
Abstract
Alzheimer’s disease (AD) is the most prevalent cause of dementia in adults. Current available drugs for AD transiently alleviate some of the symptoms, but do not modify the disease mechanism or cure it. Therefore, new drugs are desperately needed. Key contributors to AD are amyloid beta (Aβ)- and reactive oxygen species (ROS)-induced cytotoxicities. Plant-derived substances have been shown to affect various potential targets in various diseases including AD. Therefore, phytochemicals which can protect neuronal cells against these insults might help in preventing and treating this disease. In the following research, we have isolated the sesquiterpene lactone achillolide A from the plant Achillea fragrantissima and, for the first time, characterized its effects on Aβ-treated neuroblastoma cells. Aβ is a peptide derived from the sequential cleavage of amyloid precursor protein, and is part of the pathogenesis of AD. Our current study aimed to determine whether achillolide A can interfere with Aβ-induced processes in Neuro2a cells, and protect them from its toxicity. Our results show that achillolide A decreased Aβ-induced death and enhanced the viability of Neuro2a cells. In addition, achillolide A reduced the accumulation of Aβ-induced ROS and inhibited the phosphorylation of stress-activated protein kinase/c-Jun N-terminal kinase and p44/42 mitogen-activated protein kinase in these cells. We therefore suggest that achillolide A may have therapeutic potential for the treatment of AD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most prevalent cause of dementia in adults. Progressive neuronal loss takes place in specific brain areas and causes memory loss, learning difficulty, diminished recall accuracy, impaired problem-solving ability and cognitive deterioration. One of the hallmarks of the disease is the formation of amyloid plaques formed by aggregated β-amyloid (Aβ) peptides. Aβ is a 4-kDa peptide derived from the sequential cleavage of amyloid precursor protein [1] and its oligomeric form is part of the pathogenesis of AD. It is thought to exert its action through different mechanisms, including the induction of reactive oxygen species (ROS) accumulation, microglial activation, and neuronal death [2, 3]. Therefore, phytochemicals that can protect neuronal cells from Aβ toxicity and oxidative stress may assist in coping with AD. Current available drugs for AD transiently alleviate some of the symptoms, but do not modify the disease mechanism or cure it. Therefore, new drugs are desperately needed. Plant extracts and purified phytochemicals have been shown to affect various potential targets in AD such as hyperphosphorylated tau and acetyl cholinesterase activity [4,5,6,7].
Sesquiterpene lactones isolated from plants have been shown to possess diverse biological activities and to exhibit several effects, such as immunomodulation, anti-inflammatory, antitumor and antimicrobial activities [8,9,10,11,12]. Sesquiterpene lactones have also been reported to have neuroprotective effects in cultured neurons as well as in vivo, using animal stroke models and cocaine consumption studies. Thus, it is reasonable to assume that these compounds or their active metabolites can cross the blood−brain barrier [8, 13,14,15,16]. Based on these activities, sesquiterpene lactones could be promising candidates for the development of drugs for the treatment of neurodegenerative diseases [17,18,19].
We have previously shown that achillolide A, a sesquiterpene lactone that we isolated from Achillea fragrantissima (Forssk) Sch. Bip, downregulated microglial activation [20], prevented hydrogen peroxide (H2O2)-induced death of astrocytes [21], and protected neuroblastoma cells from glutamate toxicity [22]. Our current study aimed to determine whether achillolide A can interfere with Aβ-induced processes in Neuro2a (N2a) cells and protect them from its toxicity.
Materials and methods
Materials
Aβ25-35, 2′,7′-dichlorofluorescein diacetate (DCF-DA) and crystal violet were purchased from Sigma Chemical Co. (St Louis, MO, USA). Glutamine, antibiotics (10,000 IU/mL penicillin and 10,000 μg/mL streptomycin), fetal bovine serum (FBS) and Trypin-EDTA were purchased from Biological Industries (Beit Haemek, Israel). Opti-MEM and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Gibco (Paisley, UK). Dimethyl sulfoxide (DMSO) was obtained from Applichem (Darmstadt, Germany). PathScan total SAPK/JNK sandwich ELISA kit, PathScan phospho-SAPK/JNK (Thr183/Tyr185) sandwich ELISA kit, PathScan total p44/42 MAPK sandwich ELISA kit and the PathScan phospho-p44/42 MAPK (Thr202/Tyr204) sandwich ELISA kit were purchased from Cell Signaling Technology.
Plant material
The aerial parts of Achillea fragrantissima (Forssk) Sch. Bip (Af) were collected in the Arava Valley. The plant was authenticated by the botanist Mrs Mimi Ron at The Mount Scopus Botanical Garden in The Hebrew University of Jerusalem. The voucher specimen is kept as part of the Arava Rift Valley Plant Collection under the accession code AVPC0040.
Achillolide A (98% pure) was isolated as previously described from the aerial parts of Af [20].
Preparation of aged Aβ25-35
The Aβ25-35 peptide was solubilized in sterile double distilled water at a concentration of 2 mM, incubated in a capped vial at 37 °C for 48 h, aliquoted, and stored frozen at −20 °C until use. Fresh dilutions of Aβ were prepared in the growth medium just prior to each experiment, and were used immediately.
Determination of cytotoxicity
N2a cells were grown and then re-plated onto 96-well plates (5 × 103 cells/well) in 50% Opti-MEM, 43% DMEM (high glucose), 2 mM glutamine, 5% FBS, penicillin at 100 U/mL, and streptomycin at 100 μg/mL. Aβ and/or achillolide A were added 24 h later, and cytotoxicity was determined after 20 h using the lactate dehydrogenase (LDH) activity colorimetric assay (Roche Applied Science, Germany). The absorbance was measured at 492 nm in a Synergy2 Multi-Detection Microplate Reader (BioTek Instruments, Inc., Winooski, VT, USA).
Determination of cell viability
N2a cells were grown and treated as in the cytotoxicity assay. Cell viability was determined by a modification of the crystal violet assay [23], and the optical density was measured at 540 nm with a 690-nm reference filter in a Synergy2 Multi-Detection Microplate Reader (BioTek Instruments, Inc.).
Determination of intracellular ROS levels
N2a cells were grown and re-plated as in the cytotoxicity assay, using 1% instead of 5% FBS. After 24 h the cells were treated with 20 µM DCF-DA for 30 min at 37 °C. Following incubation, the cultures were rinsed with phosphate-buffered saline, and fresh medium was added to the cells. The ROS levels before and after treatment with achillolide A and Aβ were determined according to fluorescence (excitation at 485 nm and emission at 520 nm) in a Synergy2 Multi-Detection Microplate Reader (BioTek Instruments, Inc.).
Determination of total and phospho-SAPK/JNK, and total and phospho-p44/42 MAPK levels
N2a cells were treated with achillolide A and Aβ25-35. The cells were lysed after 40 or 30 min for SAPK/JNK or p44/42 MAPK, respectively, in lysis buffer that was part of the ELISA kit. Protein concentrations in cell lysates were determined with Bradford reagent (Bio-Rad, Hercules, CA, USA), and equal amounts of proteins were analyzed by sandwich ELISA kits. The optical density was determined at 450 nm using a multi-detection microplate reader (BioTek Instruments, Inc.).
Statistical analyses
The results were analyzed by one-way ANOVA followed by Tukey–Kramer multiple comparison tests, using the Graph Pad InStat 3 for Windows (GraphPad Software, San Diego, CA, USA).
Results and discussion
Achillolide A (Fig. 1) was previously shown by us to inhibit microglial activation, protect astrocytes from oxidative stress, and protect neuroblastoma cells from glutamate toxicity [22, 24]. Aβ25-35 is a neurotoxic peptide commonly used in cellular models of AD [25, 26]. We have previously shown that exposure of N2a neuroblastoma cells to Aβ25-35 resulted in their death 20 h later [22]. To examine the effect of achillolide A on the toxicity of Aβ, these cells were treated with 25 μM of the Aβ25-35 peptide together with different concentrations of achillolide A. Viability and cytotoxicity were determined 20 h later. Our results show that achillolide A reduced Aβ25-35-induced cell death by 71% at a concentration of 16 nM (Fig. 2a), as observed by the LDH method, and completely rescued viability, as observed by the crystal violet method (Fig. 2b). It should be noted that achillolide A by itself is not cytotoxic to N2a cells that were exposed to different concentrations (up to 1566 nM) of this molecule, as determined using the crystal violet assay (Fig. 2c).

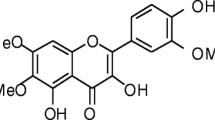
Chemical structure of achillolide A

Achillolide A protects N2a cells from Aβ -induced cytotoxicity. Cells were either untreated (‘untreated cells’) or treated with Aβ with or without various concentrations of achillolide A. Twenty hours later cell death was determined by (a) the LDH or (b, c) the crystal violet method. Cytotoxicity (a) was significantly reduced in cells treated with Aβ + achillolide A compared to Aβ-treated cells. Likewise, viability (b, c) was significantly increased in cells treated with Aβ + achillolide A compared to Aβ-treated cells. The results are the mean ± SEM of two experiments (n = 16). The maximal LDH release after disruption of the cells by Triton x-100 was A492 = 0.61 ± 0.04 as measured in two experiments (n = 5). ***P < 0.001
One mechanism by which Aβ may exert cell toxicity is the generation of ROS, leading to neuronal death [3]. Treatment of N2a cells with Aβ25-35 for 20 h results in a two-fold increase in intracellular ROS levels, as we have previously shown [22]. We therefore tested whether achillolide A could inhibit the elevated production of ROS following treatment with Aβ25-35 and, as a result, protect the cells from Aβ25-35-induced cytotoxicity. To test this possibility, cells were treated with various concentrations of achillolide A at the time of Aβ25-35 application, and ROS formation was determined 20 h later. Our results show that treatment with achillolide A inhibits 78% of the intracellular levels of Aβ25-35-induced ROS. We observed that 16 nM achillolide A is the lowest effective dose at both attenuating neuronal cell death and reducing the level of ROS production following Aβ25-35 treatment (Figs. 2, 3).

Achillolide A attenuates ROS levels induced by Aβ in N2a cells. Levels of ROS were significantly reduced in N2a cells treated with Aβ + achillolide A compared to Aβ-treated cells. The results represent the mean ± SEM of three experiments (n = 24). *p < 0.05, ***p < 0.001
The antioxidant characteristics of achillolide A demonstrated in this study in Aβ-treated N2a neuroblastoma cells support our previous observations, showing similar effects in glutamate-treated neuroblastoma N2a cells [22], H2O2-treated astrocytes and LPS-activated microglial cells [20, 24]. Although part of Aβ toxicity is mediated by glutamate [27], the mechanism underlying Aβ cytotoxicity is complex and involves many downstream targets (for review see [28]). As neuronal vulnerability in AD originates in the hippocampal formation, future experiments should examine the effect of achillolide A on these neuronal populations in vitro and in vivo.
Enhanced activation of SAPK/JNK and p44/42 MAPK was observed following treatment of cells with Aβ [29,30,31], as well as in brains of AD patients [32,33,34]. Since MAPK signaling was shown to be modulated by sesquiterpene lactones [35,36,37], we determined the effect of achillolide A on the phosphorylation of SAPK/JNK and p44/42 MAPK by Aβ25-35.
Figure 4 shows that Aβ25-35 increased the phosphorylation of SAPK/JNK by 7.5-fold in N2a cells, and achillolide A (at 158 nM) inhibited 79% of Aβ25-35-induced phosphorylation (Fig. 4a). At the same concentration, achillolide A also inhibited 78% of the Aβ25-35-induced phosphorylation of p44/42 MAPK (Fig. 4b), without affecting the total amount of these proteins in the cells (data not shown). These results suggest that inhibition of the Aβ-induced phosphorylation of SAPK/JNK and p44/42 MAPK is part of the mechanism by which achillolide A protects neurons against Aβ25-35 toxicity.

Achillolide A attenuates the phosphorylation of p44/42 MAPK and SAPK/JNK induced by Aβ in N2a cells. Cells were either untreated or treated with Aβ only (25 μM) or Aβ + achillolide A for 30 min (p44/42 MAPK) or 40 min (SAPK/JNK). The levels of phosphorylated and total SAPK/JNK (a) and p44/42 MAPK (b) in cell extracts were determined by corresponding ELISA kits. The levels of the phosphorylated proteins were normalized to the levels of the total amount of the related proteins, and are presented as the mean ± SEM of two experiments (n = 4) for SAPK/JNK, and three experiments (n = 6) for p44/42 MAPK. The levels of the phosphorylated proteins were significantly lower in cells treated with both Aβ + achillolide A compared to cells treated with Aβ only. **p < 0.01, ***p < 0.001
Conclusions
In this study, we have shown for the first time that achillolide A, a natural sesquiterpene lactone we isolated from A. fragrantissima, can protect N2a cells from Aβ25-35-induced cell death. In addition, achillolide A reduced the accumulation of Aβ-induced ROS in N2a cells and inhibited the phosphorylation of SAPK/JNK and p44/42 MAPK in these cells. Based on our results in astrocytes, microglial cells and N2a neuroblastoma cells, it is proposed that achillolide A has neuroprotective therapeutic characteristics. Further studies are warranted in order to substantiate the therapeutic potential of achillolide A for the treatment of AD.
References
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766
Liu J, Yang B, Ke J, Li W, Suen WC (2016) Antibody-based drugs and approaches against amyloid-beta species for Alzheimer’s disease immunotherapy. Drugs Aging 33:685–697
Jiang T, Sun Q, Chen S (2016) Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog Neurobiol 147:1–19
Essa MM, Vijayan RK, Castellano-Gonzalez G, Memon MA, Braidy N, Guillemin GJ (2012) Neuroprotective effect of natural products against Alzheimer’s disease. Neurochem Res 37:1829–1842
Kim MH, Kim SH, Yang WM (2014) Mechanisms of action of phytochemicals from medicinal herbs in the treatment of Alzheimer’s disease. Planta Med 80:1249–1258
Rasool M, Malik A, Qureshi MS, Manan A, Pushparaj PN, Asif M, Qazi MH, Qazi AM, Kamal MA, Gan SH et al (2014) Recent updates in the treatment of neurodegenerative disorders using natural compounds. Evid Based Complement Alternat Med. https://doi.org/10.1155/2014/979730 (Article ID 979730)
Akhondzadeh S, Abbasi SH (2006) Herbal medicine in the treatment of Alzheimer’s disease. Am J Alzheimer’s Dis Other Dement 21:113–118
Kim SK, Cho SB, Moon HI (2010) Neuroprotective effects of a sesquiterpene lactone and flavanones from Paulownia tomentosa Steud. against glutamate-induced neurotoxicity in primary cultured rat cortical cells. Phytother Res 24:1898–1900
Merfort I (2011) Perspectives on sesquiterpene lactones in inflammation and cancer. Curr Drug Targets 12:1560–1573
Choi EM, Kim GH, Lee YS (2009) Protective effects of dehydrocostus lactone against hydrogen peroxide-induced dysfunction and oxidative stress in osteoblastic MC3T3-E1 cells. Toxicol In Vitro 23:862–867
Gach K, Dugosz A, Janecka A (2015) The role of oxidative stress in anticancer activity of sesquiterpene lactones. Naunyn Schmiedebergs Arch Pharmacol 388:477–486
Song JX, Sze SC, Ng TB, Lee CK, Leung GP, Shaw PC, Tong Y, Zhang YB (2012) Anti-Parkinsonian drug discovery from herbal medicines: what have we got from neurotoxic models? J Ethnopharmacol 139:698–711
Dong LP, Qiao HM, Zhang XJ, Zhang XL, Wang CH, Wang LN, Cui LL, Zhao JR, Xing YX, Li YH et al (2013) Parthenolide is neuroprotective in rat experimental stroke model: downregulating NF-kappa B, phospho-p38MAPK, and caspase-1 and ameliorating BBB permeability. Mediat Inflamm 370804
Liu C, Zhao H, Ji ZH, Yu XY (2014) Neuroprotection of atractylenolide III from Atractylodis macrocephalae against glutamate-induced neuronal apoptosis via inhibiting caspase signaling pathway. Neurochem Res 39:1753–1758
Schwarz D, Bloom D, Castro R, Pagan OR, Jimenez-Rivera CA (2011) Parthenolide blocks cocaine’s effect on spontaneous firing activity of dopaminergic neurons in the ventral tegmental area. Curr Neuropharmacol 9:17–20
Amoah SKS, Dalla Vecchia MT, Pedrini B, Carnhelutti GL, Goncalves AE, dos Santos DA, Biavatti MW, de Souza MM (2015) Inhibitory effect of sesquiterpene lactones and the sesquiterpene alcohol aromadendrane-4 beta,10 alpha-diol on memory impairment in a mouse model of Alzheimer. Eur J Pharmacol 769:195–202
Baptista FI, Henriques AG, Silva AMS, Wiltfang J, Silva OABDE (2014) Flavonoids as therapeutic compounds targeting key proteins involved in Alzheimer’s disease. ACS Chem Neurosci 5:83–92
Williams RJ, Spencer JP (2012) Flavonoids, cognition, and dementia: actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic Biol Med 52:35–45
Orhan IE, Daglia M, Nabavi SF, Loizzo MR, Sobarzo-Sanchez E, Nabavi SM (2015) Flavonoids and dementia: an update. Curr Med Chem 22:1004–1015
Elmann A, Telerman A, Mordechay S, Erlank H, Rindner M, Kashman Y, Ofir R (2015) Downregulation of microglial activation by achillolide A. Planta Med 81:215–221
Elmann A, Telerman A, Mordechay S, Erlank H, Rindner M, Ofir R, Kashman Y (2014) 3,5,4′-Trihydroxy-6,7,3′-trimethoxyflavone protects astrocytes against oxidative stress via interference with cell signaling and by reducing the levels of intracellular reactive oxygen species. Neurochem Int 78:67–75
Elmann A, Telerman A, Ofir R, Kashman Y (2017) Glutamate toxicity to differentiated neuroblastoma N2a cells is prevented by the sesquiterpene lactone achillolide A and the flavonoid 3,5,4′-trihydroxy-6,7,3′-trimethoxyflavone from Achillea fragrantissima. J Mol Neurosci 62:99–105
Kueng W, Silber E, Eppenberger U (1989) Quantification of cells cultured on 96-well plates. Anal Biochem 182:16–19
Elmann A, Telerman A, Erlank H, Ofir R, Kashman Y, Beit-Yannai E (2016) Achillolide A protects astrocytes against oxidative stress by reducing intracellular reactive oxygen species and interfering with cell signaling. Molecules 21:301
Cuevas E, Lantz SM, Tobon-Velasco JC, Newport GD, Wu QG, Virmani A, Ali SF, Santamaria A (2011) On the in vivo early toxic properties of A beta(25-35) peptide in the rat hippocampus: involvement of the receptor-for-advanced glycation-end-products and changes in gene expression. Neurotoxicol Teratol 33:288–296
Kaminsky YG, Marlatt MW, Smith MA, Kosenko EA (2010) Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: evidence for Abeta(25-35). Exp Neurol 221:26–37
Li H, Han W, Wang H, Ding F, Xiao L, Shi R, Ai L, Huang Z (2017) Tanshinone IIA inhibits glutamate-induced oxidative toxicity through prevention of mitochondrial dysfunction and suppression of MAPK activation in SH-SY5Y human neuroblastoma cells. Ox Med Cell Longev. https://doi.org/10.1155/2017/4517486
Chen YC (2017) Impact of a discordant helix on beta-amyloid structure, aggregation ability and toxicity. Eur Biophys J 46:681–687
Braithwaite SP, Schmid RS, He DN, Sung MLA, Cho S, Resnick L, Monaghan MM, Hirst WD, Essrich C, Reinhart PH et al (2010) Inhibition of c-Jun kinase provides neuroprotection in a model of Alzheimer’s disease. Neurobiol Dis 39:311–317
Tare M, Modi RM, Nainaparampil JJ, Puli OR, Bedi S, Fernandez-Funez P, Kango-Singh M, Singh A (2011) Activation of JNK signaling mediates amyloid-beta-dependent cell death. Plos One 6(9):e24361. https://doi.org/10.1371/journal.pone.0024361
Ghasemi R, Moosavi M, Zarifkar A, Rastegar K, Maghsoudi N (2015) The interplay of Akt and ERK in A beta toxicity and insulin-mediated protection in primary hippocampal cell culture. J Mol Neurosci 57:325–334
Zeitlin R, Patel S, Burgess S, Arendash GW, Echeverria V (2011) Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer’s transgenic mice. Brain Res 1417:127–136
Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW (2002) Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci 22:3376–3385
Echeverria V, Ducatenzeiler A, Dowd E, Janne J, Grant SM, Szyf M, Wandosell F, Avila J, Grimm H, Dunnett SB et al (2004) Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience 129:583–592
Huang CC, Lin KJ, Cheng YW, Hsu CA, Yang SS, Shyur LF (2013) Hepatoprotective effect and mechanistic insights of deoxyelephantopin, a phyto-sesquiterpenelactone, against fulminant hepatitis. J Nutr Biochem 24:516–530
Chiu KY, Wu CC, Chia CH, Hsu SL, Tzeng YM (2016) Inhibition of growth, migration and invasion of human bladder cancer cells by antrocin, a sesquiterpene lactone isolated from Antrodia cinnamomea, and its molecular mechanisms. Cancer Lett 373:174–184
Kang JS, Yoon YD, Lee KH, Park SK, Kim HM (2004) Costunolide inhibits interleukin-1beta expression by down-regulation of AP-1 and MAPK activity in LPS-stimulated RAW 264.7 cells. Biochem Biophys Res Commun 313:171–177
Acknowledgements
The authors wish to thank Yardena Abudi (TAU) and Miriam Rindner (ARO) for their technical assistance. This is publication 808/18 from the Agricultural Research Organization. This work was supported by the BARD, the United States−Israel Binational Agricultural Research and Development Fund [Grant number IS-4473-11, AE and OL].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Elmann, A., Telerman, A., Ofir, R. et al. β-amyloid cytotoxicity is prevented by natural achillolide A. J Nat Med 72, 626–631 (2018). https://doi.org/10.1007/s11418-018-1191-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-018-1191-0