
Abstract
This study was carried out to evaluate a series of ceramic coatings in a sulfidation atmosphere containing 99% synthetic air and 1% H2S (vol.%) under a flow rate of 50 mL/min. The study was carried out at 773.15 K for 336 hours (heating rate 278.15 K/min). Chemically resistant glass enamels based on SiO2-B2O3-TiO2-Na2O compounds were deposited on the surface of 16Mo3 (16M) low-alloyed steel. Kinetic data were recorded periodically every 168 hours; macro- and microanalyses using scanning electron microscopy (SEM), phase analyses using x-ray diffractometry (XRD) and chemical composition using energy-dispersive x-ray spectrometry along with EDS x-ray mappings were carried out to observe the degradation process of the sulfidized coatings. The results indicated that some of the ceramic coatings exposed in the harsh atmosphere at 773.15 K for 336 hours showed a high degree of protection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thermodynamic efficiency in the ultra-supercritical (USC) plants is achieved by increasing the operating temperatures and pressures, which lowers fuel consumption and subsequently the emissions (Ref 1). Furthermore, the European goal of decreased CO2 emissions is a clear-cut requirement, which marks a historical turnover whereby Europe shall become fully neutral by 2050 (Ref 2). This can only be achieved by maximizing the efficiency of the plants and using biomass, which is considered carbon neutral. However, very little is known about the effects of the conditions in ultra-supercritical (USC) plants and the effects impurities from biomass may have on the corrosion resistance of the hot components in power plants (boiler walls, superheaters (SH), re-heaters (RH)). These components are already operating at the limit of their tolerance in less critical plants, and therefore, a radical step forward will be needed in materials technology to ensure the durability of components in such aggressive environments.
In particular, the problem is present when low-alloyed steels are used in power plant construction, even in USC technology. Low-alloyed steels such as 16Mo3 (16M) show very good thermal conductivity in contrast to austenitic steels; however, they show no corrosion resistance in aggressive atmospheres. In an air atmosphere, 16Mo3 (16M) steel with no coating protection indicates relatively high metal loss due to corrosion when a non-protective scale is formed. More severe corrosion degradation is always observed in sulfur-containing gas. Recent work published by Dudziak et al. (Ref 3) and past papers presented by Mrowec (Ref 4) and Ghaseni (Ref 5) demonstrate poor corrosion behavior of low-alloyed steels in sulfur gas. Degradation by sulfur results in the formation of a thick, porous, non-adherent sulfide scale with no protection. To secure low-alloyed steels in energy conversion systems and still use them due to high thermal conductivity, surface engineering as a coating design is a highly potential route. In the energy sector, the most popular types of coating are based on thermal spray technologies. There are many different types of thermal spray coating systems including plasma, wire, flame and kinetic; however, the most popularly used ones include high-velocity oxy-fuel (HVOF) or air, atmospheric plasma spray (APS) and others (Ref 6,7,8,9,10).
These two coating technologies are based on the same principles; a heat source melts the feedstock, where powder with an appropriate shape and diameter is used. The powder is accelerated at high temperature and impacts a substrate to form a coating with the desired thickness. In the APS process, plasma is produced via a direct current (DC) arc or radiofrequency (RF) discharge giving a temperature of over 8000 K with particle acceleration up to 20-500 m/s. These values depend on particle size and density, but higher speed is responsible for excellent deposition densities, bond strength and lower porosity of the deposited protective coating. The HVOF process was developed based on jet engine technology. The process relies on the oxygen–fuel ratio inside the combustion chamber where a highly pressurized mixture is formed. In contrast to the APS technique, in the HVOF process, particle velocity may reach 1000 m/s at a temperature of 3000 K. Similar to APS, density, bond strength and porosity directly depend on particle velocity, density and diameter.
Nevertheless, in addition to remarkable corrosion resistance, these coating types possess some degree of porosity (dependent on the technology used) where ash can accumulate. Here, it initiates, accelerates and propagates the damage and destruction of the applied coating. Therefore, it is crucial to develop new protection systems for high-temperature applications, especially when a higher temperature is essential to promote a higher efficiency of the heat conversion system.
There is one group of materials that are highly resistant to high temperature. However, these materials are often brittle and may lead to a significant reduction in heat transfer. The ceramic-based coatings are new materials that can be used as protection for the boiler steels exposed to harsh atmospheres, where aggressive gases and ash are present. In contrast to thermal spray coatings, the ceramic glaze-based coatings are free from porosity; hence, there is no issue related to the development of local corrosion products within the coating structure. Furthermore, application of the coating from both sides (outer and inner) is possible. Heat transfer characteristics are the major bottleneck of the ceramic system; unusable for energy conversion system or a coating that is too thick may reduce it significantly, making such a coating unusable for energy conversion systems. Nevertheless, the applied coating should protect a low-alloyed steel against the formation of low-melting eutectics, especially when H2S and SO3 are present in contact with Na and K salts (Ref 11,12,13). In the energy sector, ceramic-based coatings are not very popular and there are a limited number of papers related to ceramic coatings. However, there are some solutions from the past, using a mixture of a special grade of calcined aluminum oxide and a conventional type of ground-coat frit with water to appropriate fineness on a steel surface. This mixture is then dried and fired according to well-known methods in the ceramic industry. Such a type of coating reached 70 μm. Progress in ceramic coatings for high-temperature applications has been extensively enhanced in the last century (Ref 14). In 1980, ceramic coatings were applied in adiabatic engines (Ref 15). Further, ceramic coatings were applied in gas turbine wings, pistons, cylinder linings, valves and piston crowns (Ref 16). Predominantly, nowadays, ceramic coatings are mainly used to protect Ni-based alloys against hot corrosion, oxidation and wear degradation (Ref 17, 18).
To get a better understanding of advanced ceramic materials and further develop them for a particular engineering application (i.e., energy sector, boiler protection), extensive research is essential for evaluating macro- and microstructural and oxidation resistance. In this research, new types of ceramic coatings based on SiO2-Na2O-B2O3-TiO2 oxide phase were applied on a low-alloyed Cr-Mo steel 16Mo3 (16M).
In total, seven different coating types were tested. In this study, uncoated steel was not tested. It is well known that sulfidation resistance of low-alloyed steels is poor (Ref 3). The materials were tested at high temperature in sulfidation conditions using a 99% synthetic air–1% H2S gas mixture (vol.%) to monitor the degradation mechanism. The following gas mixture was acknowledged based on the literature research, where 1% of H2S is the highest % share in the overall combustion gas mixture found in a coal-fired power plant. The test was carried out for 338 hours at 773.15 K. Kinetic data were recorded periodically every 168 hours. A full spectrum of research analyses using SEM/EDS/XRD were carried out to establish corrosion resistance of the exposed materials at high temperature.
Experimental Procedure
Materials
The uncoated sample 16Mo3 (16M) was used as a reference sample and was delivered in as-received conditions. Prior to experimentation, the surface of the reference sample was ground using 600-grit SiC paper. The ceramic coatings were applied on the surface of the delivered materials, with no extra surface modification. The uncoated and the coated specimens had dimensions of 7-9 x 12 mm and a thickness of 4-5 mm in this work. The chemical composition according to Polish Norm: PN-75/H-84024 of the materials used in this work is listed in Table 1.
The enamel producing process took place in two stages. The raw materials were prepared by following the procedures given in Table 2, where individual ingredients were weighed, dry-mixed and rendered in alundum crucibles, and further, to finalize, the process was completed in an electric furnace using the following conditions:
-
Set filling temperature-1473.15 K,
-
melt pouring temperature-1633.15-1663.15 K,
-
melting time-1-2 h.
The second stage involved the production of enamels on a laboratory scale. The oxide compositions of enamels in weight percentage are presented in Table 2. For better traceability of the coatings used in this work, individual symbols were used.
Additions such as five parts by weight of clay, NaNO2 in the amount of 0.1 part by weight, Na2[B4O5(OH)4]·8H2O and MgCO3 by 0.2 part by weight and deionized water 60 parts by weight were added. All the additives were calculated in relation to 100 parts of flux weight. To ensure traceability, the enamels were numbered 1 to 10. Some of the enamels contained a different concentration of flux: Sample F contains 80% flux, marked as A with 20% chromium trioxide CrO3. The sample marked G contains 70% flux marked as B with 30% chromium trioxide CrO3. In both cases, the percentage of other additives was calculated in relation to the total mass of the flux. The flux samples in the amount of 100 g were dry-milled (to accelerate the milling process) for 30 minutes in a high-speed planetary mill. The grinding process was controlled by a granular distribution analysis as shown in Table 3, and melting points are listed in Table 4.
The selected enamels were applied to a steel substrate 16M and fired at 1153.15 K for approximately 20 minutes. The enamel symbols are not continuous, there are no coatings denoted as FCHO2, FCHO4, FCHO5, FCHO9 and finally FCHO10—these samples during preparation for high-temperature testing at 773.15 K showed weak mechanical behavior, spallation occurred and cracks on the sample surfaces were observed; therefore, the samples were rejected from the further examination. Water suspensions with densities of around 1.60 ± 0.5 g/cm3 were spread by a spray method onto the steel surface and fired in the conditions given in Table 5.
Visual and microscopic assessments of the surfaces showed the presence of holes in the structure of the coated materials (coatings C-F) as presented in Fig. 1.

Macro structures of the samples prior sulphidation test at 773.15 K for 336 hours
Sulfidation Test
Sulfidation testing was conducted in a flowing gas mixture containing synthetic air–H2S. The content of H2S in the gas mixture was 1 vol.%, with a flow rate of 50 mL/min and a heating rate of 278.15 K/min. The temperature (773.15 K) and pressure (1 bar) of the testing corresponded to the average service conditions in pulverized coal-fired power plants. The schematic of the experimental testing rig used for the high-temperature sulfidation testing is presented in Fig. 2.

Sulphidation rig using 99% synthetic air—1%H2S (vol. %) gas mixture
The testing rig contains an electric tubular furnace (CARBOLITETM) with wire heating elements, the container with reactive gases, piping for the gas supply equipped with the flow control system and the cooling unit. The container with the reactive gas (synthetic air 99%–H2S 1%) was obtained from the gas supplier (Air Products). The gas flow through the furnace was controlled using a rotameter installed between the gas cylinder and the furnace. The flow rate was set to 50 Nml/min (Nml/min at 293.15 K, 1 bar—standard conditions). The gas mixture was constantly controlled via an online camera setup using gDMSS HD Lite software. The test samples were placed onto a double-deck alumina (99.5% of Al2O3) refractory holder, on a long edge, with a spacing between each sample before being inserted into a tube furnace and then heated within the furnace. The temperature in the furnace hot zone was monitored using a programmable controller from EUROTHERM. Before the high-temperature sulfidation tests, the furnace was carefully calibrated by testing three samples at once. For safety reasons, the reaction chamber (alumina tube) of the furnace was enclosed in a 316-L stainless steel vessel capped at both ends with 316-L stainless steel flanges locked by four screws. The exiting gas mixture was passed through a NaOH solution for H2S neutralization and scrubbing and then directed to a ventilation system.
Sulfidation testing of the coated samples was conducted for 336 hours. The testing comprised two cycles of 168 hours/cycle. The samples were placed into the furnace at room temperature and were heated up to 773.15 K using a ramp rate of 278.15 K/min and held at this temperature for 168 hours; then, the samples were removed from the furnace, visually observed and pictured, and the first group of the samples (containing each material) were selected for further examination for the next 168 hours. Before sulfidation testing at high temperature, the coated samples were carefully measured by an electronic micrometer, later the samples were cleaned using an ultrasonic bath for 15 min at 313.15 K, and finally, the initial mass of each sample was recorded using a high-accuracy electronic balance Sartorius CPA225D. Post-exposure investigations were carried out using a single-lens DSLR camera Canon EOS 70D camera coupled with a Canon MP-E 65 mm f/2,8 macro lens and two scanning electron microscopes. Low-magnification images were captured using a Hitachi 3000 Tabletop microscope operated in backscatter electron mode (BSE). Higher magnifications of the cross-sectioned samples were carried out using a SCIOS FEI dual-beam SEM. Chemical analyses of the exposed samples were conducted employing energy-dispersive x-ray spectrometry (EDS) from Bruker and EDAX. The XRD phase analyses of the corroded samples were carried out using PANalytical Empyrean, PiXcel3D machine with ICDD PDF-4+2020 database.
Results and Discussion
Macro-Observations
Figure 3 shows macro-images of the unexposed and the exposed ceramic coating in the H2S gas mixture. As seen in the test, the uncoated 16M steel was not exposed. It is well known that a low-alloyed steel with as low as 1 wt.% Cr in sulfide-containing atmosphere shows no protection. Those types of steels predominantly form flaky sulfide scale with a tendency to spallate from the metallic core (Ref 3). Furthermore, two of the coatings (F (FCHO11) and G (FCHO12)) were withdrawn from the test, due to detachment of the ceramic layer from the metallic substrate (16M steel), and both coatings were formed based on a Cr2O3 phase. The other coatings showed a decent corrosion resistance in the sulfide atmosphere, and despite this, some of the coatings showed some porosity and hole formation during preparation. These holes and subsequent levels of porosity showed no big influence on the overall corrosion resistance of the applied ceramic coatings; however, the coatings with a higher concentration of holes and porosity indicated poorer sulfidation resistance (especially coatings C and D).

Macro images of the exposed coatings at 773.15 K for 336 hours in sulphidising atmosphere
The macro-observations revealed that coatings A and B indicated a smooth, non-cracked, non-delaminated surface. Both coatings also indicated a higher concentration of SiO2, B2O3, TiO2 and Na2O phases compared with the other coatings used in this work.
Kinetic
Ceramic coating kinetics are shown in Fig. 4.

Kinetic data for the ceramic coatings exposed in 99% synthetic air–1%H2S (vol.%) gas mixture
The results presented clearly show that exposure of ceramic coatings to a 99% synthetic air–1%H2S (vol.%) gas mixture indicates a relatively low mass gain; three out of four coatings showed similar results, where mass gain after 336 hours at 773.15 K/min reached around 1.9 mg/cm2. Coating D indicated over four times higher mass gain. As mentioned in this work previously, two coatings (coatings F and G) delaminated from the metallic substrate and were withdrawn from the test; there is no need to describe corrosion behavior of these coatings when delamination occurred during the laboratory exposure. Table 6 shows error calculations for the exposed systems in sulfidizing atmosphere. The calculation was done for a singular sample that was measured five times in a row. Based on the performed measurements, a standard deviation was evaluated as well as error value measurement. The calculations showed that mass gain kinetics were measured with a little error (lower than 5%).
XRD analyses
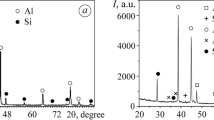
Figure 5, 6, 7, 8 and 9 shows XRD spectra of the ceramic coatings exposed in sulfidation conditions.

XRD spectra of the coating A exposed in 99% synthetic air—1%H2S (vol. %) gas mixture

XRD spectra of the coating B exposed in 99% synthetic air—1%H2S (vol. %) gas mixture

XRD spectra of the coating C exposed in 99% synthetic air–1%H2S (vol. %) gas mixture

XRD spectra of the coating D exposed in 99% synthetic air—1%H2S (vol. %) gas mixture

XRD spectra of the coating E exposed in 99% synthetic air—1%H2S (vol. %) gas mixture
In coatings A and B, the formation of Na2SO4 was predominantly responsible for the mass gain of the samples according to XRD spectra. In coating C, in addition to Na2SO4, CaSO4 phase formation increased mass gain. The results are in good agreement with kinetics, and higher mass gain was attributed to the samples with a higher number of sulfide phases developed after 336 hours of sulfidation in 99% synthetic air–1%H2S (vol.%) gas mixture. Additionally, the mass gain of the coatings is proportional to the sulfur content reacting with the reagent phases that were applied on the coating. The α-SiO2 and TiO2 were untouched in coatings A–C. However, the Na2O phase in coatings A–C reacted with H2S at high-temperature, forming Na2SO4; in contrast, the Li2O phase, with their high activity oxygen atmosphere, reacted to create Li2TiO3 phase instead of reacting with H2S. This finding shows that the formation of an Li2TiO3 phase is more favorable than the formation of Li-S-type phase or Ti-S-type phase. Further analyses of the XRD data show coating C with CaO forms a CaSO4 phase. The highest mass gain was observed in coating D where the highest number of phases (sulfides, oxides) formed; based on the results, the following phases were developed: Na3Fe(SO4)3, NaLiSO4, Co3O4 and finally CaMn3O8. The findings indicate the formation of alkali-iron tri-sulfate Na3Fe(SO4)3 and important observation since alkali-iron tri-sulfate is present in fireside corrosion atmospheres (Ref 6, 10). The mechanism of the formation of alkali-iron tri-sulfate phase is proposed in the following. In the first stage of the experiment, H2S is decomposed into H2 and S2, then, S2 reacts with O2 from the synthetic air atmosphere to form SO2, and finally, SO2 reacts with O2 according to the following reactions:
During high-temperature exposure at 773.15 K, the formed SO3 gas reacted with Na2O phase from the glaze. Simultaneously, the developed Fe oxide reacted with Na2SO4 to form the alkali-iron tri-sulfate (Na3Fe(SO4)3):
The formation of alkali-iron tri-sulfate is the principle reason for the degradation of the exposed steels with low Cr content. Corey et al. (Ref 19) found that alkali-iron tri-sulfate is stable when the concentration of SO3 is at a specific level. Below this level, the tri-sulfate phase becomes unstable and decomposes. In this case, the concentration of SO3 in flue gas reached a value of 250 ppm or higher. The formation of SO3 kept the concentration above 250 ppm to form a stable alkali-iron tri-sulfate that was found by XRD analysis.
The other phases found by XRD analyses (NaLiSO4, CaSO4) also reacted with the S2 derived from H2S decomposition. The phase formation mechanism is shown in the following:
The formation of NaLiSO4 and CaSO4 phases originates in the first step from H2S decomposition and a reaction with O2 according to reactions Eq 1, 3 and 3. In the case of NaLiSO4, it is strongly believed that firstly Na2SO4 is formed according to reaction Eq 4, due to the higher concentration of Na2O than Li2O in coating D. Further, Na2SO4 is combined with the Li2O phase according to the following reaction:
The formation of CaSO4 is derived from the combination of CaO and SO3:
The reaction of CaO with H2S to produce CaSO4 is rather impossible in the current conditions according to Allen et al. (Ref 20) where a direct reaction between CaO and H2S formulates CaS and H2O as follows:
However, in this work a CaS phase was not detected by XRD; therefore, reaction 8 showing the formation of CaSO4 is valid.
In addition to sulfide formation, performed XRD analyses indicated the formation of mixed oxides such as Co3O4 and Ca2Mn3O8 (coating D) andLi2TiO3 (coatings A and C). No new oxide phases were detected in coating B. The formation of these oxide phases is directly related to the presence of oxygen in the synthetic air. The formation of oxides, similar to the formation of sulfides, is responsible for the mass gain of the samples. The number of oxide formation is in good correlation with mass gain, and the higher the number of oxides formed, the higher the mass gain observed. The highest mass gain was presented in coating D, and this coating also developed more oxides than other coatings exposed in this work. Coating D developed Co3O4 and Ca2Mn3O8 phases according to XRD findings. The formation of Co3O4 results from oxidation of CoO according to the following reaction:
The formation of Ca2Mn3O8 structure is postulated by the following reaction:
There are a very limited number of papers indicating the formation of Ca2Mn3O8 phase. It is believed that this compound is formed due to the direct reaction between CaO and MnO in an oxygen-rich atmosphere. The coating developed during sulfidation tests the following phases: Na2SO4 (similar to coatings A–C); in addition, XRD found CaTiF6 and (KNa)AlSiO4. By the formation of CaTiF6, it is believed to originate from the direct reaction of CaO, TiO2 and F:
Based on XRD data, the coating developed during sulfidation test the following phases: Na2SO4 (similar to the coatings A–C) and CaTiF6, K,Na)AlSiO4. The formation of CaTiF6 is believed to originates from the direct reaction between CaO, TiO2, and F. Finally the formation of (K,Na)AlSiO4 is a rather more complex reaction chain similar to the formation of alkali iron tri-sulphates, as described in the previous paper (Ref 21).
Because the performed XRD analyses were unable to provide concentration of phases on XRD diffractogram directly, the Rietveld analyses were carried out. The concentration of the phases developed during sulfidation test is shown in Table 7. The analyses were performed based on PDF-4+ 2020 database. Table 7 shows model fit parameters (quality): Rp [%], wRp [%] and x2. The lower the values of those parameters, the analyses have a better quality of the model fit. Furthermore, Table 7 shows concentration of crystalline phases for the individual sample and unit cell parameters. The performed analyses clearly showed that the highest concentration of Na2SO4 was indicated in coating E (57 %), coating B (39 %), coating C (26 %) and coating A (24 %), but in coating D no Na2SO4 was found according to the report. The highest concentration of the formation of TiO2 was observed in coatings B and A, respectively, and the other coatings show no TiO2 (anatase) formation. (In coating D, concentration of TiO2 (anatase ) reached 0.9 %.) Furthermore, SiO2 (cristobalite) was observed only in coating A (15 %), but α-SiO2 was shown in coating A (15 %) and coating E (38 %). The formation of alkali-iron tri-sulfate Na3Fe(SO4)3 was observed in coating D (57 %), and this coating achieved the highest mass gain of the exposed materials in this work. Finally, the analyses found α-Fe concentration in coating B (26 %), in the other coatings, α-Fe concentration was much lower, in coating C, only CaSO4 was indicated, and no CaSO4 was discovered in the other exposed coatings in this study. To summarize, the most commonly known phases are as follows: Na2SO4: coatings: A, B, C and E, TiO2 (anatase): coatings: A, B (D traces) and SiO2: coatings: A and E.
Microanalyses
Cross-sectioned SEM Images of the Uncoated 16Mo3 (16M) Steel
Figure 10 shows cross-sectional images of the uncoated 16Mo3 (16M) steel exposed in sulfidation conditions at 773.15 K for 100 hours.

Cross sectioned images of the uncoated 16Mo3 (16M) steel exposed in sulphidation test conditions at 773.15 K for 100 hours
The figures were captured during different researches carried out in the past (Ref 22). As shown, the uncoated steel shows very poor corrosion resistance and a thick scale formed where plate- and needle-like structures rich in Fe-S phase are developed. The thickness of the sulfide scale reached around 500 µm (measured from the top of the sulfide scale to the substrate interface). Based on EDS analyses (not shown here) on cross-sectioned samples performed after exposure at 773.15 K, it was found that the outermost part of the sulfide scale consisted of a high concentration of S reaching 70 and 30 wt.% Fe. The middle part of the formed scale possessed high concentrations of S equivalent to 35 wt.% and Fe concentrations reaching 65 wt.%, indicating the formation of a non-stoichiometric Fe1-XS phase. According to the findings presented by Mrowec (Ref 23) in the past, a lower concentration of Cr than 4 wt.% in the exposed steel was identified, revealing the sulfidation rate as for pure iron. In sulfide-rich scale, the growth depends on the diffusion of Fe2+/Fe3+ ions from the metal matrix. Furthermore, when low-alloyed steel with up to 4 wt.% Cr such as 16Mo3 (16M) is exposed to a sulfur-rich atmosphere, S-rich phases are likely to develop such as FeS. The FeS phase possesses some degree of non-stoichiometry in the crystal lattice according to work performed by Mrowec (Ref 19), Gesmundo et al. (Ref 24) and McCammon (Ref 25); thus, the real formula of FeS should be written as Fe1-XS, where x = 0-0.2 (Ref 26). The phase also possesses p-type semiconductor properties, where holes are the majority carriers and electrons are the minority carriers. Defects in Fe1-XS develop due to the absorption of sulfur from the gaseous phase onto the Fe1-XS crystal structure; furthermore, diffusion of iron cations from the metal matrix occurs via chemisorption with S2- anions.
Cross-Sectioned SEM Images of the Coated 16Mo3 (16M) Steel
Figure 11(a-e) shows cross-sectioned SEM images of the coated 16Mo3 (16M) steel, and the steel was coated using different coatings to reduce sulfidation degradation. The images present only samples: A, B, C, D and E. Samples F and G were withdrawn from the test due to spallation of the coating from the substrate.

Cross sectioned images of the coated 16Mo3 (16M) steel with different coatings exposed in 99% synthetic air and 1% H S (% vol.) conditions at 773.15 K for 336 hours
The present results clearly indicate a high degree of protectiveness of the ceramic coatings deposited on 16Mo3 (16M) steel. Among the exposed coatings, coatings A and B showed high resistance against sulfidation, where no cracks perpendicular or horizontal to the substrate appeared. The two coatings showed the lowest mass gains recorded; on the other hand, coating E despite low mass gain showed horizontal cracks nearby substrate interface. It is believed that cracks observed in cross-sectional image were derived from cutting process. Similar cracks were observed in coating C. Such observations lead to conclusion that those two types of chemical composition in the coatings are prone to hydrogen embrittlement; however, no analyses in this direction were carried out. The highest degree of cracks and degradation of the coating was observed in coating D, and this type of coating showed the highest mass gain related to the coated material. The results presented in cross-sectional captures are in good correlation with mass gain data presented earlier in this work.
Furthermore, the coating deposited on the 16Mo3 (16M) steels showed a high degree of non-reactivity with the highly aggressive 99% synthetic air and 1% H2S (vol.%) gas mixture at 773.15 K. The results clearly indicated no corrosion products on the external coating surface exposed to harsh conditions. All three coatings showed under cross-sectioned x-ray mapping investigations a concentration of S equivalent to 2 (wt.%). The frit composition contained a mixture of a special grade of calcined aluminum oxide and a conventional type of ground-coat frit with water to appropriate fineness on a steel surface, and drying and firing led to high degree of protectiveness in comparison with the uncoated steel. In general, frit-based coatings exposed to harsh conditions with a high content of SiO2 showed a high degree of non-reactivity as mentioned earlier. Nevertheless, according to the previous work published by Swaroop (Ref 27), when a coating is rich in SiO2 in a highly concentrated H2 system (99% synthetic air and 1% H2S (vol.%) as in this work, silica glass tends to lose silicon, by the formation of SiO at low O2 partial pressure. The loss of Si may act by the direct dissociation of SiO2 by reaction with silicon in the disilicide. Furthermore, this process may accelerate highly, because of H2 and water vapor in the system. The presence of H2 in contrast to S2 in the atmosphere is the main issue, and as S2 identified shows no impact on coating degradation, there are no thick corrosion products or coating cracks, where S2 could ingress and react with the substrate. On the other hand, due to a high ability for penetration, H2 can easily ingress into the coating structure and lead to brittleness of the coating structure, and this phenomenon is called hydrogen embrittlement. It is believed that this process was found in the coatings presented in Fig. 11(c, d). Hydrogen embrittlement is a well-known phenomenon. This process leads to a reduction in the ductility of the structure and load bearing capacity that finally provokes cracking, delamination and serious degradation of the coating system. Unfortunately, a detailed process is not well understood and still there is still ongoing experimentation with regard to the exact mechanisms. In short, the process is governed by the diffusion of H2 throughout grain boundaries of the structured materials. (In steels, the diffused H2 reacts with C producing CH4.) The gas is not mobile and collects in small voids along the grain boundaries where it builds up enormous pressures that initiate cracks in the structure or leads to the formation of brittle phases that initiate crack propagation under external loading (cutting process). Similar observations could be detected in SiO2-based glassy coatings exposed to H2-rich atmosphere (Ref 28). Since H2 possesses the smallest radius of atoms, a high number of defects in glassy materials provide easy access for H2 atoms into the inner part of the coating. On the other hand, no corrosion products related to S2 were found in this study, suggesting an impermeable character of the developed coating for S2 atoms. Similar results were achieved by Kurkowa et al. (Ref 29), indicating CrSi2-Ni and CoNiCrAlY-Si alloys in atmospheres containing NaCl and Na2SO4 showing a high degree of protection against S2 inward diffusion due to SiO2 scale formation. The SiO2 scale plays a significant role in suppressing the inward diffusion of sulfur, resulting in the remarkable suppression of the formation of sulfide at the scale/substrate interface and inside of the substrate. The work was conducted on alloys able to individually form SiO2; however, SiO2 that possesses such properties opens new possibilities for the alloys structures that may be used in atmospheres containing H2S.
EDS Mapping of the Exposed Samples
Figure 12(a-c) presents the EDS x-ray mapping performed on the best performing samples in the test carried out in this study.

EDS X-ray mappings of the samples exposed in 99% synthetic air and 1% HS (% vol.) at 773.15 K for 336 hours: (a) coating A, (b) coating B and (c) coating E
In general, the coatings analyzed under EDS x-ray mapping system showed a very similar concentration of the elements. The coatings mainly consisted of Si and O. The base material indicated the highest concentration of Fe and Cr was not detected due to a very low concentration of this element within the metal matrix. The interface observed between the coating and the substrate was occupied by Mn. The element diffused from the metal matrix of 16Mo3 (16M material). As mentioned Cr was not detected, neither Mo nor S2 was observed by carrying out investigation, suggesting a high resistance of the developed coating against S2 diffusion from the S2-rich atmosphere (H2S atmosphere). In addition, S2 was not observed on the surface of the exposed coatings.
Conclusions
The aim of this work was to investigate a series of ceramic coatings exposed to 99% synthetic air and 1% H2S (vol.%) under a flow rate of 50 mL/min for 336 hours at 773.15 K. The sulfidation study showed that coatings based on SiO2-B2O3-TiO2-Na2O compounds indicated an overall good sulfidation resistance, and some spallation was observed in F and G coatings. Horizontal cracks were observed in C, D and E coatings, most probably due to the H2-rich atmosphere and residual H2O from synthetic air. After 500 hours of testing, the coatings showed no S2 inward diffusion or S2 presence on the substrate–coating interface observed.
References
J.M. Beer, High Efficiency Electric Power Generation: The Environmental Role, Prog. Energy Combust. Sci., 2007, 33(2), p 107–134.
Making the EU climate-neutral by 2050, European Commission - Press release, 2020
T. Dudziak, K. Jura, P. Dudek, L. Boron and J. Rutkowska, Sulphidation of Low-Alloyed Steels Used in Power Industry, Oxid. Met., 2019, 92(5–6), p 379–399.
S. Mrowec, The Problem of Sulfur in High-Temperature Corrosion, Oxid. Met., 1995, 44, p 177–209.
H. Ghaseni, High Temperature Sulfidation of Carbon Steel Heater Tubes in Gas Condensate Containing Sulfur Compounds, Eng. Fail. Anal., 2011, 18(3), p 980–987.
T. Hussain, T. Dudziak, N.J. Simms and J.R. Nicholls, Fireside Corrosion Behaviour of Hvof & Plasma Sprayed Coatings in Advanced Coal/Biomass Co-Fired Power Plants, J. Therm. Spray Tech., 2013, 22(5), p 797–807.
D. Tejero-Martin, M. Rezvani Rad, A. McDonald and T. Hussain, Beyond Traditional Coatings: A Review on Thermal-Sprayed Functional and Smart Coatings, J. Therm. Spray Tech., 2019, 28, p 598–644.
Ch. Liang-Yu, W. Haiyang, Z. Sheng Cuihu, Z.-X. Lu, W. Jin, S. Shujin and C. Lai-Chang Zhang, Automatic Remelting and Enhanced Mechanical Performance of A Plasma Sprayed NiCrBSi Coating”, Surf Coat Technol, 2019, 369, p 31–43.
B. Wielage, A. Wank, H. Pokhmurska, T. Grand, Ch. Rupprecht, G. Reisel and E. Friesen, Development and Trends in HVOF Spraying Technology, Surf. Coat. Technol., 2006, 201, p 2032–2037.
L.-Y. Chen, Xu. Tianxiang, Lu. Sheng, Z.-X. Wang, S. Chen and L.-C. Zhang, Improved Hardness and Wear Resistance of Plasma Sprayed Nanostructured Nicrbsi Coating Via Short-Time Heat Treatment, Surf. Coat. Technol., 2018, 350, p 436–444.
N.S. Harding and D.C. O’Connor, Ash Deposition Impacts in the Power Industry, Fuel Process. Technol., 2007, 88(11–12), p 1082–1093.
E. Labuda, K. J. Shields, D. A. Cline, “Fireside corrosion in coal- and oil-fired units: failure mechanisms and methods of prevention”, Corrosion, Paper 00031, 2000
A.U. Syed, N.J. Simms and J.E. Oakey, Fireside Corrosion of Superheaters: Effects of Air and Oxy-Firing of Coal and Biomass, Fuel, 2012, 101, p 62–73.
C.M. Pavan and B.R. Narendra Babu, Review of Ceramic Coating on Mild Steel Methods, Applications and Opportunities, Int. J. Adv. Sci. Technol., 2018, 4(7), p 44–49.
K. Srinivasulu and S. Manisha Vidyavathy, Advanced Ceramic Coatings on Stainless Steel, A Review of Research, Methods, Materials, Applications and Opportunities, Int. J. Adv. Engg. Tech., 2016, 7(3), p 126–141.
Y. Adraider, Y.X. Pang, F. Nabhani, S.N. Hodgson, M.C. Sharp and A. Al-Waidh, Laser-Induced Deposition of Alumina Ceramic Coating on Stainless Steel From Dry Thin Films for Surface Modification, Ceram. Int., 2014, 40, p 6151–6156.
M. Seraffon, N.J. Simms, J. Sumner and J.R. Nicholls, The Development of New Bond Coat Compositions for Thermal Barrier Coating Systems Operating Under Industrial Gas Turbine Conditions, Surf. Coat. Technol., 2011, 7, p 1529–1537.
M. Seraffon, N.J. Simms, J.R. Nicholls, J. Sumner and J. Nunn, Performance of Thermal Barrier Coatings in Industrial Gas Turbine Conditions, Mater. High Temp., 2011, 28(4), p 309–314.
R.C. Corey, B.J. Cross and W.T. Reid, Trans. Am. Soc. Mech. Eng., 1945, 67, p 289–319.
D. Allen and A.N. Hayhurst, The Kinetics of the Reaction Between Calcium Oxide and Hydrogen Sulphide at the Temperatures of Fluidized Bed Combustors, Symp. Combust Proc., 1991, 23(1), p 935–941.
T. Dudziak, T. Hussain, N. J. Simms, A. U. Syed, J. E. Oakey, Fireside corrosion degradation of ferritic alloys at 600 °C in oxy-fired conditions, Corrosion Science, 79 (2014), 184–191
T. Dudziak, K. Jura, P. Dudek, L. Boron and J. Rutkowska, Sulphidation of Low-Alloyed Steels Used in Power Industry, Oxid. Met., 2019, 98, p 379–399.
S. Mrowec, The Problem Of Sulfur in High-Temperature Corrosion, Oxid. Met., 1995, 44(1/2), p 177–209.
F. Gesmundo, D.J. Young and S.K. Roy, The High Temperature Corrosion of Metals in Sulfidizing-Oxidizing Environments: A Critical Review, High Temp. Mater. Process, 1989, 8, p 149–188.
C.A. McCammon and L. Lin-gun, The Effects of Pressure and Temperature on Nonstoichiometric Wüstite, Fexo: The Iron-Rich Phase Boundary”, Phys. Chem. Miner., 1984, 10, p 106–113.
E.J. Fasiska, Some Defect Structures of Iron Sulphide, Phys. Status Solidi, 1972, 10(1), p 169–173.
R. Swaroop, “Ceramic coatings for components exposed to coal-gas environments: A review”, Argonne National Laboratory, Argonne, Illinois, ANL-76-124, 1976
X. Li, X. Ma, J. Zhang et al., Review of Hydrogen Embrittlement in Metals: Hydrogen Diffusion, Hydrogen Characterization, Hydrogen Embrittlement Mechanism and Prevention, Acta Metall. Sin. (Engl. Lett.), 2020, 33, p 759–773.
K. Kurokawa, T. Sudiro, T. Sano, S. Kyo, O. Ishibashi and M. Nakamori, High-Temperature Corrosion Resistance of SiO2-Forming Materials, Corros. Rev., 2018, 36(1), p 65–74.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rząd, E., Dudziak, T., Polczyk, T. et al. Sulfidation of Ceramic-Based Coatings Deposited on Low-Alloyed Steel 16Mo3 Exposed at High Temperature. J. of Materi Eng and Perform 30, 8538–8550 (2021). https://doi.org/10.1007/s11665-021-06070-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11665-021-06070-y