
Abstract
Purpose
The growing availability of new psychoactive substances with unknown toxicity is alarming all over the world. The simplicity of acquiring these drugs of abuse from internet markets has caused an increase in the total number of seizures. We present the first systematic spectroscopic study of methylone and pentylone, which are in the class of synthetic cathinones.
Methods
High performance liquid chromatography (HPLC) was used for the enantioseparation of methylone and pentylone. The substances were further analysed by the conventional methods of ultraviolet (UV) and infrared (IR) absorption and chiroptical methods, specifically electronic circular dichroism (ECD) and vibrational circular dichroism (VCD). The obtained data were supported with the density functional theory (DFT) calculations using the B3LYP or B3PW91 functional and 6-311++G(d,p) basis sets, including the solvent effects.
Results
The 3D structure of methylone and pentylone in solution was revealed. Moreover, the chiral separation of both studied substances was achieved and the absolute configuration of the respective enantiomers was determined.
Conclusion
Vibrational circular dichroism spectroscopy seems to be the best of the methods employed to distinguish structurally similar chiral substances, especially in combination with quantum chemical calculations. According to the structural analysis, 5 and 9 stable conformers of methylone and pentylone, respectively, were revealed in an aqueous solution. Finally, very good agreement between the experimental and simulated spectra was achieved.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
New psychoactive substances (NPS), compounds structurally similar to already legislatively controlled illicit drugs recently appearing on the drug scene with increasing frequency, pose an increasing global problem. The EU Early Warning System for NPS currently monitors over 670 substances, with 51 reported in 2017 for the first time and 101 in 2014 [1, 2]. In addition, the total number of NPS all over the world monitored by the United Nations Office on Drugs and Crime is 803 individual NPS [3]. In 2016, about 71,000 seizures of NPS were reported through the EU Early Warning and almost one-third of these seizures are of synthetic cathinones [1].
New psychoactive substances are often considered by users as a safe alternative to illicit narcotics and psychotropic substances, despite the fact that they possess similar structures and psychoactive effects. Structural modification of the parent NPS may often lead to the enhancement of hallucinogenic or psychostimulative effects, e.g. in case of synthetic cannabinoids (THC analogues) and cathinones (cathinone analogues) [4, 5]. Synthetic cathinones (often sold as “legal highs”, “bath salts” or “research chemicals”) [6] are the second most frequently intercepted NPS. Their effects include empathy, openness, increased energy and libido of consumers, but also dangerous neurological, cardiac and psychiatric signs and symptoms [1, 7]. Methylone and pentylone (Fig. 1) studied here are homologues in the class of synthetic cathinones. Unlike natural cathinone, methylone and pentylone both possess the methylenedioxy functional group on the phenyl ring, similarly to the well-known psychoactive substance 3,4-methylenedioxyamphetamine (MDMA), better known as ecstasy. Due to this structural similarity, methylone and pentylone combine the effects of cathinone and MDMA [8, 9]. Their mechanism of action is based primarily on inhibiting the uptake of norepinephrine, dopamine and serotonin by monoamine neurotransmitter transporters [10]. However, the full metabolism in the human body is not yet completely understood.

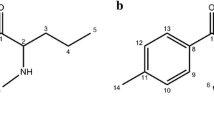
Structures of methylone hydrochloride (left) and pentylone hydrochloride (right) with labelled dihedral angles α1–α5
Methylone and pentylone differ in the length of the α-alkyl chain; methylone contains a methyl group and pentylone a propyl group. According to pharmacokinetic studies on rats [9, 11], a longer α-alkyl chain causes higher lipophilicity, hence, pentylone exhibits a higher plasma concentration than methylone after dosing the same amount of the pure substance. However, safety data concerning their toxicity on humans as well as information about the side effects of long-term usage are unavailable [3].
The current trends in the control of NPS demand a fast and reliable analysis. Moreover, methods for a detailed description of their structure are required. However, most studies [12,13,14] are focused on the detection of NPS in biological samples (blood, plasma, urine, hair) performed by high-performance liquid chromatography (HPLC) or gas chromatography (GC) with mass spectrometry (MS). For studies on identification of real NPS samples, vibrational spectroscopy techniques such as infrared (IR) or Raman spectroscopy are often used. The advantage of this approach is a fast and reliable analysis, which can even be performed by portable spectrometers [15, 16].
Molecular spectroscopy is one of the effective tools for the identification of the NPS. Since synthetic cathinones are chiral, chiroptical methods [17] can be used for their detailed study. The chiroptical methods, specifically vibrational circular dichroism (VCD), electronic circular dichroism (ECD) and Raman optical activity (ROA), in combination with theoretical quantum chemical predictions can provide further 3D structural information on NPS in solution, which is crucial, e.g. for the understanding of their biological activity and toxicity [17,18,19]. The comparative analysis of experimental chiroptical properties and DFT calculations also allow the determination of the absolute configuration of individual separated enantiomers [20, 21]. Analyses performed by different independent chiroptical spectroscopic methods provide more reliable structural characterizations of the studied molecules [22,23,24].
The precise structural characterization of enantiomers, enabled by the combination of chiroptical spectroscopic methods and DFT calculations, may be helpful for understanding differences in their toxicity. These methods can also be used to determine the presence of a particular drug enantiomer in a mixture of different compounds—a typical feature of synthetic cathinone products available on the Internet [7]. There are many examples that enantiomers of licit as well as illicit drugs have different properties and toxicity. Apart from the widely known Contergan affair [25], it has already been shown in literature that enantiomers of some widely used drugs of abuse, e.g. ketamine and deschloroketamine possess significantly different properties in terms of pharmacological effect and toxicity [26, 27]. With the increasing number of synthetic cathinones, the need for detailed information on the structure of individual enantiomers and structure-properties relationships is essential. This is due to the fact that naturally occurring active substance is (S)-cathinone, and it has already been documented that (S)-enantiomers of synthetic cathinones are in many cases more potent/toxic than (R)-enantiomers [28,29,30]. It is therefore evident that not only simple identification of a substance by analytical methods, but also more in depth structural information is required, and in this case, DFT calculations represent an important tool.
In the continuation of our systematic study of NPS [27, 31], we introduce a comparison of two NPS homologues—methylone and pentylone—in an aqueous solution. Employing the methods of VCD, ECD, IR spectroscopy and ultraviolet (UV) absorption spectroscopy supported by the DFT calculations at B3LYP/6-311++G(d,p) or B3PW91/6 311++G(d,p) levels, including solvent effects, we elucidated in detail the 3D structure of both drugs and compared their structural features. Moreover, we performed chiral separation of the studied substances and determined the absolute configuration of the respective enantiomers.
Materials and methods
Chemicals, reagents
Racemic standards of methylone hydrochloride and pentylone hydrochloride with purity exceeding 98% were obtained from Alfarma (Černošice, Czech Republic). Deuterium oxide (D2O; 99.9% D) for the dissolution of methylone and pentylone was purchased from ISOSAR GmbH, Germany. HPLC grade solvents (hexane, heptane, propan-2-ol IPA) were obtained from Labicom (Olomouc, Czech Republic) and ethanol was purchased from Merck (Darmstadt, Germany). Diethylamine, used as a basic additive, was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Enantioseparation
Both the analytical and the preparative chiral separation of the drugs were performed using the Waters AutoPurification System (Milford, MA, USA) equipped with a UV–Vis detector (detection wavelength of 254 nm). The analytical separation was performed on a chiral polysaccharide column ChiralArt Amylose-SA (250 × 4.6 mm i.d., 5 μm, YMC Europe GmbH, Dinslaken, Germany) with an analyte concentration of 1 mg ml−1 and a flow rate of 1 ml min−1 at ambient temperature. For the preparative chromatography, an analogous polysaccharide column available in our laboratory—Chiralpak IA (250 × 20 mm i.d., 5 μm, Chiral Technologies Europe, Illkirch, France)—was used. For methylone, the sample concentration was 17.5 mg ml−1, while in case of pentylone, for which a different mobile phase was required (vide infra), the sample concentration was 4 mg ml−1. In both cases, at flow rate of 15 ml min−1 and ambient temperature conditions were employed. For analytical chromatography screening, commercially available drugs were dissolved in the mobile phase containing 0.1% of diethylamine. The sample for preparative chromatography was dissolved in a mixture of 1% diethylamine in propan-2-ol (0.45 ml) and a total volume of 1 ml was adjusted by the addition of heptane. In both cases, the addition of diethylamine ensured the liberation of a free base of the drug from the corresponding hydrochloric acid salt.
Electronic circular dichroism
The experimental ECD spectra were acquired using a J-815 spectrometer (JASCO Corporation, Tokyo, Japan) purged with nitrogen gas (purity 99.99%, Siad, Prague, Czech Republic) during the measurement. To measure the ECD spectra, solutions of individual enantiomers of a concentration of 15 mg l−1 in demineralized water (UCT Prague, Czech Republic) were prepared. These sample solutions (3 ml) were pipetted into a quartz cuvette (Hellma, Müllheim, Germany) with a pathlength of 1 cm and measured in a spectral range of 185–380 nm at ambient temperature with a 20 nm min−1 scanning speed, 8-s response time, 3 accumulations, and 1 nm band width. The baseline was corrected by subtracting the spectra of demineralized water obtained under identical experimental conditions. The corresponding UV absorption was calculated from the detector HT voltage using the Spectra Analysis module of the Spectra Manager software (JASCO Corporation, Tokyo, Japan).
Vibrational circular dichroism
The VCD and IR absorption spectra were acquired using the FTIR IFS 66/S spectrometer equipped with a VCD/IRRAS PMA 37 module (Bruker, Bremen, Germany), a ZnSe beam splitter (Hinds Instrument, Hillsboro, OR, USA), a BaF2 polarizer, and an MCT detector (InfraRed Associates, Stuart, FL, USA) [32]. The solutions of individual enantiomers of methylone hydrochloride and pentylone hydrochloride in D2O (100 mg ml−1) were placed individually into a BioCell cuvette with CaF2 windows (BioTools, Inc., Jupiter, FL, USA) and an optical pathlength of 27.3 μm. The spectra were measured at ambient temperature in a spectral range of 1750 to 1250 cm−1 with a resolution of 8 cm−1. The VCD spectra were averaged from 9 to 12 blocks, each measured for 20 min and containing 3680 interferometric records. The baseline of each enantiomer was corrected by the subtraction of the solvent (D2O) spectra measured under identical experimental conditions. The VCD noise spectra were offset from zero for clarity.
Quantum chemical calculations
The starting geometries of (R)-methylone and (R)-pentylone hydrochlorides were optimized using the DFT method at the B3LYP/6-31G(d) level. Reoptimization of the final lowest energy conformers, considering their Gibbs free energies, was performed by the DFT method at several higher levels of theory (B3LYP/6-311++G(d,p), B3PW91/aug-cc-pVDZ, B3PW91/6-311++G(d,p), CAM-B3LYP/6-311++G(d,p), CAM-B3LYP/aug-cc-pVDZ, wB97XD/6-311++G (d,p), wB97XD/aug-cc-pVDZ, and wB97XD/TZVP, Electronic Supplementary Material, Table S1 and S2) using the Gaussian 09 program package [33]. In this work, only the results providing the best level of agreement with the experimental spectra are presented in detail (B3LYP/6-311++G(d,p), B3PW91/6-311++G(d,p)). All the calculations were carried out on a supercomputer Altix UV 2000 (UCT Prague, Czech Republic). In general, 12 processors with total memory of 64 GB were used for individual computation tasks, which took 40–48 h.
The D2O solvation effect was considered via the conductor-like polarizable continuum model (CPCM). The DFT calculations of VCD spectra included the effect of deuteration of the amine hydrogens. The population weighted average VCD, ECD, IR and UV spectra were calculated using the Boltzmann distribution based on Gibbs free energy at a temperature of 298 K. The similarity overlaps of the experimental and calculated spectra were undertaken using the CDSpecTech program, based on the similarity of the dissymmetry factor to determine quantitatively the agreement between both spectra and to choose the optimal scaling factor [34, 35]. A scaling factor of 1.0 marks the maximum similarity.
For visualization of the simulated VCD and IR spectra, a Lorentz profile function with a 10 cm−1 bandwidth was assumed. For ECD and UV absorption, the 30 lowest singlet states of each conformer were calculated and Gaussian band profiles with a 12 nm bandwidth were assumed.
Results and discussion
Enantioseparation
Since methylone and pentylone are in the cathinone family, they can be enantiomerically resolved by several different methods including, for example, capillary electrophoresis, supercritical fluid chromatography, and HPLC [36,37,38]. However, only some methods from this list can provide pure enantiomers in preparative amounts of the milligram to gram scale. The most prominent method for the separation of chiral substances nowadays is preparative HPLC. We have already employed this method in our previous study focused on the determination of the absolute configuration of butylone [31], thus HPLC was selected as the method of choice in this study.
Starting from the previously optimized conditions, we have found that although they are in the same family of NPS, methylone, butylone and pentylone exhibit significantly different chromatographic behaviours. Therefore, different methods of chiral separation for methylone and pentylone have been developed; more detailed information on the screening of the mobile phase conditions can be found in the Electronic Supplementary Material. Methylone, as the more polar substance, was resolved using a mobile phase composed of a mixture of hexane/EtOH (4/1, v/v) with diethylamine (0.1%) (Fig. 2), whereas the more lipophilic pentylone was successfully resolved using a mixture of heptane/IPA (95/5, v/v) with diethylamine (0.1%) (Fig. 3).

Preparative enantioseparation of methylone with the marked boundaries for the collection of methylone 1 and methylone 2 peaks. Insets show the analytical control of optical purity of the collected enantiomers: methylone 1 (lower) and methylone 2 (upper)

Preparative enantioseparation of pentylone with the marked boundaries for the collection of pentylone 1 and pentylone 2 peaks. Insets show the analytical control of optical purity of the collected enantiomers: pentylone 1 (lower) and pentylone 2 (upper)
In both cases, automatic fraction collection was set to ensure high purity of the collected enantiomers. In order to assure the stability of the separated enantiomers and to avoid a cyclization reaction occurring for the cathinone free base [39], the separated substances were transformed to hydrochlorides by adding an etheric solution of hydrogen chloride. Both enantiomers (methylone 1 and methylone 2) were found to be of comparable optical purity with the enantiomeric excess of > 96.3%. This value was still acceptable for the subsequent spectroscopic measurements. For pentylone, even better values of > 99.4% and > 98.9% for the first and the second eluting enantiomers, respectively, were found.
Conformational analysis
The individual (R)-enantiomers of methylone and pentylone hydrochloride were selected for the conformational analysis. Energetically preferred geometries of these molecules were determined by three or five dihedral angles α1, α2, α3, and optionally α4 and α5 (Fig. 1) for methylone and pentylone, respectively. The dihedral angle α1 was systematically changed by 180° rotation about a single bond and the remaining angles α2, α3 and possibly α4 and α5 by 120° using the MCM software [40]. As a result, 18 and 162 starting geometries for methylone and pentylone, respectively, were obtained. Those geometries were subsequently optimized by the DFT method at the B3LYP/6-31G(d) level of theory. Since most starting structures were converging to the same geometry or their population was too low, conformational analysis revealed all the relevant stable conformers (relative energy < 2 kcal mol−1): 5 and 9 for methylone and pentylone, respectively. These stable conformers were reoptimized at a more refined level of theory: B3LYP/6-311++G(d,p) or B3PW91/6-311 ++G(d,p) to provide the final structures (Fig. 4) and their relative abundances based on the Boltzmann distribution (Table 1).

Structures of the five stable conformers of (R)-methylone hydrochloride (top: M I–M V) and nine stable conformers of (R)-pentylone hydrochloride (bottom: P I–P IX) simulated at the B3LYP/6-311++G(d,p) level. The structures are displayed with the same orientation of the aromatic ring. Blue: nitrogen, red: oxygen, green: chlorine, grey: carbon, white: hydrogen (color figure online)
All the conformers were stabilized by the hydrogen bonding between the oxygen atom of the carbonyl group and the hydrogen atom of the amino group. From all the conformers of methylone, this interaction most stabilized the structure of the conformer M I, probably resulting in its high relative abundance (85%). The distance of hydrogen and oxygen in the case of the conformer M I was ~ 2.1 Å, for other conformers of methylone this distance was 2.2–2.5 Å. As for the individual dihedral angles (Table 1), the angle α1 occupied predominantly two positions that differed by ~ 180°. The total number of stable conformers of pentylone was higher than methylone due to the elongation of the alkyl chain and the associated increase in the number of dihedral angles in the molecule. All 9 conformers of pentylone were also stabilized by the hydrogen bonding between the oxygen of the carbonyl group and the hydrogen of the amino group, with the distance of these atoms being in the range of 2.0–2.2 Å for all the stable conformers. Relatively small differences in the length of this stabilizing interaction may be the reason for the more uniform relative abundances of conformers (33, 26 and 19%) than in the case of methylone.
Electronic spectroscopy
Since the experimental ECD spectra of the first and the second eluting compounds (Fig. 5) were mirror images, the enantiomeric character of the separated products was confirmed. The comparison of the experimental and simulated spectra determined the absolute configuration of the individual enantiomers: methylone 1 and 2 were determined as (R)- and (S)-enantiomer, respectively. On the contrary, pentylone 1 and 2 were determined as (S)- and (R)-enantiomer, respectively. These results could be confirmed further using the similarity overlap plots (Electronic Supplementary Material, Figs. S3 and S4). For the correct assignment of the bands in the experimental and simulated spectra, the simulated spectra of methylone were scaled by a factor of 0.96 in order to reach the maximum value of the similarity index. After scaling, a similarity index of 0.85 was achieved in this case. For pentylone, a scaling factor of 0.97 was applied and the corresponding index of similarity reached 0.75. Both results were more than sufficient to reliably determine the absolute configuration of individual enantiomers, which was subsequently confirmed by a detailed VCD spectra analysis.

ECD spectra of methylone hydrochloride (left) and pentylone hydrochloride (right): the B3LYP/6-311++G(d,p) and B3PW91/6-311++G(d,p) calculated ECD spectra of stable conformers (top), their Boltzmann averaged spectra (middle) and experimental ECD spectra (bottom)
In the experimental ECD spectra of methylone and pentylone hydrochloride (Fig. 5), six very similar bands reflecting the combination of electron transitions π → π *, n → π * and n → σ * were observed. The bands at 330 nm and partially at 278 nm were mainly an expression of an electron transition from the highest occupied to the lowest unoccupied molecular orbitals (HOMO → LUMO, Fig. 6). Three positive and three negative bands in the spectra of (R)-enantiomers were also present in the simulated weighted average spectra of (R)-enantiomers. The spectra of individual conformers of the (R)-enantiomers differed considerably from one another (Fig. 5, top), indicating the exceptional sensitivity of ECD to the 3D structure of the molecules compared to non-polarized methods (UV, IR). However, the experimental ECD spectra did not allow reliable differentiation between the two studied compounds.

Highest occupied (bottom) and lowest unoccupied molecular orbitals (top) for conformer I of (R)-methylone (a) and (R)-pentylone (b) hydrochloride. Blue: nitrogen, red: oxygen, green: chlorine, grey: carbon, white: hydrogen, red and green shadows: electronic molecular orbitals (color figure online)
The simulated UV spectra of (R)-methylone and (R)-pentylone hydrochloride correctly predicted four significant bands of the experimental spectra (Fig. 7). The simulated spectra were scaled by a factor of 0.97 and 0.98 for methylone and pentylone, respectively, and the final similarity overlap reached 0.82 and 0.78 for methylone and pentylone, respectively (Electronic Supplementary Material, Figs. S3 and S4). However, this method does not seem to be suitable either for the differentiation of substances differing in the number of –CH2 groups (since the experimental spectra of both studied compounds did not significantly differ) nor for the studies of the 3D structural changes of the individual conformers. The shapes of the partial spectra (Fig. 7, top) were very similar, with slight differences up to 10 nm observed in the change of the position of the band maximum at 330 nm. Differences between the intensities of band at ~ 234 nm in the simulated spectra of (R)-pentylone were caused mainly by the change of dihedral angle α1: if its value was positive, the intensity of the band was lower. In contrast, the intensity of the band at 270 nm was higher for positive values of α1.

UV absorption spectra of methylone hydrochloride (left) and pentylone hydrochloride (right): the calculated B3LYP/6-311++G(d,p) UV absorption spectra of stable conformers (top), their Boltzmann averaged spectra (middle) and experimental UV absorption spectra (bottom)
Vibrational spectroscopy
The shape of the experimental IR absorption spectra of methylone and pentylone in the spectral range 1750–1250 cm−1 was very similar with a total number of 11 and 9 bands for methylone and pentylone, respectively (Fig. 8). For the correct assignment of the individual bands and their respective vibrational modes (Tables 2, 3), the spectra were scaled by a factor of 0.99 and 0.98 and resulted in a very convincing index of spectra similarity: 0.85 and 0.91 for methylone and pentylone, respectively (Electronic Supplementary Material, Figs. S3 and S4). Looking at the individual spectra of the conformers (Fig. 6, top), it is worth noting the large influence of the dihedral angle α1 on the spectral shape. Band 1 was shifted to the lower wavenumbers in the spectra of all the conformers with a positive value of angle α1 (~ 5°) compared with the spectra of the other conformers. Band 2 was apparent only in the spectra of the conformers with the positive value of angle α1 (~ 5°), hence the intensity of this band was lower in the averaged spectra and appeared only as a shoulder. In contrast, bands 9 in the spectra of methylone and 8 in the spectra of pentylone were not present in the spectra of those conformers. The bands in this area reflected the bending vibration of the C–H group, including the umbrella bending vibration of –CH3. Band 7 in the spectra of methylone and band 6 in the spectra of pentylone were more intense in the case of the conformers with positive value of α1. Band 10 in the spectra of methylone reflected the bending vibration of the C–H group in CH–CH3 and was correctly predicted by the DFT calculations. This group was not present in the structure of pentylone, therefore we did not expect a band reflecting the bending vibration of the C–H group in CH–CH3 to occur in its IR spectra. This was confirmed by the experimental spectra as well. Furthermore, slight differences in the spectral shapes were observed between the experimental IR absorption spectra of methylone and pentylone hydrochloride (Fig. 8, bottom), particularly in the spectral range of 1420–1280 cm−1. Therefore, this method can be used to distinguish structurally similar substances, but, unfortunately, with low sensitivity.

IR absorption spectra of (R)-methylone hydrochloride (left) and (R)-pentylone hydrochloride (right): the calculated B3LYP/6-311++G(d,p) IR absorption spectra of the stable conformers (top), their Boltzmann averaged spectra (middle) and experimental IR absorption spectra (bottom)
The experimental VCD spectra (Fig. 9, bottom) were presented in the spectral range of 1750–1263 cm−1 because of the high level of VCD noise caused by overabsorption at lower wavenumbers. The simulated VCD spectra were scaled by a factor of 0.98 and 0.99 for methylone and pentylone, respectively, for the correct assignment of the individual bands (Tables 4, 5). The resulting index of spectra overlap reached convincing values of 0.86 and 0.70 for methylone and pentylone, respectively (Electronic Supplementary Material, Figs. S3 and S4). In the experimental VCD spectra of (R)-methylone and (R)-pentylone hydrochloride (Fig. 9, bottom), there were several areas in which individual substances could be reliably distinguished, mainly in a spectral range of 1400–1263 cm−1, where the molecules not only had a different number of bands but also different spectral shapes. Moreover, that was predicted correctly by the quantum chemical simulations (Fig. 9, middle). In addition, in the spectral range of 1700–1580 cm−1, there were different intensities of bands 1 and 2, which was also correctly predicted by the simulations. Our results demonstrating a higher propensity of distinguishing between two structurally similar compounds by VCD than ECD are in very good agreement with the literature [22]. The conformers of methylone and pentylone with a positive value of dihedral angle α1 again had a distinct spectral shape different from the rest of the conformers, e.g. band 3 in the VCD spectra of both (R)-methylone and (R)-pentylone as well as bands 5, 6 and 7.

VCD spectra of (R)-methylone hydrochloride (left) and (R)-pentylone hydrochloride (right): the B3LYP/6-311++G(d,p) calculated VCD spectra of the stable conformers (top), their Boltzmann averaged spectra (middle) and experimental VCD spectra with VCD noise spectrum (bottom)
Conclusions
The enantioseparation of two cathinone homologues—methylone and pentylone—by HPLC with UV–Vis detection has been successfully carried out using different methods of chiral separation for individual substances. Furthermore, a detailed structural analysis of both drugs of abuse has been achieved by the methods of electronic and vibrational spectroscopy, specifically conventional UV and IR absorption with ECD and VCD in combination with DFT calculations at the B3LYP/6-311++G(d,p) or B3PW91/6-311++G(d,p) levels of theory, including solvent effects. According to this study, these homologues can be difficult to distinguish solely by ECD or UV absorption. VCD spectroscopy appeared to be the best of the employed methods for distinguishing structurally similar chiral substances, not only in a small spectral range such as IR absorption spectroscopy provides, but essentially across the spectrum, especially in combination with quantum chemical calculations. Structural analysis suggested the existence of 5 and 9 stable conformers of methylone and pentylone, respectively, in an aqueous solution. Finally, very good agreement between the experimental and the corresponding simulated spectra was achieved. Thus, we were able not only to determine the absolute configuration of the individual conformers of methylone and pentylone, but also to describe the molecular structure of both compounds. In conclusion, chiroptical spectroscopic methods confirmed the high potential for structural analysis of molecules significant in the forensic sciences. These data can be helpful in considering the forensic evidence in criminal proceedings. Moreover, the structural analysis performed by the quantum chemical calculations can be used for the understanding of the biological activity and toxicity of the NPS. A detailed knowledge of the 3D structure of the substances may allow not only the detailed study of the binding properties, metabolism, transport or distribution of these substances in the human organism, but also the development of antagonist drugs for overdoses, addictions or poisoning.
References
European Monitoring Centre for Drugs and Drug Addiction (2018) European drug report 2018: trends and developments. Publications Office of the European Union, Luxembourg
European Monitoring Centre for Drugs and Drug Addiction (2014) European drug report 2014: trends and developments. Publications Office of the European Union, Luxembourg
United Nations Office on Drugs and Crime (2018) World drug report 2018. United Nations publication, Vienna
Barceloux DG (2012) Medical toxicology of drug abuse: synthesized chemicals and psychoactive plants. Wiley, Hoboken
Al-Hebshi N, Skaug N (2005) Khat (Catha edulis)—an updated review. Addict Biol 10:299–307. https://doi.org/10.1080/13556210500353020
European Monitoring Centre for Drugs and Drug Addiction (2016) The internet and drug markets. Publications Office of the European Union, Luxembourg
Mas-Morey P, Visser MHM, Winkelmolen L, Touw DJ (2013) Clinical toxicology and management of intoxications with synthetic cathinones (“Bath Salts”). J Pharm Pract 26:353–357. https://doi.org/10.1177/0897190012465949
Calinski DM, Kisor DF, Sprague JE (2018) A review of the influence of functional group modifications to the core scaffold of synthetic cathinones on drug pharmacokinetics. Psychopharmacology. https://doi.org/10.1007/s00213-018-4985-6
Štefková K, Židková M, Horsley RR, Pinterová N, Šíchová K, Uttl L, Balíková M, Danda H, Kuchař M, Páleníček T (2017) Pharmacokinetic, ambulatory, and hyperthermic effects of 3,4-methylenedioxy-N-methylcathinone (methylone) in rats. Front Psychiatry 8:232. https://doi.org/10.3389/fpsyt.2017.00232
Sogawa C, Sogawa N, Ohyama K, Kikura-Hanajiri R, Goda Y, Sora I, Kitayama S (2011) Methylone and monoamine transporters: correlation with toxicity. Curr Neuropharmacol 9:58–62. https://doi.org/10.2174/157015911795017425
Grecco GG, Kisor DF, Magura JS, Sprague JE (2017) Impact of common clandestine structural modifications on synthetic cathinone “bath salt” pharmacokinetics. Toxicol Appl Pharmacol 328:18–24. https://doi.org/10.1016/j.taap.2017.05.010
Montesano C, Vannutelli G, Gregori A, Ripani L, Compagnone D, Curini R, Sergi M (2016) Broad screening and identification of novel psychoactive substances in plasma by high-performance liquid chromatography–high-resolution mass spectrometry and post-run library matching. J Anal Toxicol 40:519–528. https://doi.org/10.1093/jat/bkw043
Olesti E, Pujadas M, Papaseit E, Pérez-Mañá C, Pozo ÓJ, Farré M, de la Torre R (2017) GC–MS quantification method for mephedrone in plasma and urine: application to human pharmacokinetics. J Anal Toxicol 41:100–106. https://doi.org/10.1093/jat/bkw120
Salomone A, Gazzilli G, Di Corcia D, Gerace E, Vincenti M (2016) Determination of cathinones and other stimulant, psychedelic, and dissociative designer drugs in real hair samples. Anal Bioanal Chem 408:2035–2042. https://doi.org/10.1007/s00216-015-9247-4
Assi S, Guirguis A, Halsey S, Fergus S, Stair JL (2015) Analysis of ‘legal high’ substances and common adulterants using handheld spectroscopic techniques. Anal Methods 7:736–746. https://doi.org/10.1039/c4ay02169j
Guirguis A, Girotto S, Berti B, Stair JL (2017) Identification of new psychoactive substances (NPS) using handheld Raman spectroscopy employing both 785 and 1064 nm laser sources. Forensic Sci Int 273:113–123. https://doi.org/10.1016/j.forsciint.2017.01.027
Berova N, Polavarapu PL, Nakanishi K, Woody RW (2012) Comprehensive chiroptical spectroscopy: applications in stereochemical analysis of synthetic compounds, natural products, and biomolecules. Wiley, Hoboken
Nafie LA (2011) Vibrational optical activity: principles and applications. Wiley, Chichester
Fagan P, Kocourková L, Tatarkovič M, Králík F, Kuchař M, Setnička V, Bouř P (2017) Cocaine hydrochloride structure in solution revealed by three chiroptical methods. ChemPhysChem 18:2258–2265. https://doi.org/10.1002/cphc.201700452
Julínek O, Setnička V, Řezáčová A, Dohnal J, Vosátka V, Urbanová M (2010) Product of alaptide synthesis: determination of the absolute configuration. J Pharm Biomed Anal 53:958–961. https://doi.org/10.1016/j.jpba.2010.07.007
Stephens PJ, Pan JJ, Devlin FJ, Urbanová M, Julínek O, Hájíček J (2008) Determination of the absolute configurations of natural products via density functional theory calculations of vibrational circular dichroism, electronic circular dichroism, and optical rotation: the iso-schizozygane alkaloids isoschizogaline and isoschizogamine. Chirality 20:454–470. https://doi.org/10.1002/chir.20466
Polavarapu PL, Donahue EA, Shanmugam G, Scalmani G, Hawkins EK, Rizzo C, Ibnusaud I, Thomas G, Habel D, Sebastian D (2011) A single chiroptical spectroscopic method may not be able to establish the absolute configurations of diastereomers: dimethylesters of hibiscus and garcinia acids. J Phys Chem A 115:5665–5673. https://doi.org/10.1021/jp202501y
Nicu VP, Mándi A, Kurtán T, Polavarapu PL (2014) On the complementarity of ECD and VCD techniques. Chirality 26:525–531. https://doi.org/10.1002/chir.22330
Kohout M, Vandenbussche J, Roller A, Tůma J, Bogaerts J, Bultinck P, Herrebout W, Lindner W (2016) Absolute configuration of the antimalarial erythro-mefloquine—vibrational circular dichroism and X-ray diffraction studies of mefloquine and its thiourea derivative. RSC Adv 6:81461–81465. https://doi.org/10.1039/c6ra19367f
Kim JH, Scialli AR (2011) Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol Sci 122:1–6. https://doi.org/10.1093/toxsci/kfr088
Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI et al (2016) NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533:481–486. https://doi.org/10.1038/nature17998
Jurásek B, Králík F, Rimpelová S, Čejka J, Setnička V, Ruml T, Kuchař M, Kohout M (2018) Synthesis, absolute configuration and in vitro cytotoxicity of deschloroketamine enantiomers: rediscovered and abused dissociative anaesthetic. New J Chem 42:19360–19368. https://doi.org/10.1039/c8nj03107j
Silva B, Fernandes C, Tiritan ME, Pinto MMM, Valente MJ, Carvalho M et al (2016) Chiral enantioresolution of cathinone derivatives present in “legal highs”, and enantioselectivity evaluation on cytotoxicity of 3,4-methylenedioxypyrovalerone (MDPV). Forensic Toxicol 34:372–385. https://doi.org/10.1007/s11419-016-0324-y
Gregg RA, Baumann MH, Partilla JS, Bonano JS, Vouga A, Tallarida CS et al (2015) Stereochemistry of mephedrone neuropharmacology: enantiomer-specific behavioural and neurochemical effects in rats. Br J Pharmacol 172:883–894. https://doi.org/10.1111/bph.12951
Gannon BM, Williamson A, Suzuki M, Rice KC, Fantegrossi WE (2016) Stereoselective effects of abused “bath salt” constituent 3,4-methylenedioxypyrovalerone in mice: drug discrimination, locomotor activity, and thermoregulation. J Pharmacol Exp Ther 356:615–623. https://doi.org/10.1124/jpet.115.229500
Spálovská D, Králík F, Kohout M, Jurásek B, Habartová L, Kuchař M, Setnička V (2017) Structure determination of butylone as a new psychoactive substance using chiroptical and vibrational spectroscopies. Chirality 30:548–559. https://doi.org/10.1002/chir.22825
Urbanová M, Setnička V, Volka K (2000) Measurements of concentration dependence and enantiomeric purity of terpene solutions as a test of a new commercial VCD spectrometer. Chirality 12:199–203. https://doi.org/10.1002/(sici)1520-636x(2000)12:4%3c199:aid-chir6%3e3.0.CO;2-L
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09 citation. Gaussian Inc, Wallingford
Covington CL, Polavarapu PL (2013) Similarity in dissymmetry factor spectra: a quantitative measure of comparison between experimental and predicted vibrational circular dichroism. J Phys Chem A 117:3377–3386. https://doi.org/10.1021/jp401079s
Polavarapu PL, Covington CL (2014) Comparison of experimental and calculated chiroptical spectra for chiral molecular structure determination. Chirality 26:539–552. https://doi.org/10.1002/chir.22316
Geryk R, Kalíková K, Schmid MG, Tesařová E (2016) Enantioselective separation of biologically active basic compounds in ultra-performance supercritical fluid chromatography. Anal Chim Acta 932:98–105. https://doi.org/10.1016/j.aca.2016.04.044
Wolrab D, Frühauf P, Moulisová A, Kuchař M, Gerner C, Lindner W, Kohout M (2016) Chiral separation of new designer drugs (Cathinones) on chiral ion-exchange type stationary phases. J Pharm Biomed Anal 120:306–315. https://doi.org/10.1016/j.jpba.2015.12.023
Albals D, Heyden YV, Schmid MG, Chankvetadze B, Mangelings D (2016) Chiral separations of cathinone and amphetamine-derivatives: comparative study between capillary electrochromatography, supercritical fluid chromatography and three liquid chromatographic modes. J Pharm Biomed Anal 121:232–243. https://doi.org/10.1016/j.jpba.2015.12.007
Szendrei K (1980) The chemistry of khat. Bull Narc 32:5–35
Bouř P, Maloň P (1995–2009) The MCM program. Academy of Sciences Prague, Czech Republic
Acknowledgements
The work was supported by the Ministry of the Interior of the Czech Republic (MV0/VI20172020056). Partial support was provided by the Operational Program Prague—Competitiveness (CZ.2.16/3.1.00/24503 and CZ.2.16/3.1.00/21537), the National Program of Sustainability (NPU MSMT—LO1601; 43760/2015), and the Specific University Research (MSMT No. 21-SVV/2018 and 21-SVV/2019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Spálovská, D., Maříková, T., Kohout, M. et al. Methylone and pentylone: structural analysis of new psychoactive substances. Forensic Toxicol 37, 366–377 (2019). https://doi.org/10.1007/s11419-019-00468-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-019-00468-z