
Abstract
Mephedrone (4-methyl-meth-cathinone) or 4-MMC and its derivative 3-MMC are analogs of cathinone which act as non-selective substrates for monoamine transporters, facilitating a neurotransmitter release via the reversal of normal transporter fluxes. The pharmacological effects like paranoia, hallucinations, euphoria, aggressiveness, and psychosis make these molecules “abusive designer drugs.” In the present study, conformations of all isomers of 3- and 4-MMC have been studied at the quantum-chemical level. Calculations have been performed using Møller-Plesset second-order (MP2) methods with 6-31G(d, p) basis sets. Present study shows that there are low-energy conformers for the meta (3-MMC) and para (4-MMC) isomers that are connected by way of low-barrier transition states. Natural bond orbital (NBO) analyses have been also performed to corroborate the results as estimated from the MP2 calculations. The molecular electrostatic potential surface data for each molecule has been calculated, revealing likely that reaction sites and probable metabolism pathway for both derivatives have been proposed. IR spectra for MMC derivatives have been compared with experimental data, and rotational transition states connecting all conformers have been reported.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cathinone (β-keto-amphetamine), a naturally occurring psychoactive constituent obtained from the leaves of khat (Catha edulis), is a basis for the development of a series of synthetic cathinone derivatives used recreationally [1]. Designer cathinones easily permeate the blood-brain barrier (BBB) layer, eliciting sympathomimetic and psychostimulant actions by acting as a central nervous system stimulant by promoting the release of monoamine neurotransmitters and likely inhibiting their uptake. Their effects in humans include paranoia, hallucinations, euphoria, aggressiveness, psychosis, and an increment of libido, in addition to the more serious occurrences of hypertension, hyperthermia, seizures, respiratory distress, together with reports of fatal outcomes [2, 3]. The higher number of intoxication reports during the last 10 years has led to laws that banned the use and sale of synthetic cathinones throughout different countries, although they continue to be commercialized worldwide illegally as components of illicit bath salts or legal highs [4]. Because of its amphetamine-like effects and its resultant high-abuse potential, cathinone derivatives were assigned schedule I status by the US Drug Enforcement Administration (DEA) in 1993 and class B status under the UK Misuse of Drugs Act in 1998 [5].
Mephedrone (4-methylmethcathinone)/4-MMC is para-substituted meth-cathinone analog, one of the most commonly employed designer drug in “legal highs” drug moiety, most commonly available cathinone sold on recreational market in the period up to 2010 that was first time synthesized in 1929 [6]. 4-MMC was mainly used for illicit recreational consumption due to their psychoactive effects, which primarily act as substrate for monoamine transporters, facilitating the release of neurotransmitters via the reversal of normal transporter fluxes [2]. Of note, recent reports [7, 8] have demonstrated that 4-MMC display similar potencies to increase the release of monoamines in vitro, with a low selectivity for dopamine (DA) versus serotonin (5-HT) transporters (DAT and SERT, respectively). 4-MMC was widely used in East Africa and the Arab Peninsula as an amphetamine-like stimulant and popularized in Europe and the USA as one of the primary components of bath salts during the beginning of twenty-first century [9]. Because of its rapid rise to international notoriety as a designer drug of abuse, as well as its cocaine- and methamphetamine-like discriminative stimulus effects, 4-MMC was added to the list of controlled drugs in both the USA and the UK in 2010–2011 [10].
Mephedrone has two major derivatives, 2-methyl meth-cathinone (2-MMC) and 3-methyl meth-cathinone (3-MMC) [11–13]. Due to structural similarity with 4-MMC, comparable pharmacological actions are noticed for both derivatives. Though, 4-MMC is controlled in the Republic of Ireland since May 2010 but 2-MMC and 3-MMC are not controlled. All derivatives of cathinone are controlled in Poland since 2010, but they are legal in Czech Republic. The banned 4-MMC leads to create more demand of 3-MMC in the “legal highs” market, and the major concern is the limited analytical data available about 3-MMC derivatives and their metabolites. These legal and analytical problems prompted us for quantum chemical analysis of 3-MMC along with major cathinone derivative 4-MMC molecule.
The primary aim of this study is to identify all possible conformers of 3- and 4-MMC (Fig. 1), so that these structures may be compared to cathinone structure. These derivatives are considered for ab initio treatments for energy profiling along with that molecular electrostatic potential surfaces (MEPSs) are also calculated. The natural bond orbital (NBO) analyses have been also performed to corroborate the results as estimated from the Møller-Plesset second-order (MP2) calculations. Additionally, the possible metabolism path has been also introspected.

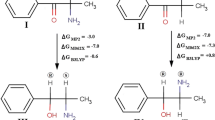
Structures of 3-methylmethcathinone (3-MMC, meta) and 4-methylmethcathinone (4-MMC, para)
Computational methods and analysis
The obtained results from our previous work [14] helped us to choose the best optimization method and basis set; therefore, full geometry optimization has been carried out using MP2 calculations [15] with 6-31G(d,p) basis sets employing Gaussian 09 package [16]. Potential energy surfaces were scanned, and all minima were optimized to find all possible conformers of 3-MMC and 4-MMC (mephedrone). The transition states have been obtained with one imaginary frequency. The molecular energy potential surface has been prepared using GaussView program.
The thermodynamic parameters of all compounds were calculated using statistical mechanic expressions utilizing the ideal gas, rigid rotor, and harmonic oscillator approximations. Solvation-free energies (ΔG solv) were calculated at the MP2 level, using COSMO [17] in water solvent. The ΔG solv values were calculated in aqueous continuum (ε = 78.39), and the molecular cavities were built using the united atom for the Hartree-Fock (UAHF) procedure, as used in COSMO. In principle, the free energy surfaces are determined by rigorous combination of free energy perturbation umbrella sampling approaches [18, 19]. These surfaces are very rigorous, as they reflect non-equilibrium solvation. However, such calculations are very challenging when performed within the ab initio framework. Thus, a more practical, simplified approach has been made. The relative free energies (ΔG) are determined as [20]
In Eq. (1), ΔH gas (298 K) is the enthalpy at 298 K, ΔS is the gas-phase entropy, ΔG gas (298 K) is the Gibb’s free energy at 298 K, and ΔG solv is the relative free energy of solvation. The contribution of the RT ln (ω) term is zero, as the electronic degeneracy term, ω, for the singlet state is unity. The ΔG values are used to calculate the relative population of various conformers of the molecules, under study, in aqueous medium.
Since the molecules are much more floppy in solution than in the gas phase, we have used another definition of free energy (Δg flex) to compare the intrinsic flexibility of different conformers, when embedded in aqueous medium. This is the practical implementation of the more general expression used by Warshel and coworkers [20, 21]. The Δg flex could be expressed as
where ΔE solute and ΔZPE in Eq. (2) are the relative gas-phase energy separations and the zero-point energies of the various conformers. The scale factor α is usually taken as zero [21], and Eq. (2) can be simplified as
All terms in Eqs. (1) and (3) are available from the thermochemical analyses based on statistical mechanic expressions using the ideal gas, rigid rotator, and harmonic oscillator approximations [22]. Population has been calculated using Boltzmann equation. The thermodynamic parameters of all compounds were calculated using statistical mechanic expressions utilizing the ideal gas, rigid rotor, and harmonic oscillator approximations.
The NBO analysis was generated using NBO 6.0 program [23]. NBO 6.0 is a critical detection program for studying complicated wave functions like interactions between bonds [24].
Results and discussions
3-MMC
All conformers of 3-MMC (Fig. 2) have been calculated using MP2 level of theory. Results show that 3-MMC has six different low-energy conformers connected through transition states. All computed data have been represented in Table 1 for gas phase, which suggested that the difference between the lowest- and the highest-energy conformer is 1.96 kcal/mol for MP2 (conformer e). The highest barrier for rotation between two conformers is 7.07 kcal/mol (rotation c↔e). The lowest rotational barrier is 3.64 kcal/mol for transition a↔b. The most populated conformers are a and f, and they do not change with the change of different level of theory. Transition barrier between conformers a and f is equal to 6.55 kcal/mol. The energy profile calculated on MP2 level with 6-31G(d,p) basis set for gas and solution phase is shown in Fig. 3a, b, respectively. In Fig. 3b, the most propagated conformer is conformer f, whose propagation changed from 41.08 to 50.17% in solution. However, conformer a has changed its propagation from 47.02 to 40.20%. Energy difference between highest- and lowest-energy conformer is equal to 1.98 kcal/mol. The highest barrier is 7.07 kcal/mol between conformers d and f in gas phase and 11.49 kcal/mol between conformers b and c. The lowest barrier in solution is 2.59 kcal/mol for transition between conformers a and b. Solution lowers energy difference between conformers, which is true for all conformers. The solution affects rotational barriers by raising them significantly. All calculated energies for solvent and population data have been reported in Table 1. Structural data for 3-MMC has been provided in Table S2 in supplementary materials.

All six conformers of 3-MMC calculated with 6-31G(d,p) basis set on MP2 level

Energy profile in gas (a) and solution (b) for rotational transitions calculated on MP2 level of theory with 6-31G(d,p) basis set for 3-MMC
In Fig. 4a, we proposed probable metabolic pathway and metabolites of 3-MMC, which is matched with the proposed pathway by Meyer and Maurer [25]. Figure 5 presents the MEPSs of all rotational conformers of 3-MMC. In all cases, the most negative surface region is around oxygen atom. Conformers a, d, and f have negative region around nitrogen atom as well. It suggests that attacks of hydrogen go to the oxygen atom first as it is supported from the proposed metabolic pathway.

Main probable metabolites of 3-MMC and 4-MMC [26]

Molecular electrostatic potential surfaces (MEPSs) of all rotational conformers of 3-MMC. Order of increasing electron density is blue < green < yellow < orange < red
Experimental IR [11] is compared with the calculated IR spectrum of 3-MMC and presented in Fig. 6a. The shape of calculated spectrum is almost similar to calculated one. In Table 2, wave numbers of characteristic vibrations of 3-MMC for both experimental and calculated spectrum are reported. The carbonyl C = O stretching band for 4-MMC is approximately at 1686 cm−1 for experiment and 1738 cm−1 for calculated one. The aromatic C = C ring vibration or ring “breathing” bands is approximately at 1603 cm−1 for experiment and 1678 cm−1 for computed one, and it is important to note that the peak is very strong for 3-MMC in both cases. The out-of-plane C–H bending bands are in the region of 675–894 cm−1 for experiment and 673–932 cm−1 for calculated IR. The N–H stretching band is approximately 3569 and 3434 cm−1 for calculated and experiment IR, respectively.

Calculated IR spectrum in solution for a 3-MMC and b 4-MMC
The NBO analysis revealed that 3-MMC has two main resonance structures. The 3-MMC has three lonely pairs (LPs) located on nitrogen and oxygen atoms, where two of them are located on oxygen atom. Oxygen LP donate electrons to C–C* antibonding with energy 23.32 and 22.98 kcal/mol. A double bond between the atoms O and C results from the overlap of sp2.30d0.01 hybrid on C with sp1.32d0.01 hybrid on O and sp. hybrid on C with sp. hybrid on O for the second bond. The steric properties of molecule show total steric exchange energy (SXE) of 1160.10 kcal/mol with pairwise sum of 388.40 kcal/mol. The strongest interaction of SXE is 19.87 kcal/mol for O-LP/C-C. The overlapping orbital has been shown in Fig. 7. Total dipole moment is equal to 3.31 D, where Lewis and non-Lewis contributions to the dipole moment are 3.54 and 1.17 D, respectively.

Overlapping orbitals of a 3-MMC and b 4-MMC, where light blue/yellow is the lonely pair of oxygen atom and dark blue/yellow is the C-C binding
4-MMC
The 4-MMC (Fig. 1) has three low-energy conformers (Fig. 8) connected through three transition states. Figure 9a, b shows the energetic profile for gas phase and solution calculated on MP2 level using 6-31G(d,p) basis set. The highest conformer lays about 1.02 kcal/mol above the lowest-energy conformer. The highest barrier is between conformer 2 and 3, and it is 11.42 kcal/mol above the lowest-energy conformer. The most populated conformer is conformer 1 with 48.26%, and the second most populated energy conformer is conformer 3.

All three conformers of 4-MMC calculated with 6-31G(d,p) basis set on MP2 level

Energy profile for 4-MMC calculated at MP2 level with 6-31G(d,p) basis set in a gas phase and b solution

Molecular electropotential surfaces of all conformers of 4-MMC calculated with 6-31G(d,p) basis set on MP2 level. Order of increasing electron density is blue < green < yellow < orange < red
All computed thermodynamic data for 4-MMC in gas phase and in solvent have been represented in Table 3. In solution, the lowest-energy conformer is, like in gas phase, conformer 1. Shape of energy profile changes in solvent, and energies of conformers 2 and 3 have been also changed in solvent phase. Therefore, conformer 3 becomes the highest-energy one in solvent phase. The most populated energy conformer is conformer 1 with 88.90% of population. Population of conformer 3 has decreased to 4.67% in solution. Dipole moment in solution, as it has been expected, has increased in solution (Table 3). Structural data for 4-MMC has been provided in Table S2 in supplementary materials.
Experimental IR [11] is compared with the calculated IR spectrum of 4-MMC and presented in Fig. 6b. Most interestingly, the shape of calculated spectrum is almost similar to the experimental one. In Table 2, wave numbers of characteristic vibrations of 4-MMC for both experimental and calculated spectrum are reported. The carbonyl C = O stretching band for 4-MMC is approximately at 1685 cm−1 for experiment and 1720 cm−1 for calculated one. The aromatic C = C ring vibration or ring “breathing” bands is approximately at 1600 cm−1 for experiment and 1672 cm−1for computed one, and it is important to note that the peak is very strong for 4-MMC in both cases. The out-of-plane C–H bending bands are in the region of 675–900 cm−1 for experiment and 695–907 cm−1 for calculated IR. In experimental IR, spectra are visible in overtone bands (1700–2000 cm−1), which have not been calculated in theoretical one. The N–H stretching band is approximately 3549 and 3416 cm−1 for calculated and experiment IR, respectively.
Para-mephedrone (4-MMC) has highly delocalized structure with two most important resonance structures. The 4-MMC molecule has three LPs, where two LPs localized on oxygen and one LP localized on nitrogen atoms. Total SXE for 4-MMC is equal to 1160.52 kcal/mol, where pairwise sum is only 391.00 kcal/mol. A double bond between the atoms O and C results from the overlap of a sp2.29d0.01 hybrid on C with a sp1.34d0.01 hybrid on O and a sp. hybrid on C with a sp. hybrid on O for the other bond. The overlapping orbitals have been shown in Fig. 7. The highest overlapping energy is between lonely pair localized on oxygen atom, and C–C bond is equal to 19.14 kcal/mol.
Analysis of MEPS (Fig. 10) of 4-MMC shows that the most reactive site of mephedrone moiety is present in part of nitrogen and oxygen atoms, and the most reactive one is molecule number 1 (Fig. 10), which is the lowest-energy conformer and specially visible in proposed metabolites of 4-MMC in Fig. 4b. In one metabolism reaction path, H+ ion attacks nitrogen atom and breaks bond between CH4 group and moiety resulting release of CH4 [25]. Calculation shows that for this reaction, enzyme is necessary to decrease barrier, which is 142 kcal/mol high for this reaction. Experiment shows that mephedrone reacts with dopamine and serotonine [26]. Dopamine is one of the neurotransmitters, a chemical messenger heavily active in the mesolimbic and mesocortical reward pathways. As discussed in the “Introduction” section, cathinone and amphetamine target various neurotransmitters involved in various pathways of the central nervous system functions [27, 28]. According to our quantum-mechanical study, we believe that mephedrone acts the same way as amphetamine and cathinone and further introspection is required, which is under investigation.
Conclusion
In this work, we carried out conformational analysis of isomers of mephedrone derivatives at the MP2/6-31G(d,p) level. The dependence of the low-energy conformers of mephedrone derivatives on aqueous solvation was studied using the MP2/COSMO model, and the MEP surfaces were generated for these low-energy surfaces to interpret the nature of practical reaction regions. Our calculations indicate that 3-MMC and 4-MMC have several low-energy conformers in the gas as well as in solvent medium. The transition states of 4-MMC low-energy conformers are higher than the 3-MMC derivative. The number of conformers and energy barrier height could explain different influence of 3-MMC and 4-MMC in human body as low rotational barriers and numbers of conformers for 3-MMC due to molecular shape could not always fit to the neurotransmitter, and therefore, inhibition is weaker. In solution, there is no transition state between conformers 1 and 3 (4-MMC). These low-energy conformers are connected through low-barrier transition states, indicating that they can be converted from one form to the other. The calculated IR spectra for both MMCs are similar to experimental one; thus, we are sure that the calculated molecules correspond good with reality. From steric evaluation of both derivatives by NBO analysis, we find a difference in steric exchange energy of 0.42 kcal/mol and in the sum of pairwise contributions of 2.60 kcal/mol between 3-MMC and 4-MMC. Both values indicate that steric repulsions are favoring the 4-MMC. The MEP surfaces revealed potential regions of 3- and 4-MMC, where reaction can take place in the body. This study also reveals the importance of low-energy conformers of mephedrone derivatives in exhibiting its specific biological properties. Understanding the structure offers a starting point for defining the mechanism of action and provides for the development of antidotes for drug poisoning, overdose, or addiction.
References
Baumann MH, Ayestas MA, Partilla JS, et al. (2011) The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37:1192–1203. doi:10.1038/npp.2011.304
Gregg RA, Rawls SM (2014) Behavioral pharmacology of designer cathinones: a review of the preclinical literature. Life Sci 97:27–30. doi:10.1016/j.lfs.2013.10.033
Valente MJ, Guedes de Pinho P, de Lourdes BM, et al. (2014) Khat and synthetic cathinones: a review. Arch Toxicol 88:15–45. doi:10.1007/s00204-013-1163-9
Green AR, King MV, Shortall SE, Fone KCF (2014) The preclinical pharmacology of mephedrone; not just MDMA by another name. Br J Pharmacol 171:2251–2268. doi:10.1111/bph.12628
Bonano JS, Glennon RA, De Felice LJ, et al. (2014) Abuse-related and abuse-limiting effects of methcathinone and the synthetic “bath salts” cathinone analogs methylenedioxypyrovalerone (MDPV), methylone and mephedrone on intracranial self-stimulation in rats. Psychopharmacology 231:199–207. doi:10.1007/s00213-013-3223-5
de Buranga Sanchez MJS (1929) Bull Soc Chim Fr 45:284–286
Bonano JS, Banks ML, Kolanos R, et al. (2015) Quantitative structure-activity relationship analysis of the pharmacology of para-substituted methcathinone analogues. Br J Pharmacol 172:2433–2444. doi:10.1111/bph.13030
Sakloth F, Kolanos R, Mosier PD, et al. (2015) Steric parameters, molecular modeling and hydropathic interaction analysis of the pharmacology of para-substituted methcathinone analogues. Br J Pharmacol 172:2210–2218. doi:10.1111/bph.13043
Hall C, Heyd C, Butler C, Yarema M (2014) “Bath salts” intoxication: a new recreational drug that presents with a familiar toxidrome. Can J Emerg Med 16:171–176. doi:10.2310/8000.2013.131042
Gatch MB, Taylor CM, Forster MJ (2013) Locomotor stimulant and discriminative stimulus effects of “bath salt” cathinones. Behav Pharmacol 24:437–447. doi:10.1097/FBP.0b013e328364166d
Power JD, McGlynn P, Clarke K, et al. (2011) The analysis of substituted cathinones. Part 1: chemical analysis of 2-, 3- and 4-methylmethcathinone. Forensic Sci Int 212:6–12
Torrance H, Cooper G (2010) The detection of mephedrone (4-methyl methcathinone) in 4 fatalities in Scotland. Forensic Sci Int 202:e62–e63. doi:10.1016/j.forsciint.2010.07.014
Carhart-Harris RL, King LA, Nutt DJ (2011) A web-based survey on mephedrone. Drug Alcohol Depend 2–5. doi:10.1016/j.drugalcdep.2011.02.011
Kolodziejczyk W, Kar S, Hill GA, Leszczynski J (2016) A comprehensive computational analysis of cathinone and its metabolites using quantum mechanical approaches and docking studies. Struct Chem 27:1291–1302. doi:10.1007/s11224-016-0779-9
Møller C, Plesset MS (1943) Note on an approximation treatment for many-electron systems. Phys Rev 46:618. doi:10.1103/PhysRev.56.841.2
Frisch MJ, Trucks GW, Schlegel HB, et al. (2009) Gaussian 2009
Barone V, Cossi M (1998) Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A 102:1995–2001
Hwang J-K, King G, Creighton S, Warshel A (1988) Simulation of free energy relationships and dynamics of SN2 reactions in aqueous solution. J Am Chem Soc 110:5297–5311
Warshel A (1991) Comput Model Chem React Enzym Solut
Florián J, Warshel A (1998) Phosphate ester hydrolysis in aqueous solution: associative versus dissociative mechanisms. J Phys Chem B 102:719–734
Florián J, Štrajbl M, Warshel A (1998) Conformational flexibility of phosphate, phosphonate, and phosphorothioate methyl esters in aqueous solution. J Am Chem Soc 120:7959–7966
Davidson N (1962) Stat Mech
Glendening ED, Badenhoop JK, Reed AE, et al. (2013) NBO 6.0
Tobergte DR, Curtis S (2013) Discovering chemistry with natural bond orbitals. J Chem Inf Model. doi:10.1017/CBO9781107415324.004
Meyer MR, Maurer HH (2010) Metabolism of designer drugs of abuse: an updated review. Curr Drug Metab 11:468–482
Martínez-Clemente J, Escubedo E, Pubill D, Camarasa J (2011) Interaction of mephedrone with dopamine and serotonin targets in rats. Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2011.07.009
Moore KE (1977) The actions of amphetamine on neurotransmitters: a brief review. Biol Psychiatry 12:451–462
Kalix P (1985) The khat alkaloid (−)cathinone acts like amphetamine on physiological catecholamine stores. NIDA Res Monogr Ser NO 55:286
Acknowledgements
The authors would like to thank the Wroclaw Supercomputing and Networking Center (WCSS) for the generous allotment of computer time. We would also like to thank the Mississippi Center for Supercomputing Research (MCSR) for providing the computing time. Authors want to thank the National Science Foundation (NSF/CREST HRD-1547754) for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 22 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kolodziejczyk, W., Kar, S. & Hill, G.A. Conformational analysis, energy profile, and structural-electronic properties evaluation of mephedrone derivatives employing quantum-mechanical models. Struct Chem 28, 791–799 (2017). https://doi.org/10.1007/s11224-016-0878-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-016-0878-7