatic Liver Injury by Targeting Mixed-Lineage Leukemia-4 Methyltransferase in Mice. Hepatology 71:2118–2134
Yokota A, Fukiya S, Islam KB, Ooka T, Ogura Y, Hayashi T et al (2012) Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut microbes 3:455–459
Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S et al (2011) Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60:463–472
Thibaut MM, Bindels LB (2022) Crosstalk between bile acid-activated receptors and microbiome in entero-hepatic inflammation. Trends Mol Med 28:223–236
Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J et al (2019) Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 576:143–148
Paik D, Yao L, Zhang Y, Bae S, D’Agostino GD, Zhang M et al (2022) Human gut bacteria produce Tau(Eta)17-modulating bile acid metabolites. Nature 603:907–912
Di Ciaula A, Bonfrate L, Baj J, Khalil M, Garruti G, Stellaard F et al (2022) Recent advances in the digestive, metabolic and therapeutic effects of farnesoid x receptor and fibroblast growth factor 19: from cholesterol to bile acid signaling. Nutrients 14(23):4950
Jakubczyk D, Leszczynska K, Gorska S (2020) The effectiveness of probiotics in the treatment of inflammatory bowel disease (IBD)-a critical review. Nutrients 12(7):1973
Li N, Zhan S, Tian Z, Liu C, Xie Z, Zhang S et al (2021) Alterations in Bile Acid Metabolism Associated With Inflammatory Bowel Disease. Inflamm Bowel Dis 27:1525–1540
Russell DW (2003) The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72:137–174
Trauner M, Boyer JL (2003) Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev 83:633–671
Wong MH, Oelkers P, Craddock AL, Dawson PA (1994) Expression cloning and characterization of the hamster ileal sodium-dependent bile acid transporter. J Biol Chem 269:1340–1347
Chen M-J, Liu C, Wan Y, Yang L, Jiang S, Qian D-W et al (2021) Enterohepatic circulation of bile acids and their emerging roles on glucolipid metabolism. Steroids 165:108757
Gong YZ, Everett ET, Schwartz DA, Norris JS, Wilson FA (1994) Molecular cloning, tissue distribution, and expression of a 14-kDa bile acid-binding protein from rat ileal cytosol. Proc Natl Acad Sci USA 91:4741–4745
Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV et al (2005) The heteromeric organic solute transporter α-β, Ostα-Ostβ, is an ileal basolateral bile acid transporter. J Biol Chem 280:6960–6968
Hagenbuch B, Meier PJ (1994) Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Investig 93:1326–1331
Stellaard F, Lutjohann D (2021) Dynamics of the enterohepatic circulation of bile acids in healthy humans. Am J Physiol Gastrointest Liver Physiol 321:G55–G66
Dowling RH (1973) The enterohepatic circulation of bile acids as they relate to lipid disorders. J Clin Pathol Suppl (Ass Clin Path) 5:59
Stellaard F, Paumgartner G (1987) A new model to assess deoxycholic acid metabolism in health using stable isotope dilution technique. Eur J Clin Invest 17:63–67
Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M (2017) Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology 65:350–362
Wang DQH, Neuschwander-Tetri BA, Portincasa P (2017) The biliary system, 2nd edn. Morgan & Claypool Life Sciences Ed, San Rafael, CA (USA). https://doi.org/10.4199/c00147ed2v01y201611isp071
Li T, Chiang JY (2014) Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 66:948–983
Vantrappen G, Ghoos Y, Rutgeerts P, Janssens J (1977) Bile acid studies in uncomplicated Crohn’s disease. Gut 18:730–735
Nyhlin H, Merrick MV, Eastwood MA (1994) Bile acid malabsorption in Crohn’s disease and indications for its assessment using SeHCAT. Gut 35:90–93
Yang ZH, Liu F, Zhu XR, Suo FY, Jia ZJ, Yao SK (2021) Altered profiles of fecal bile acids correlate with gut microbiota and inflammatory responses in patients with ulcerative colitis. World J Gastroenterol: WJG 27:3609–3629
Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E et al (2013) Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62:531–539
Gnewuch C, Liebisch G, Langmann T, Dieplinger B, Mueller T, Haltmayer M et al (2009) Serum bile acid profiling reflects enterohepatic detoxification state and intestinal barrier function in inflammatory bowel disease. World J Gastroenterol: WJG 15:3134–3141
Wilson A, Almousa A, Teft WA, Kim RB (2020) Attenuation of bile acid-mediated FXR and PXR activation in patients with Crohn’s disease. Sci Rep 10:1866
Suchy FS, Balistreri WF (1981) Ileal dysfunction in Crohn’s disease assessed by the postprandial serum bile acid response. Gut 22:948–952
Vijayvargiya P, Gonzalez Izundegui D, Calderon G, Tawfic S, Batbold S, Saifuddin H et al (2022) Increased Fecal Bile Acid Excretion in a Significant Subset of Patients with Other Inflammatory Diarrheal Diseases. Dig Dis Sci 67:2413–2419
Chen L, Jiao T, Liu W, Luo Y, Wang J, Guo X et al (2022) Hepatic cytochrome P450 8B1 and cholic acid potentiate intestinal epithelial injury in colitis by suppressing intestinal stem cell renewal. Cell Stem Cell 29(1366–81):e9
Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW et al (2019) Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569:655–662
Yan JB, Luo MM, Chen ZY, He BH (2020) The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J Immunol Res 2020:8813558
Gonzalez FJ (2012) Nuclear receptor control of enterohepatic circulation. Compr Physiol 2:2811–2828
Matsubara T, Li F, Gonzalez FJ (2013) FXR signaling in the enterohepatic system. Mol Cell Endocrinol 368:17–29
Sun L, Cai J, Gonzalez FJ (2021) The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat Rev Gastroenterol Hepatol 18:335–347
Di Ciaula A, Baj J, Garruti G, Celano G, De Angelis M, Wang HH et al (2020) Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. J Clin Med 9:2648
Ma K, Tang D, Yu C, Zhao L (2021) Progress in research on the roles of TGR5 receptor in liver diseases. Scand J Gastroenterol 56:717–726
Wang Y, Aoki H, Yang J, Peng K, Liu R, Li X et al (2017) The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 65:2005–2018
Nagahashi M, Yuza K, Hirose Y, Nakajima M, Ramanathan R, Hait NC et al (2016) The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J Lipid Res 57:1636–1643
Garruti G, Wang HH, Bonfrate L, de Bari O, Wang DQ, Portincasa P (2012) A pleiotropic role for the orphan nuclear receptor small heterodimer partner in lipid homeostasis and metabolic pathways. J Lip 2012:304292
Zollner G, Trauner M (2008) Mechanisms of cholestasis. ClinLiver Dis 12(1–26):vii
Di Ciaula A, Wang DQ, Molina-Molina E, Lunardi Baccetto R, Calamita G, Palmieri VO et al (2017) Bile Acids and Cancer: Direct and Environmental-Dependent Effects. Ann Hepatol 16:s87–s105
Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K (2011) The bile acid membrane receptor TGR5: a valuable metabolic target. Dig Dis 29:37–44
Broeders EP, Nascimento EB, Havekes B, Brans B, Roumans KH, Tailleux A et al (2015) The Bile Acid Chenodeoxycholic Acid Increases Human Brown Adipose Tissue Activity. Cell Metab 22:418–426
Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G et al (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10:167–177
Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B (2003) Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol 17:259–272
Sirvent A, Claudel T, Martin G, Brozek J, Kosykh V, Darteil R et al (2004) The farnesoid X receptor induces very low density lipoprotein receptor gene expression. FEBS Lett 566:173–177
Claudel T, Inoue Y, Barbier O, Duran-Sandoval D, Kosykh V, Fruchart J et al (2003) Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology 125:544–555
Schoenfield LJ, Lachin JM (1981) Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: the National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann Int Med 95:257–282
Perino A, Schoonjans K (2015) TGR5 and Immunometabolism: Insights from Physiology and Pharmacology. Trends Pharmacol Sci 36:847–857
Schaap FG, Trauner M, Jansen PL (2014) Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 11:55–67
Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W (2008) Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48:1632–1643
Zhang S, Wang J, Liu Q, Harnish DC (2009) Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol 51:380–388
Renga B, Mencarelli A, Migliorati M, Cipriani S, D’Amore C, Distrutti E et al (2011) SHP-dependent and -independent induction of peroxisome proliferator-activated receptor-gamma by the bile acid sensor farnesoid X receptor counter-regulates the pro-inflammatory phenotype of liver myofibroblasts. Inflamm Res 60:577–587
Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L et al (2005) Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor γ contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther 315:58–68
Li M, Cai SY, Boyer JL (2017) Mechanisms of bile acid mediated inflammation in the liver. Mol Aspects Med 56:45–53
Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L et al (2011) Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochem Biophys Acta 1812:851–858
Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S (2009) The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183:6251–6261
Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M et al (2006) Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 103:3920–3925
Fiorucci S, Zampella A, Ricci P, Distrutti E, Biagioli M (2022) Immunomodulatory functions of FXR. Mol Cell Endocrinol 551:111650
Wang L, Gong Z, Zhang X, Zhu F, Liu Y, Jin C et al (2020) Gut microbial bile acid metabolite skews macrophage polarization and contributes to high-fat diet-induced colonic inflammation. Gut Microbes 12:1–20
Cao H, Xu M, Dong W, Deng B, Wang S, Zhang Y et al (2017) Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int J Cancer 140:2545–2556
Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ (2007) Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol 292:G1114–G1122
Langmann T, Moehle C, Mauerer R, Scharl M, Liebisch G, Zahn A et al (2004) Loss of detoxification in inflammatory bowel disease: dysregulation of pregnane X receptor target genes. Gastroenterology 127:26–40
Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G et al (2011) The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 6:e25637
Fiorucci S, Distrutti E (2019) The Pharmacology of Bile Acids and Their Receptors. Handb Exp Pharmacol 256:3–18
Biagioli M, Marchiano S, Roselli R, Di Giorgio C, Bellini R, Bordoni M et al (2022) GLP-1 mediates regulation of colonic ACE2 expression by the bile acid receptor GPBAR1 in inflammation. Cells 11(7):1187
Garg M, Royce SG, Tikellis C, Shallue C, Batu D, Velkoska E et al (2020) Imbalance of the renin-angiotensin system may contribute to inflammation and fibrosis in IBD: a novel therapeutic target? Gut 69:841–851
Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG et al (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2:217–225
Liu H, Hu C, Zhang X, Jia W (2018) Role of gut microbiota, bile acids and their cross-talk in the effects of bariatric surgery on obesity and type 2 diabetes. J Diabetes Investig 9:13–20
Wahlström A, Sayin SI, Marschall H-U, Bäckhed F (2016) Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 24:41–50
Ory DS (2004) Nuclear receptor signaling in the control of cholesterol homeostasis: have the orphans found a home? CircRes 95:660–670
Manichanh C, Borruel N, Casellas F, Guarner F (2012) The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 9:599–608
Turpin W, Lee SH, Raygoza Garay JA, Madsen KL, Meddings JB, Bedrani L et al (2020) Increased Intestinal Permeability Is Associated With Later Development of Crohn’s Disease. Gastroenterology 159(2092–100):e5
Torres J, Petralia F, Sato T, Wang P, Telesco SE, Choung RS et al (2020) Serum Biomarkers Identify Patients Who Will Develop Inflammatory Bowel Diseases Up to 5 Years Before Diagnosis. Gastroenterology 159:96–104
Ni J, Wu GD, Albenberg L, Tomov VT (2017) Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol 14:573–584
Matsuoka K, Kanai T (2015) The gut microbiota and inflammatory bowel disease. Semin Immunopathol 37:47–55
Blaser M, Bork P, Fraser C, Knight R, Wang J (2013) The microbiome explored: recent insights and future challenges. Nat Rev Microbiol 11:213–217
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR (2007) Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA 104:13780–13785
Takaishi H, Matsuki T, Nakazawa A, Takada T, Kado S, Asahara T et al (2008) Imbalance in intestinal microflora constitution could be involved in the pathogenesis of inflammatory bowel disease. Int J Med Microbiol 298:463–472
Hirano A, Umeno J, Okamoto Y, Shibata H, Ogura Y, Moriyama T et al (2018) Comparison of the microbial community structure between inflamed and non-inflamed sites in patients with ulcerative colitis. J Gastroenterol Hepatol. https://doi.org/10.1111/jgh.14129
Prosberg M, Bendtsen F, Vind I, Petersen AM, Gluud LL (2016) The association between the gut microbiota and the inflammatory bowel disease activity: a systematic review and meta-analysis. Scand J Gastroenterol 51:1407–1415
Zuo T, Ng SC (2018) The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front Microbiol 9:2247
De Hertogh G, Aerssens J, Geboes KP, Geboes K (2008) Evidence for the involvement of infectious agents in the pathogenesis of Crohn’s disease. World J Gastroenterol: WJG 14:845–852
Kalischuk LD, Buret AG (2010) A role for Campylobacter jejuni-induced enteritis in inflammatory bowel disease? Am J Physiol Gastrointest Liver Physiol 298:G1-9
Li J, Butcher J, Mack D, Stintzi A (2015) Functional impacts of the intestinal microbiome in the pathogenesis of inflammatory bowel disease. Inflamm Bowel Dis 21:139–153
Xu N, Bai X, Cao X, Yue W, Jiang W, Yu Z (2021) Changes in intestinal microbiota and correlation with TLRs in ulcerative colitis in the coastal area of northern China. Microb Pathog 150:104707
Sokol H, Jegou S, McQuitty C, Straub M, Leducq V, Landman C et al (2018) Specificities of the intestinal microbiota in patients with inflammatory bowel disease and Clostridium difficile infection. Gut Microbes 9:55–60
Qiu X, Zhang M, Yang X, Hong N, Yu C (2013) Faecalibacterium prausnitzii upregulates regulatory T cells and anti-inflammatory cytokines in treating TNBS-induced colitis. J Crohns Colitis 7:e558–e568
Nemoto H, Kataoka K, Ishikawa H, Ikata K, Arimochi H, Iwasaki T et al (2012) Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig Dis Sci 57:2955–2964
Mankowska-Wierzbicka D, Stelmach-Mardas M, Gabryel M, Tomczak H, Skrzypczak-Zielinska M, Zakerska-Banaszak O et al (2020) The effectiveness of multi-session fmt treatment in active ulcerative colitis patients: a pilot study. Biomedicines 8(8):268
Jia W, Xie G, Jia W (2018) Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol 15:111–128
Lavelle A, Sokol H (2020) Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol 17:223–237
Lajczak-McGinley NK, Porru E, Fallon CM, Smyth J, Curley C, McCarron PA et al (2020) The secondary bile acids, ursodeoxycholic acid and lithocholic acid, protect against intestinal inflammation by inhibition of epithelial apoptosis. Physiol Rep 8:e14456
Beaumont M, Paes C, Mussard E, Knudsen C, Cauquil L, Aymard P et al (2020) Gut microbiota derived metabolites contribute to intestinal barrier maturation at the suckling-to-weaning transition. Gut Microbes 11:1268–1286
Portincasa P, Bonfrate L, Vacca M, De Angelis M, Farella I, Lanza E et al (2022) Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int J Mol Sci 23:1105
Golubeva AV, Joyce SA, Moloney G, Burokas A, Sherwin E, Arboleya S et al (2017) Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 24:166–178
Lindhardt K, Bechgaard E (2003) Sodium glycocholate transport across Caco-2 cell monolayers, and the enhancement of mannitol transport relative to transepithelial electrical resistance. Int J Pharm 252:181–186
Sun Y, Fihn BM, Sjövall H, Jodal M (2004) Enteric neurones modulate the colonic permeability response to luminal bile acids in rat colon in vivo. Gut 53:362–367
Wildt S, Nørby Rasmussen S, Lysgård Madsen J, Rumessen JJ (2003) Bile acid malabsorption in patients with chronic diarrhoea: Clinical value of SeHCAT test. Scand J Gastroenterol 38:826–830
Craven PA, Pfanstiel J, DeRubertis FR (1987) Role of activation of protein kinase C in the stimulation of colonic epithelial proliferation and reactive oxygen formation by bile acids. J Clin Investig 79:532–541
Xiao ZL, Rho AK, Biancani P, Behar J (2002) Effects of bile acids on the muscle functions of guinea pig gallbladder. Am J Physiol Gastroint Liver Physiol 283:G87–G94
Glinghammar B, Rafter J (2001) Colonic luminal contents induce cyclooxygenase 2 transcription in human colon carcinoma cells. Gastroenterology 120:401–410
van Hengel J, Gohon L, Bruyneel E, Vermeulen S, Cornelissen M, Mareel M et al (1997) Protein kinase C activation upregulates intercellular adhesion of α-catenin–negative human colon cancer cell variants via induction of desmosomes. J Cell Biol 137(5):1103–1116
Qiao D, Stratagouleas ED, Martinez JDJC (2001) Activation and role of mitogen-activated protein kinases in deoxycholic acid-induced apoptosis. Carcinog 22(1):35–41
Kurz AK, Graf D, Schmitt M, Vom Dahl S, Häussinger DJG (2001) Tauroursodesoxycholate-induced choleresis involves p38MAPK activation and translocation of the bile salt export pump in rats. Gastroenterol 121(2):407–419
Rust C, Karnitz LM, Paya CV, Moscat J, Simari RD, Gores GJ (2000) The bile acid taurochenodeoxycholate activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. J Biol Chem 275(26):20210–20216
Takikawa Y, Miyoshi H, Rust C, Roberts P, Siegel R, Mandal PK et al (2001) The bile acid–activated phosphatidylinositol 3-kinase pathway inhibits Fas apoptosis upstream of bid in rodent hepatocytes. Gastroenterol 120(7):1810–1817
Araki Y, Katoh T, Ogawa A, Bamba S, Andoh A, Koyama S et al (2005) Bile acid modulates transepithelial permeability via the generation of reactive oxygen species in the Caco-2 cell line. Free Radical Biol Med 39:769–780
Raimondi F, Santoro P, Barone MV, Pappacoda S, Barretta ML, Nanayakkara M et al (2008) Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol 294:G906–G913
Stenman LK, Holma R, Korpela R (2012) High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J Gastroenterol: WJG 18:923–929
Stenman LK, Holma R, Eggert A, Korpela R (2013) A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol 304:G227–G234
Stenman LK, Holma R, Forsgard R, Gylling H, Korpela R (2013) Higher fecal bile acid hydrophobicity is associated with exacerbation of dextran sodium sulfate colitis in mice. J Nutr 143:1691–1697
Li DK, Chaudhari SN, Lee Y, Sojoodi M, Adhikari AA, Zukerberg L et al (2022) Inhibition of microbial deconjugation of micellar bile acids protects against intestinal permeability and liver injury. Sci Adv 8:2794
Li S, Zhuge A, Wang K, Lv L, Bian X, Yang L et al (2021) Ketogenic diet aggravates colitis, impairs intestinal barrier and alters gut microbiota and metabolism in DSS-induced mice. Food Funct 12:10210–10225
Hernandez-Rocha C, Borowski K, Turpin W, Filice M, Nayeri S, Raygoza Garay JA et al (2021) Integrative Analysis of Colonic Biopsies from Inflammatory Bowel Disease Patients Identifies an Interaction Between Microbial Bile Acid-inducible Gene Abundance and Human Angiopoietin-like 4 Gene Expression. J Crohns Colitis 15:2078–2087
Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S et al (2019) Author Correction: Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4:898
Lepage P, Hasler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A et al (2011) Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 141:227–236
Hasler R, Sheibani-Tezerji R, Sinha A, Barann M, Rehman A, Esser D et al (2017) Uncoupling of mucosal gene regulation, mRNA splicing and adherent microbiota signatures in inflammatory bowel disease. Gut 66:2087–2097
Quraishi MN, Acharjee A, Beggs AD, Horniblow R, Tselepis C, Gkoutos G et al (2020) A Pilot Integrative Analysis of Colonic Gene Expression, Gut Microbiota, and Immune Infiltration in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Association of Disease With Bile Acid Pathways. J Crohns Colitis 14:935–947
Phua T, Sng MK, Tan EH, Chee DS, Li Y, Wee JW et al (2017) Angiopoietin-like 4 Mediates Colonic Inflammation by Regulating Chemokine Transcript Stability via Tristetraprolin. Sci Rep 7:44351
Cabrera D, Arab JP, Arrese M (2019) UDCA, NorUDCA, and TUDCA in liver diseases: a review of their mechanisms of action and clinical applications. Handb Exp Pharmacol 256:237–264
Hofmann AF (1999) The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med 159:2647–2658
Bernardes-Silva CF, Damiao AO, Sipahi AM, Laurindo FR, Iriya K, Lopasso FP et al (2004) Ursodeoxycholic acid ameliorates experimental ileitis counteracting intestinal barrier dysfunction and oxidative stress. Dig Dis Sci 49:1569–1574
Ward JBJ, Lajczak NK, Kelly OB, O’Dwyer AM, Giddam AK, Ni Gabhann J et al (2017) Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol Gastrointest Liver Physiol 312:G550–G558
Van den Bossche L, Hindryckx P, Devisscher L, Devriese S, Van Welden S, Holvoet T et al (2017) Ursodeoxycholic acid and its taurine- or glycine-conjugated species reduce colitogenic dysbiosis and equally suppress experimental colitis in mice. Appl Environ Microbiol 83(7):e02766–16
Wang Z, Chen J, Chen Z, Xie L, Wang W (2021) Clinical effects of ursodeoxycholic acid on patients with ulcerative colitis may improve via the regulation of IL-23-IL-17 axis and the changes of the proportion of intestinal microflora. Saudi J Gastroenterol 27:149–157
Wax J, Clinger WA, Varner P, Bass P, Winder CV (1970) Relationship of the enterohepatic cycle to ulcerogenesis in the rat small bowel with flufenamic acid. Gastroenterology 58:772–780
Kafil TS, Nguyen TM, Patton PH, MacDonald JK, Chande N, McDonald JW (2017) Interventions for treating collagenous colitis. Coch Database Syst Rev 11:003575
Pavlidis P, Heneghan M, Hayee B (2015) Cholestyramine treats primary sclerosing cholangitis-associated inflammatory bowel disease. J Crohns Colitis 9:210
Gonzalez-Lozano E, Garcia-Garcia J, Galvez J, Hidalgo-Garcia L, Rodriguez-Nogales A, Rodriguez-Cabezas ME et al (2022) Novel horizons in postbiotics: lactobacillaceae extracellular vesicles and their applications in health and disease. Nutrients 14(24):5296
Shadnoush M, Hosseini RS, Khalilnezhad A, Navai L, Goudarzi H, Vaezjalali M (2015) Effects of Probiotics on Gut Microbiota in Patients with Inflammatory Bowel Disease: A Double-blind. Placebo-controlled Clinical Trial Korean J Gastroenterol 65:215–221
Kato K, Mizuno S, Umesaki Y, Ishii Y, Sugitani M, Imaoka A et al (2004) Randomized placebo-controlled trial assessing the effect of bifidobacteria-fermented milk on active ulcerative colitis. Aliment Pharmacol Ther 20:1133–1141
Bourreille A, Cadiot G, Le Dreau G, Laharie D, Beaugerie L, Dupas JL et al (2013) Saccharomyces boulardii does not prevent relapse of Crohn’s disease. Clin Gastroenterol Hepatol 11:982–987
Guslandi M, Mezzi G, Sorghi M, Testoni PA (2000) Saccharomyces boulardii in maintenance treatment of Crohn’s disease. Dig Dis Sci 45:1462–1464
Zocco MA, dal Verme LZ, Cremonini F, Piscaglia AC, Nista EC, Candelli M et al (2006) Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment Pharmacol Ther 23:1567–1574
Bjarnason I, Sission G, Hayee B (2019) A randomised, double-blind, placebo-controlled trial of a multi-strain probiotic in patients with asymptomatic ulcerative colitis and Crohn’s disease. Inflammopharmacology 27:465–473
Prantera C, Scribano ML, Falasco G, Andreoli A, Luzi C (2002) Ineffectiveness of probiotics in preventing recurrence after curative resection for Crohn’s disease: a randomised controlled trial with Lactobacillus GG. Gut 51:405–409
Schultz M, Timmer A, Herfarth HH, Sartor RB, Vanderhoof JA, Rath HC (2004) Lactobacillus GG in inducing and maintaining remission of Crohn’s disease. BMC Gastroenterol 4:5
Marteau P, Lemann M, Seksik P, Laharie D, Colombel JF, Bouhnik Y et al (2006) Ineffectiveness of Lactobacillus johnsonii LA1 for prophylaxis of postoperative recurrence in Crohn’s disease: a randomised, double blind, placebo controlled GETAID trial. Gut 55:842–847
Van Gossum A, Dewit O, Louis E, de Hertogh G, Baert F, Fontaine F et al (2007) Multicenter randomized-controlled clinical trial of probiotics (Lactobacillus johnsonii, LA1) on early endoscopic recurrence of Crohn’s disease after lleo-caecal resection. Inflamm Bowel Dis 13:135–142
Tursi A, Brandimarte G, Giorgetti GM, Forti G, Modeo ME, Gigliobianco A (2004) Low-dose balsalazide plus a high-potency probiotic preparation is more effective than balsalazide alone or mesalazine in the treatment of acute mild-to-moderate ulcerative colitis. Med Sci Monit Int Med J Experiment Clin Res 10:I126–I131
Sood A, Midha V, Makharia GK, Ahuja V, Singal D, Goswami P et al (2009) The probiotic preparation, VSL#3 induces remission in patients with mild-to-moderately active ulcerative colitis. Clin Gastroenterol Hepatol 7(1202–9):91
Petersen AM, Mirsepasi H, Halkjaer SI, Mortensen EM, Nordgaard-Lassen I, Krogfelt KA (2014) Ciprofloxacin and probiotic Escherichia coli Nissle add-on treatment in active ulcerative colitis: a double-blind randomized placebo controlled clinical trial. J Crohns Colitis 8:1498–1505
Matthes H, Krummenerl T, Giensch M, Wolff C, Schulze J (2010) Clinical trial: probiotic treatment of acute distal ulcerative colitis with rectally administered Escherichia coli Nissle 1917 (EcN). BMC Complement Altern Med 10:13
Kruis W, Schutz E, Fric P, Fixa B, Judmaier G, Stolte M (1997) Double-blind comparison of an oral Escherichia coli preparation and mesalazine in maintaining remission of ulcerative colitis. Aliment Pharmacol Ther 11:853–858
Li L, Liu T, Gu Y, Wang X, Xie R, Sun Y et al (2022) Regulation of gut microbiota-bile acids axis by probiotics in inflammatory bowel disease. Front Immunol 13:974305
Martoni CJ, Labbe A, Ganopolsky JG, Prakash S, Jones ML (2015) Changes in bile acids, FGF-19 and sterol absorption in response to bile salt hydrolase active L. reuteri NCIMB 30242. Gut Microbes 6:57–65
Zhai Q, Liu Y, Wang C, Qu D, Zhao J, Zhang H et al (2019) Lactobacillus plantarum CCFM8661 modulates bile acid enterohepatic circulation and increases lead excretion in mice. Food Funct 10:1455–1464
Degirolamo C, Rainaldi S, Bovenga F, Murzilli S, Moschetta A (2014) Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep 7:12–18
Matsuoka K, Uemura Y, Kanai T, Kunisaki R, Suzuki Y, Yokoyama K et al (2018) Efficacy of Bifidobacterium breve Fermented Milk in Maintaining Remission of Ulcerative Colitis. Dig Dis Sci 63:1910–1919
Wildt S, Nordgaard I, Hansen U, Brockmann E, Rumessen JJ (2011) A randomised double-blind placebo-controlled trial with Lactobacillus acidophilus La-5 and Bifidobacterium animalis subsp. lactis BB-12 for maintenance of remission in ulcerative colitis. J Crohns Colitis 5:115–121
Chen M, Feng Y, Liu W (2021) Efficacy and safety of probiotics in the induction and maintenance of inflammatory bowel disease remission: a systematic review and meta-analysis. Ann Palliat Med 10:11821–11829
Lorentz A, Muller L (2022) Probiotics in the treatment of inflammatory bowel disease in adulthood: a systematic review. J Gastr Liver Dis JGLD 31:74–84
Bamola VD, Dubey D, Samanta P, Kedia S, Ahuja V, Madempudi RS et al (2022) Role of a probiotic strain in the modulation of gut microbiota and cytokines in inflammatory bowel disease. Anaerobe 78:102652
Zhang T, Li P, Wu X, Lu G, Marcella C, Ji X et al (2020) Alterations of Akkermansia muciniphila in the inflammatory bowel disease patients with washed microbiota transplantation. Appl Microbiol Biotechnol 104:10203–10215
Bajer L, Kverka M, Kostovcik M, Macinga P, Dvorak J, Stehlikova Z et al (2017) Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J Gastroenterol: WJG 23:4548–4558
Bian X, Wu W, Yang L, Lv L, Wang Q, Li Y et al (2019) Administration of Akkermansia muciniphila Ameliorates Dextran Sulfate Sodium-Induced Ulcerative Colitis in Mice. Front Microbiol 10:2259
Zhai R, Xue X, Zhang L, Yang X, Zhao L, Zhang C (2019) Strain-Specific Anti-inflammatory Properties of Two Akkermansia muciniphila Strains on Chronic Colitis in Mice. Front Cell Infect Microbiol 9:239
Wang L, Tang L, Feng Y, Zhao S, Han M, Zhang C et al (2020) A purified membrane protein from Akkermansia muciniphila or the pasteurised bacterium blunts colitis associated tumourigenesis by modulation of CD8(+) T cells in mice. Gut 69:1988–1997
Brennan JJ, Gilmore TD (2018) Evolutionary Origins of Toll-like Receptor Signaling. Mol Biol Evol 35:1576–1587
Blasius AL, Beutler B (2010) Intracellular toll-like receptors. Immunity 32:305–315
Liu Y, Yang M, Tang L, Wang F, Huang S, Liu S et al (2022) TLR4 regulates RORgammat(+) regulatory T-cell responses and susceptibility to colon inflammation through interaction with Akkermansia muciniphila. Microbiome 10:98
Juarez-Fernandez M, Porras D, Petrov P, Roman-Saguillo S, Garcia-Mediavilla MV, Soluyanova P et al (2021) The synbiotic combination of akkermansia muciniphila and quercetin ameliorates early obesity and NAFLD through gut microbiota reshaping and bile acid metabolism modulation. Antioxidants (Basel) 10(12):2001
Vandeputte D, Falony G, Vieira-Silva S, Wang J, Sailer M, Theis S et al (2017) Prebiotic inulin-type fructans induce specific changes in the human gut microbiota. Gut 66:1968–1974
Scott KP, Martin JC, Duncan SH, Flint HJ (2014) Prebiotic stimulation of human colonic butyrate-producing bacteria and bifidobacteria, in vitro. FEMS Microbiol Ecol 87:30–40
Valcheva R, Koleva P, Martinez I, Walter J, Ganzle MG, Dieleman LA (2019) Inulin-type fructans improve active ulcerative colitis associated with microbiota changes and increased short-chain fatty acids levels. Gut Microbes 10:334–357
Alexander C, Cross TL, Devendran S, Neumer F, Theis S, Ridlon JM et al (2018) Effects of prebiotic inulin-type fructans on blood metabolite and hormone concentrations and faecal microbiota and metabolites in overweight dogs. Br J Nutr 120:711–720
Hafer A, Kramer S, Duncker S, Kruger M, Manns MP, Bischoff SC (2007) Effect of oral lactulose on clinical and immunohistochemical parameters in patients with inflammatory bowel disease: a pilot study. BMC Gastroenterol 7:36
Benjamin JL, Hedin CR, Koutsoumpas A, Ng SC, McCarthy NE, Hart AL et al (2011) Randomised, double-blind, placebo-controlled trial of fructo-oligosaccharides in active Crohn’s disease. Gut 60:923–929
Kanauchi O, Mitsuyama K, Homma T, Takahama K, Fujiyama Y, Andoh A et al (2003) Treatment of ulcerative colitis patients by long-term administration of germinated barley foodstuff: multi-center open trial. Int J Mol Med 12:701–704
Hanai H, Kanauchi O, Mitsuyama K, Andoh A, Takeuchi K, Takayuki I et al (2004) Germinated barley foodstuff prolongs remission in patients with ulcerative colitis. Int J Mol Med 13:643–647
Hallert C, Kaldma M, Petersson BG (1991) Ispaghula husk may relieve gastrointestinal symptoms in ulcerative colitis in remission. Scand J Gastroenterol 26:747–750
Fernandez-Banares F, Hinojosa J, Sanchez-Lombrana JL, Navarro E, Martinez-Salmeron JF, Garcia-Puges A et al (1999) Randomized clinical trial of Plantago ovata seeds (dietary fiber) as compared with mesalamine in maintaining remission in ulcerative colitis. Spanish Group for the Study of Crohn’s Disease and Ulcerative Colitis (GETECCU). Am J Gastroenterol 94:427–433
Casellas F, Borruel N, Torrejon A, Varela E, Antolin M, Guarner F et al (2007) Oral oligofructose-enriched inulin supplementation in acute ulcerative colitis is well tolerated and associated with lowered faecal calprotectin. Aliment Pharmacol Ther 25:1061–1067
Arifuzzaman M, Won TH, Li TT, Yano H, Digumarthi S, Heras AF et al (2022) Inulin fibre promotes microbiota-derived bile acids and type 2 inflammation. Nature 611:578–584
Bouhnik Y, Attar A, Joly FA, Riottot M, Dyard F, Flourie B (2004) Lactulose ingestion increases faecal bifidobacterial counts: a randomised double-blind study in healthy humans. Eur J Clin Nutr 58:462–466
Bouhnik Y, Neut C, Raskine L, Michel C, Riottot M, Andrieux C et al (2004) Prospective, randomized, parallel-group trial to evaluate the effects of lactulose and polyethylene glycol-4000 on colonic flora in chronic idiopathic constipation. Aliment Pharmacol Ther 19:889–899
van Berge Henegouwen GP, van der Werf SD, Ruben AT (1987) Effect of long term lactulose ingestion on secondary bile salt metabolism in man: potential protective effect of lactulose in colonic carcinogenesis. Gut 28:675–680
van Dokkum W, Wezendonk B, Srikumar TS, van den Heuvel EG (1999) Effect of nondigestible oligosaccharides on large-bowel functions, blood lipid concentrations and glucose absorption in young healthy male subjects. Eur J Clin Nutr 53:1–7
Bouhnik Y, Flourie B, Riottot M, Bisetti N, Gailing MF, Guibert A et al (1996) Effects of fructo-oligosaccharides ingestion on fecal bifidobacteria and selected metabolic indexes of colon carcinogenesis in healthy humans. Nutr Cancer 26:21–29
Kanauchi O, Serizawa I, Araki Y, Suzuki A, Andoh A, Fujiyama Y et al (2003) Germinated barley foodstuff, a prebiotic product, ameliorates inflammation of colitis through modulation of the enteric environment. J Gastroenterol 38:134–141
Araki Y, Andoh A, Fujiyama Y, Kanauchi O, Takenaka K, Higuchi A et al (2001) Germinated barley foodstuff exhibits different adsorption properties for hydrophilic versus hydrophobic bile acids. Digestion 64:248–254
Chaplin MF, Chaudhury S, Dettmar PW, Sykes J, Shaw AD, Davies GJ (2000) Effect of ispaghula husk on the faecal output of bile acids in healthy volunteers. J Steroid Biochem Mol Biol 72:283–292
Gelissen IC, Brodie B, Eastwood MA (1994) Effect of Plantago ovata (psyllium) husk and seeds on sterol metabolism: studies in normal and ileostomy subjects. Am J Clin Nutr 59:395–400
Romero AL, West KL, Zern T, Fernandez ML (2002) The seeds from Plantago ovata lower plasma lipids by altering hepatic and bile acid metabolism in guinea pigs. J Nutr 132:1194–1198
Welters CF, Heineman E, Thunnissen FB, van den Bogaard AE, Soeters PB, Baeten CG (2002) Effect of dietary inulin supplementation on inflammation of pouch mucosa in patients with an ileal pouch-anal anastomosis. Dis Colon Rectum 45:621–627
Xu M, Shen Y, Cen M, Zhu Y, Cheng F, Tang L et al (2021) Modulation of the Gut Microbiota-farnesoid X Receptor Axis Improves Deoxycholic Acid-induced Intestinal Inflammation in Mice. J Crohns Colitis 15:1197–1210
Liu HM, Liao JF, Lee TY (2017) Farnesoid X receptor agonist GW4064 ameliorates lipopolysaccharide-induced ileocolitis through TLR4/MyD88 pathway related mitochondrial dysfunction in mice. Biochem Biophys Res Commun 490:841–848
Mroz MS, Lajczak NK, Goggins BJ, Keely S, Keely SJ (2018) The bile acids, deoxycholic acid and ursodeoxycholic acid, regulate colonic epithelial wound healing. Am J Physiol Gastrointest Liver Physiol 314:G378–G387
Stidham RW, Takenaka K (2022) Artificial Intelligence for Disease Assessment in Inflammatory Bowel Disease: How Will it Change Our Practice? Gastroenterology 162:1493–1506
Liu XY, Tang H, Zhou QY, Zeng YL, Chen D, Xu H et al (2023) Advancing the precision management of inflammatory bowel disease in the era of omics approaches and new technology. World J Gastroenterol 29:272–285
Zhu C, Wang X, Li J, Jiang R, Chen H, Chen T et al (2022) Determine independent gut microbiota-diseases association by eliminating the effects of human lifestyle factors. BMC Microbiol 22:4
Seyed Tabib NS, Madgwick M, Sudhakar P, Verstockt B, Korcsmaros T, Vermeire S (2020) Big data in IBD: big progress for clinical practice. Gut 69:1520–1532
Vitek L (2015) Bile acid malabsorption in inflammatory bowel disease. Inflamm Bowel Dis 21:476–483
Fitzpatrick LR, Jenabzadeh P (2020) IBD and Bile Acid Absorption: Focus on Pre-clinical and Clinical Observations. Front Physiol 11:564
Neimark E, Chen F, Li X, Magid MS, Alasio TM, Frankenberg T et al (2006) c-Fos is a critical mediator of inflammatory-mediated repression of the apical sodium-dependent bile acid transporter. Gastroenterology 131:554–567
Chen F, Ma L, Sartor RB, Li F, Xiong H, Sun AQ et al (2002) Inflammatory-mediated repression of the rat ileal sodium-dependent bile acid transporter by c-fos nuclear translocation. Gastroenterology 123:2005–2016
Jahnel J, Fickert P, Hauer AC, Hogenauer C, Avian A, Trauner M (2014) Inflammatory bowel disease alters intestinal bile acid transporter expression. Drug Metabol Disposit Biol Fate Chem 42:1423–1431
Lenicek M, Duricova D, Komarek V, Gabrysova B, Lukas M, Smerhovsky Z et al (2011) Bile acid malabsorption in inflammatory bowel disease: assessment by serum markers. Inflamm Bowel Dis 17:1322–1327
Torres J, Bao X, Iuga AC, Chen A, Harpaz N, Ullman T et al (2013) Farnesoid X receptor expression is decreased in colonic mucosa of patients with primary sclerosing cholangitis and colitis-associated neoplasia. Inflamm Bowel Dis 19:275–282
Attinkara R, Mwinyi J, Truninger K, Regula J, Gaj P, Rogler G et al (2012) Association of genetic variation in the NR1H4 gene, encoding the nuclear bile acid receptor FXR, with inflammatory bowel disease. BMC Res Notes 5:461
Das P, Marcisauskas S, Ji B, Nielsen J (2019) Metagenomic analysis of bile salt biotransformation in the human gut microbiome. BMC Genomics 20:517
Heinken A, Ravcheev DA, Baldini F, Heirendt L, Fleming RMT, Thiele I (2019) Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 7:75
Kleessen B, Kroesen AJ, Buhr HJ, Blaut M (2002) Mucosal and invading bacteria in patients with inflammatory bowel disease compared with controls. Scand J Gastroenterol 37:1034–1041
Ansari I, Raddatz G, Gutekunst J, Ridnik M, Cohen D, Abu-Remaileh M et al (2020) The microbiota programs DNA methylation to control intestinal homeostasis and inflammation. Nat Microbiol 5:610–619
Yang S, Shang J, Liu L, Tang Z, Meng X (2022) Strains producing different short-chain fatty acids alleviate DSS-induced ulcerative colitis by regulating intestinal microecology. Food Funct 13:12156–12169
Grellier N, Suzuki MT, Brot L, Rodrigues AMS, Humbert L, Escoubeyrou K et al (2022) Impact of IBD-associated dysbiosis on bacterial quorum sensing mediated by acyl-homoserine lactone in human gut microbiota. Int J Mol Sci 23(23):15404
Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M et al (2015) Interleukin-12 and -23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 43:146–160
Castellanos JG, Longman RS (2020) Innate lymphoid cells link gut microbes with mucosal T cell immunity. Gut Microbes 11:231–236
Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F (2019) ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis 10:315
Acknowledgements
This paper is dedicated to the memory of Dr. David Q.-H. Wang, Professor of Medicine Genetics in the Departments of Medicine and Genetics, and Director of Molecular Biology and Next Generation Technology Core in the Marion Bessin Liver Research Center at Albert Einstein College of Medicine in New York, USA. Dr Wang suddenly and prematurely passed away on March 19th, 2023, at the age of 60. Dr. Wang was appointed in 2018 as Associate Editor of Internal and Emergency Medicine. As the Editor-in-Chief of the Journal, on behalf of the whole Board, and with the coauthors of this paper, we want to express our condolences to his wife Helen and their two sons for such unexpected loss. Dr. Wang made huge contributions to understanding of molecular and genetic mechanisms of cholesterol homeostasis and the pathophysiology of cholesterol-related diseases with particular attention to the translational value of research. He contributed to the identification of gallstone (LITH) genes by performing genotype phenotype studies in mice and humans, while dissecting the fundamental molecular, cellular, genetic, and physical–chemical mechanisms of cholesterol gallstone disease. This paper will be included in the Topical Collection on “Gut Inflammation” in Internal and Emergency Medicine, 2023. The authors acknowledge the longstanding professional collaboration and personal interaction with Helen H. Wang, Department of Medicine, Division of Gastroenterology and Liver Diseases, Marion Bessin Liver Research Center, Albert Einstein College of Medicine, Bronx, NY 10461, USA. They are indebted to Paola De Benedictis, Rosa De Venuto, Vito Di Ceglie for excellent longstanding technical and longstanding secretarial.
Funding
Open access funding provided by Università degli Studi di Bari Aldo Moro within the CRUI-CARE Agreement. This paper has been partly supported by funding to P.P. from the European Union’s Horizon 2020 Research and Innovation program under the Marie Skłodowska-Curie Grant Agreement No. 722619 (FOIE GRAS), Grant Agreement No. 734719 (mtFOIE GRAS), Grant Regione Puglia, CUP H99C20000340002 (Fever Apulia), and Grant EUROSEEDS Uniba—S56—By-products Sustainable Recovery 4 Health (BSR-4H): University of Bari Aldo Moro, 2022. The project SYS-TEMIC “an integrated approach to the challenge of sustainable food systems: adaptive and mitigatory strategies to address climate change and malnutrition", Knowledge hub on Nutrition and Food Security, has received funding from national research funding parties in Belgium (FWO), France (INRA), Germany (BLE), Italy (MIPAAF), Latvia (IZM), Norway (RCN), Portugal (FCT), and Spain (AEI) in a joint action of JPI HDHL, JPI-OCEANS and FACCE-JPI launched in 2019 under the ERA-NET ERA-HDHL (n° 696295). PP is grant recipient and MK is assistant researcher (RTDA) in Project funded under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3—Call for tender No. 341 of 15/03/2022 of Italian Ministry of University and Research funded by the European Union—NextGenerationEU Award Number: Project code PE0000003, Concession Decree No. 1550 of 11/10/2022 adopted by the Italian Ministry of University and Research, CUP D93C22000890001, Project title “Research and innovation network on food and nutrition Sustainability, Safety and Security—Working ON Foods” (ONFoods). PP is recipient of HORIZON-HLTH-2022-STAYHLTH-01–05-two-stage Project 101080329—PAS GRAS with the following partners Universidade De Coimbra, Portugal; Uppsala Universitet, Sweden; Universidade Nova De Lisboa, Portugal; Fundacio Eurecat (Eurecat), Barcelona, Spain; Consiglio Nazionale Delle Ricerche (Cnr), Roma Italy; Instituto Politecnico De Viana De Castelo, Viana Do Castelo 4900–347, Portugal; Technische Universitaet Muenchen (Tum), Muenchen, Germany; Instytut Biologii Doswiadczalnej Im. M. Nenckiego Polskiej Akademii Nauk (Nencki), Warszawa, Poland; Instituto Pedro Nunes Associacao Para A Inovacao E Desenvolvimento Em Ciencia E Tecnologia (Ipn), Coimbra, Portugal; The European Society For Clinical Investigation (Esci), Utrecht, Netherlands; Mediagnost Gesellschaft Fur Forschung Und Herstellung Von Diagnostika Gmbh (Mediagnost), Reutlingen, Germany; Martin-Luther-Universitat Halle-Wittenberg (Mlu), Halle, Germany; Associacao Protectora Dos Diabeticos De Portugal (Apdp), Lisboa 1250–203, Portugal; Agdcentro Associacao De Ginastica Do Centro (Agcentro), Coimbra, Portugal. The authors are indebted to the Consortium of Mediterranean Universities (CMU), Bari (www.cmungo.eu) for helpful discussion. The figures were partly generated using materials from Servier Medical Art (Servier) under consideration of a Creative Commons Attribution 3.0 Unported License. The authors acknowledge the longstanding technical and secretarial support by Paola De Benedictis, Rosa De Venuto, Vito Di Ceglie.
Author information
Authors and Affiliations
Contributions
Conceptualization, ADC; literature review, ADC, LB, MK; draft preparation ADC, LB, PP; review and editing, ADC, PP. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Human and animal rights
Not applicable.
Informed consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Di Ciaula, A., Bonfrate, L., Khalil, M. et al. The interaction of bile acids and gut inflammation influences the pathogenesis of inflammatory bowel disease. Intern Emerg Med 18, 2181–2197 (2023). https://doi.org/10.1007/s11739-023-03343-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-023-03343-3