
Abstract
Background
Alzheimer’s disease is a neurodegenerative disease, characterized by progressive decline in memory and cognitive functions, that results from loss of neurons in the brain. Amyloid beta (Aβ) protein and oxidative stress are major contributors to Alzheimer’s disease, therefore, protecting neuronal cells against Aβ-induced toxicity and oxidative stress might form an effective approach for treatment of this disease. 3,5,4′-trihydroxy-6,7,3′-trimethoxyflavone (TTF) is a flavonoid we have purified from the plant Achillea fragrantissima; and the present study examined, for the first time, the effects of this compound on Aβ-toxicity to neuronal cells.
Methods
Various chromatographic techniques were used to isolate TTF from the plant Achillea fragrantissima, and an N2a neuroblastoma cell line was used to study its activities. The cellular levels of total and phosphorylated stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and of total and phosphorylated extracellular signal-regulated kinase (ERK 1/2) were determined by enzyme-linked immunosorbent assay (ELISA). Intracellular reactive oxygen species (ROS) levels were measured by using 2′,7′-dichlorofluorescein diacetate (DCF-DA). Cytotoxicity and cell viability were assessed by using lactate dehydrogenase (LDH) activity in cell-conditioned media, or by crystal violet cell staining, respectively.
Results
TTF prevented the Aβ-induced death of neurons and attenuated the intracellular accumulation of ROS following treatment of these cells with Aβ. TTF also inhibited the Aβ-induced phosphorylation of the signaling proteins SAPK/JNK and ERK 1/2, which belong to the mitogen-activated protein kinase (MAPK) family.
Conclusion
TTF should be studied further as a potential therapeutic means for the treatment of Alzheimer’s disease.
Similar content being viewed by others


Background
Alzheimer’s disease (AD), the most common form of dementia in adults, is characterized by widespread loss of neurons in the brain, which results in progressive memory loss and cognitive decline. AD is characterized by intraneuronal neurofibrillary tangles (aggregates of cytoskeletal hyperphosphorylated Tau protein) and extraneuronal senile plaques, formed mainly by aggregated amyloid beta (Aβ) peptides [1]. Aβ is a 39- to 43-amino acid peptide derived from the cleavage of amyloid precursor protein (APP), and it is involved in the pathogenesis of AD through several different mechanisms, including oxidative stress, microglial activation, neuronal dysfunction, and neuronal cell death [2, 3]. Thus, protecting neuronal cells against Aβ-induced cytotoxicity might form an effective approach to therapeutic treatment of Alzheimer’s disease.
An increasing body of evidence shows the therapeutic potential of phytochemicals against various diseases, including AD [4, 5]. Flavonoids are phytochemicals that exhibit a broad range of biological functions, including anti-inflammatory, antioxidative and neuroprotective activities [6,7,8,9,10,11]. In light of these activities, they seem to be promising candidates for development as drugs for treatment of neurodegenerative diseases [8, 12, 13].
In a previous study, we have shown that 3,5,4′-trihydroxy-6,7,3′-trimethoxyflavone (TTF), a flavonoid that we have isolated from Achillea fragrantissima, prevented the hydrogen peroxide (H2O2)-induced death of astrocytes, and inhibited the phosphorylation of cell-signaling proteins that belong to the mitogen-activated protein kinase (MAPK) family [14]. TTF also scavenges free radicals and mitigates intracellular accumulation of ROS following treatment of these cells with H2O2 or with the peroxyl radicals-generating molecule 2,2′-azobis(amidinopropane) (ABAP) [14]; it is characterized by a relatively low polarity and low molecular weight of 360.3, therefore it might cross the blood-brain barrier and could be used for further investigations in cellular and animal models of AD.
In the present study, we aimed to determine whether TTF could counteract Aβ toxicity in N2a neuroblastoma cells, and to elucidate the molecular mechanisms involved.
Methods
Chemicals and reagents
Aβ25–35, crystal violet, and 2′,7′-dichlorofluorescein diacetate (DCF-DA) were purchased from Sigma Chemical Co. (St Louis, MO, USA); Dulbecco’s modified Eagle’s medium (DMEM) and Opti-MEM were purchased from Gibco (Paisley, UK); glutamine, antibiotics (10,000 IU/mL penicillin and 10,000 μg/mL streptomycin), fetal bovine serum (FBS) and trypin-EDTA were purchased from Biological Industries (Beit Haemek, Israel); dimethyl sulfoxide (DMSO) was obtained from Applichem (Darmstadt, Germany).
Plant material
The aerial parts of A. fragrantissima were collected in the Arava Valley, and the voucher specimens have been kept and authenticated as part of the Arava Rift Valley Plant Collection under accession code AVPC0040.
Extraction and isolation
Isolation of TTF was carried out as previously described [14]. Briefly: the sun-dried A. fragrantissima plant was homogenized and extracted with ethyl acetate twice and with ethyl acetate:methanol (9:1) once. The organic extracts were combined and evaporated. The latter residue was chromatographed on a Sephadex LH-20 column that was eluted with methanol: CH2Cl2 (1:1). A fraction yielded by the Sephadex LH-20 column, which was monitored by thin layer chromatography (TLC) and 1H–nuclear magnetic resonance (NMR) and that protected astrocytes from H2O2-induced cell death, was further purified by repeated chromatography over silica gel, with hexane that contained increasing proportions of ethyl acetate used as eluent. The active compound was afforded by elution with 50% ethyl acetate in hexane.
Treatment of neuronal cells
The original medium was aspirated from the cells and replaced with fresh medium. Fresh dilutions of TTF, first in DMSO and then in the growth medium, were prepared from stock solution just prior to each experiment, and were used immediately. The final concentration of DMSO in the medium was 0.2%. The Aβ25–35 peptide was dissolved in DDW and incubated at 37 °C for 48 h. Fresh dilutions of Aβ in the growth medium were prepared just prior to each experiment and were used immediately. Each treatment was performed in replicates.
Determination of cytotoxicity and cell viability
Cytotoxicity – N2a cells were grown in a medium containing 43% DMEM (high glucose), 50% Opti-MEM, 5% FBS, 2 mM glutamine, penicillin at 100 U/mL, and streptomycin at 100 μg/mL. The cells were re-plated in 96-well plates at a density of 5 × 103 cells/well, in a similar medium; Aβ and/or TTF were added concomitantly 24 h later, and cytotoxicity was determined 20 h later with a commercial colorimetric assay (Roche Applied Science, Germany) based on the measurement of lactate dehydrogenase (LDH) activity released into the incubation medium from the cytosol of damaged cells. The absorbance was measured at 492 nm in a Synergy2 multi-detection microplate reader (BioTek Instruments, Inc., Winooski, VT, USA). The percentage of cytotoxicity was calculated according to:
in which the term “ATriton-x treated cells” is the maximum releasable LDH from the cells.
Cell viability – N2a cells were grown and treated as in the cytotoxicity assay, except that replating was at a density of 5 × 103 cells/well. Cell viability was determined by a modification of the crystal violet assay [15], as follows. At the end of their treatments the cells were fixed with 150 μL of 5% (v/v) formaldehyde in PBS for 15 min at room temperature. The plates were washed by immersion in deionized water, dried and stained for 15 min with 150 μL of a 1% crystal violet solution. Following aspiration of the crystal violet solution the plates were washed with deionized water and dried, and the bound dye was solubilized with 150 μL of 33% aqueous solution of glacial acetic acid. The optical density was measured at 540 nm, by comparison with a 690-nm reference filter, in a Synergy2 multi-detection microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Evaluation of intracellular ROS levels
Intracellular ROS levels were detected by using the non-fluorescent cell-permeating compound, 2′,7′-dichlorofluorescein diacetate (DCF-DA). The N2a cells were plated onto 96-well plates at 10,000 cells per 0.2-mL well, in 43% DMEM (high glucose), 50% Opti-MEM, 1% FBS, 2 mM glutamine, penicillin at 100 U/mL, and streptomycin at 100 mg/mL. After 24 h the cells were treated with 20 μM DCF-DA for 30 min at 37 °C. Following incubation with DCF-DA, the cultures were rinsed twice with PBS, which was then replaced with fresh medium. The. ROS levels at time zero were evaluated according to fluorescence in a Synergy2 multi-detection microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) with excitation at 485 nm and emission at 520 nm. The cells were then treated with TTF and Aβ, and the ROS levels were measured at the indicated time points.
The percentage of ROS levels was calculated according to:
in which F is the fluorescence.
Enzyme-linked immunosorbent assays (ELISA) for total and phosphoSAPK/JNK, and total and phosphoERK (phospho-p44/42 MAPK)
N2a cells were treated with TTF concomitantly with addition of Aβ25–35. The cells were lysed after 40 or 30 min, for SAPK/JNK or ERK 1/2, respectively, in lysis buffer that was part of the PathScan Sandwich ELISA kit (Cell Signaling Technology, Danvers, MA, USA) according to the manufacturer’s protocol. Protein concentrations in cell lysates were determined with Bradford reagent (Bio-Rad, Hercules, CA, USA), and equal amounts of proteins were subjected to ELISA. To measure the amounts of total and of phosphoSAPK/JNK in cell lysates of the N2a cells, ELISA was performed according to the manufacturer’s protocols with the PathScan total SAPK/JNK sandwich ELISA kit and the PathScan phosphoSAPK/JNK (Thr183/Tyr185) sandwich ELISA kit, respectively (both kits from Cell Signaling Technology, Danvers, MA, USA). To measure the amounts of total and phosphoERK 1/2, i.e., phospho-p44/42 MAPK, in cell lysates, ELISA was performed according to the manufacturer’s protocol with Cell Signaling Technology’s PathScan total p44/42 MAPK (ERK 1/2) sandwich ELISA kit and the PathScan phospho-p44/42 MAPK (Thr202/Tyr204) sandwich ELISA kit, respectively. The optical density was measured at 450 nm with a Synergy2 multi-detection microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Statistical analysis
The results were subjected to one-way ANOVA followed by Tukey-Kramer multiple comparison tests, by means of Graph Pad InStat 3 for Windows (GraphPad Software, San Diego, CA, USA).
Results
Protective effect of TTF against Aβ 25–35 -induced neuronal cell death
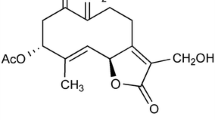
Phytochemicals that can protect neuronal cells against Aβ toxicity and oxidative stress may assist in coping with Alzheimer’s disease. In order to assess the ability of TTF (The structure of TTF is presented in Fig. 1) to counteract Aβ toxicity in N2a neuroblastoma cells, we have used the Aβ25–35 peptide, which represents the neurotoxic fragments of Aβ1–40 and Aβ1–42, and is used in cell models to mimic their toxicity. Exposure of neuronal cells to Aβ25–35 resulted in their death after 20 h, as reflected in a fivefold increase in the LDH assay (Fig. 2a), and a 55% reduction in the crystal violet assay (Fig. 2b). To assess the ability of TTF to protect neuronal cells against Aβ25–35, and to determine the optimal concentrations of TTF required to induce a protective effect, cells were treated with Aβ25–35 at 25 μM and with various concentrations of TTF. Cytotoxicity and viability were determined after 20 h by means of the LDH assay (Fig. 2c) and the crystal violet assay (Fig. 2d), respectively. Our results show that TTF exhibited a protective effect against Aβ25–35-induced cell death, with maximal efficacy (96% protection) at a concentration of 140 nM (Fig. 2c, d). It should be noted that at all concentrations tested, up to 700 nM, the crystal violet assay did not show TTF alone to be cytotoxic to neuronal cells (Fig. 2e).

Chemical structure of 3,5,4′-trihydroxy-6,7,3′-trimethoxyflavone (TTF)

TTF protects neuronal cells from Aβ-induced cell death. Neuronal cells were treated with Aβ25–35 at 25 μM, and cell death was determined 20 h later by (a) the LDH and (b) the crystal violet methods. Neuronal cells were treated with various concentrations of TTF, Aβ was added and cell death was determined 20 h later by (c) the LDH and (d) the crystal violet methods. The results are means ± SEM of two experiments (n = 16); *** P < 0.001, for comparison of treated with untreated cells. The maximal LDH release after disruption of cells by Triton x-100 was A492 = 0.61 ± 0.04 as measured in two experiments (n = 5). (e) Cells were treated with TTF alone, at various concentrations, and cell viability was determined 20 h later by the crystal violet method. The results are means ± SEM of two experiments (n = 16). *** P < 0.001, for comparison with cells that were treated with Aβ alone
Inhibitory effect of TTF on Aβ 25–35 -induced generation of ROS
The role of free radicals in AD has been reported in many studies [16]. Moreover, it has been shown that Aβ induced generation of reactive oxygen species (ROS), leading to neuronal death [3]. To investigate the effect of TTF on ROS levels, which are elevated in response to Aβ treatment, the levels of intracellular ROS were determined. Treatment of cells with Aβ25–35 for 20 h resulted in a twofold increase in intracellular ROS levels (Fig. 3), but no significant elevation in ROS levels was observed one or five hours after stimulation (Fig. 3). We therefore tested the possibility that TTF could protect neuronal cells from Aβ25–35-induced cell death by inhibiting the Aβ25–35-induced production of ROS. Cells were treated with various concentrations of TTF, simultaneously with application of Aβ25–35. Changes in intracellular levels of ROS were detected with the ROS indicator DCF-DA, and ROS formation was determined by examining fluorescence after 20 h. Our results show that treatment with TTF at a concentration of 25 nM – similar to that used to protect cells from Aβ25–35-induced cell death – inhibited the intracellular levels of Aβ25–35-induced ROS by 60% (Table 1).

Aβ-induced elevation in ROS levels in N2a cells. Intracellular ROS levels (Fluorescence units, FU) were measured 1, 5, and 20 h after treatment. The results are the mean ± SEM of one experiment (n = 8)
Inhibitory effect of TTF on Aβ-induced phosphorylation of SAPK/JNK and ERK 1/2 in N2a cells
Mitogen-activated protein kinases (MAPKs) – a family of serine/threonine protein kinases – which initiate many cellular responses to external stress signals, were shown to be modulated by flavonoids [12, 17]. Enhanced activation of SAPK/JNK and ERK 1/2, which belong to the MAPK cascade, was observed in AD-affected brains [18,19,20]. Moreover, Aβ has been reported to stimulate SAPK/JNK and ERK 1/2 [21,22,23,24]. Therefore, we determined the effect of TTF on Aβ25–35-induced phosphorylation of SAPK/JNK and ERK 1/2.
As can be seen in Fig. 4, treatment with Aβ25–35 increased cellular levels of phosphorylated SAPK/JNK and ERK 1/2 by 2.5-fold. Treatment of cells with TTF at 3 and 32 nM inhibited the Aβ25–35-induced phosphorylation of ERK 1/2 and SAPK/JNK, respectively, by 84 and 60%, respectively (Figs 4a and b, respectively). The total amounts of these proteins in the cells were not affected by TTF. It should be noted that TTF was more effective in inhibiting phosphorylation of ERK 1/2 than that of SAPK/JNK: at 3 nM it inhibited induced phosphorylation of ERK 1/2 by 84%, whereas at this concentration it inhibited that of SAPK/JNK by only 26%. These results suggest that inhibition of Aβ-induced phosphorylation of SAPK/JNK and ERK 1/2 is part of the mechanism by which TTF protects cells against Aβ25–35 toxicity.

TTF suppresses Aβ-induced phosphorylation of SAPK/JNK, and ERK 1/2 in N2a cells. Cells were treated concomitantly with 25 μM of Aβ and TTF for 30 min (ERK 1/2) or 40 min (SAPK/JNK). Cells were extracted and the levels of phosphorylated and total ERK 1/2 (a) and SAPK/JNK (b) were determined with specific ELISA kits. The levels of each of the phosphorylated proteins were normalized to the levels of the total amount of the related proteins, and are presented as means ± SEM of two experiments (n = 4) for SAPK/JNK, and of three experiments (n = 6) for ERK 1/2. **p < 0.01, ***p < 0.001, for comparison with cells that were treated with Aβ only
Discussion
In the present study we showed, for the first time, that TTF, a natural flavonoid that we isolated from A. fragrantissima [14], could protect neuronal cells against Aβ-induced cell death, and that it inhibited phosphorylation of MAP kinases and attenuated the intracellular accumulation of ROS following treatment with Aβ. To the best of our knowledge, this is the first study that investigated the effects of TTF on neuronal cells and on Aβ-induced cytotoxicity.
Drugs currently used for treating AD improve patients’ functions symptomatically, but do not modify the disease mechanism; thus, development of new and more effective drugs is required. Herbs and medicinal plants have been demonstrated in animal and cellular models to exhibit various interventions against multiple targets related to AD, including anti-cholinesterase activity, anti-amyloid, anti-oxidant and anti-inflammatory, and therefore might affect disease progression [25,26,27]; thus, they might affect disease mechanisms. Nutraceuticals that can protect neuronal cells from Aβ toxicity and oxidative stress are potential candidates for the treatment of Alzheimer’s disease. It previously has been suggested that new strategies based on memantine combined with antioxidants, could provide a multitargeted therapy to enhance neuronal protection and prevent disease progression [28]. The antioxidant activities of TTF demonstrated in neuronal cells in the present study, and in astrocytes in our previous study [14] indicate that TTF is a potential component of combined therapies.
Most flavonoids are metabolized in the gastro-intestinal tract and liver and are absorbed into the bloodstream; and some of them were shown to cross the blood/brain barrier (BBB) into the central nervous system [29,30,31,32,33]. Flavonoids were shown to have neuroprotective effects, to impact neuronal function, to modulate neurotransmission, and to improve synaptic plasticity and cognition. Moreover, although astrocytes were proposed to mediate the actions of flavonoids in the brain, little is known about their cellular targets [32]. Aβ-induced neurotoxicity might be mediated through several Aβ-binding proteins that were identified on neuron plasma membranes. They include: N-methyl-D-aspartate receptor (NMDAR); receptor for advanced glycation end products (RAGE); α7-nicotinic acetylcholine receptor (α7 nAChR); ephrin type B receptor 2; cellular prion protein (PrPc); immunoglobulin G Fc gamma receptor IIb (FcgRIIb); and paired immunoglobulin-like receptor B (PirB) [34]. Accordingly, TTF might manifest its protective effects by antagonizing these receptors. Our results that relate to inhibition of MAPK are compatible with other findings that showed flavonoids to modulate MAP-kinase signaling, or to directly bind to some protein kinases [35, 36].
Conclusions
Taken together, our findings suggest that by interfering with Aβ-induced signaling events and inhibiting elevation of intracellular ROS, TTF protects cells against Aβ-toxicity. In light of our previous results in astrocytes and our present results in neuronal cells, as well as the results of oral safety studies of extracts prepared from A. fragrantissima [37], further studies should be conducted, to substantiate the potential of TTF for therapeutic treatment of AD.
Abbreviations
- A. fragrantissima :
-
Achillea fragrantissima (Forssk) Sch. Bip
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid beta
- DCF-DA:
-
2′,7′-dichlorofluorescein diacetate
- ERK:
-
Extracellular signal regulated kinase
- MAPK :
-
Mitogen-activated protein kinase
- ROS :
-
Reactive oxygen species
- SAPK/JNK :
-
Stress-activated protein kinase /c-Jun N-terminal kinase
- TTF :
-
3,5,4′-trihydroxy-6,7,3′-trimethoxyflavone
References
Lloret A, Fuchsberger T, Giraldo E, Vina J. Molecular mechanisms linking amyloid beta toxicity and tau hyperphosphorylation in Alzheimer's disease. Free Radic Biol Med. 2015;83:186–91.
Liu J, Yang B, Ke J, Li W, Suen WC. Antibody-based drugs and approaches against amyloid-beta species for Alzheimer's disease immunotherapy. Drugs Aging. 2016;33(10):685–97.
Jiang T, Sun Q, Chen S. Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson's disease and Alzheimer's disease. Prog Neurobiol. 2016;147:1–19.
Kim MH, Kim SH, Yang WM. Mechanisms of action of phytochemicals from medicinal herbs in the treatment of Alzheimer's disease. Planta Med. 2014;80(15):1249–58.
Rasool M, Malik A, Qureshi MS, Manan A, Pushparaj PN, Asif M, et al. Recent Updates in the Treatment of Neurodegenerative Disorders Using Natural Compounds. Evid-Based Compl Alt. 2014;979730 http://dx.doi.org/10.1155/2014/979730
Afanas'ev IB, Dorozhko AI, Brodskii AV, Kostyuk VA, Potapovitch AI. Chelating and free radical scavenging mechanisms of inhibiotory action of rutin and quercetin in lipid peroxidation. Biochem Pharmacol. 1989;38(11):1763–9.
Song JX, Sze SC, Ng TB, Lee CK, Leung GP, Shaw PC, et al. Anti-parkinsonian drug discovery from herbal medicines: what have we got from neurotoxic models? J Ethnopharmacol. 2012;139(3):698–711.
Williams RJ, Spencer JP. Flavonoids, cognition, and dementia: actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic Biol Med. 2012;52(1):35–45.
Youdim KA, Qaiser MZ, Begley DJ, Rice-Evans CA, Abbott NJ. Flavonoid permeability across an in situ model of the blood-brain barrier. Free Radic Biol Med. 2004;36(5):592–604.
Serafini M, Peluso I, Raguzzini A. Session 1: antioxidants and the immune system flavonoids as anti-inflammatory agents. P Nutr Soc. 2010;69(3):273–8.
Kumar S, Pandey AK. Chemistry and Biological Activities of Flavonoids: An Overview. Sci World J. 2013; 2013: Article ID 162750.doi: 10.1155/2013/162750.
Baptista FI, Henriques AG, Silva AMS, Wiltfang J, Silva OABDE. Flavonoids as therapeutic compounds targeting key proteins involved in Alzheimer's disease. ACS Chem Neurosci. 2014;5(2):83–92.
Orhan IE, Daglia M, Nabavi SF, Loizzo MR, Sobarzo-Sanchez E, Nabavi SM. Flavonoids and dementia: an update. Curr Med Chem. 2015;22(8):1004–15.
Elmann A, Telerman A, Mordechay S, Erlank H, Rindner M, Ofir R, et al. 3,5,4′-Trihydroxy-6,7,3′-trimethoxyflavone protects astrocytes against oxidative stress via interference with cell signaling and by reducing the levels of intracellular reactive oxygen species. Neurochem Int. 2014;78:67–75.
Kueng W, Silber E, Eppenberger U. Quantification of cells cultured on 96-well plates. Anal Biochem. 1989;182(1):16–9.
Wojtunik-Kulesza KA, Oniszczuk A, Oniszczuk T, Waksmundzka-Hajnos M. The influence of common free radicals and antioxidants on development of Alzheimer's disease. Biomed Pharmacother. 2016;78:39–49.
Kaminska B, Gozdz A, Zawadzka M, Ellert-Miklaszewska A, Lipko M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat Rec. 2009;292(12):1902–13.
Zeitlin R, Patel S, Burgess S, Arendash GW, Echeverria V. Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer's transgenic mice. Brain Res. 2011;1417:127–36.
Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition. J Neurosci. 2002;22(9):3376–85.
Echeverria V, Ducatenzeiler A, Dowd E, Janne J, Grant SM, Szyf M, et al. Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neurosci. 2004;129(3):583–92.
Braithwaite SP, Schmid RS, He DN, Sung MLA, Cho S, Resnick L, et al. Inhibition of c-Jun kinase provides neuroprotection in a model of Alzheimer's disease. Neurobiol Dis. 2010;39(3):311–7.
Tare M, Modi RM, Nainaparampil JJ, Puli OR, Bedi S, Fernandez-Funez P, et al. Activation of JNK Signaling Mediates Amyloid-beta-Dependent Cell Death. Plos One. 2011;6(9)
Andrade JP, Assuncao M. Protective effects of chronic green tea consumption on age-related neurodegeneration. Curr Pharm Des. 2012;18(1):4–14.
Ghasemi R, Moosavi M, Zarifkar A, Rastegar K, Maghsoudi N. The interplay of Akt and ERK in a beta toxicity and insulin-mediated protection in primary hippocampal cell culture. J Mol Neurosci. 2015;57(3):325–34.
Ayaz M, Junaid M, Ahmed J, Ullah F, Sadiq A, Ahmad S, et al. Phenolic contents, antioxidant and anticholinesterase potentials of crude extract, subsequent fractions and crude saponins from Polygonum hydropiper L. BMC Complement Altern Med. 2014;14:145.
Ahmad S, Ullah F, Ayaz M, Sadiq A, Imran M. Antioxidant and anticholinesterase investigations of Rumex hastatus D. Don: potential effectiveness in oxidative stress and neurological disorders Biol Res. 2015;48:20.
Bowirrat A, Yassin M, Abir M, Bisharat B. Armaly Z. Traditional and Modern Medicine Harmonizing the Two Approaches in the Treatment of Neurodegeneration. In: Complementary Therapies for the Contemporary Healthcare. edn. Novi Sad: InTech Prepress. 2012:181–212.
Annweiler C, Beauchet O. Possibility of a new anti-alzheimer's disease pharmaceutical composition combining memantine and vitamin D. Drugs Aging. 2012;29(2):81–91.
Romagnoli R, Baraldi PG, Carrion MD, Cara CL, Cruz-Lopez O, Preti D, et al. Design, synthesis, and biological evaluation of thiophene analogues of chalcones. Bioorg Med Chem Lett. 2008;16(10):5367–76.
Abd El Mohsen MM, Kuhnle G, Rechner AR, Schroeter H, Rose S, Jenner P, et al. Uptake and metabolism of epicatechin and its access to the brain after oral ingestion. Free Radic Biol Med. 2002;33(12):1693–702.
Ferruzzi MG, Lobo JK, Janle EM, Cooper B, Simon JE, Wu QL, et al. Bioavailability of Gallic acid and Catechins from grape seed polyphenol extract is improved by repeated dosing in rats: implications for treatment in Alzheimer's disease. J Alzheimers Dis. 2009;18(1):113–24.
Matias I, Buosi AS, Gomes FCA. Functions of flavonoids in the central nervous system: astrocytes as targets for natural compounds. Neurochem Int. 2016;95:85–91.
Jager AK, Saaby L. Flavonoids and the CNS. Molecules. 2011;16(2):1471–85.
Kam TI, Gwon Y, Jung YK. Amyloid beta receptors responsible for neurotoxicity and cellular defects in Alzheimer's disease. Cell Mol Life Sci. 2014;71(24):4803–13.
Murakami A, Ohnishi K. Target molecules of food phytochemicals: food science bound for the next dimension. Food Funct. 2012;3(5):462–76.
Hou DX, Kumamoto T. Flavonoids as protein kinase inhibitors for cancer chemoprevention: direct binding and molecular modeling. Antiox Redox Signal. 2010;13(5):691–719.
Mandour MA, Al-Shami SA, Al-Eknah MM, Hussein YA, El-Ashmawy IM. The acute and long-term safety evaluation of aqueous, Methanolic and Ethanolic extracts of Achillea Fragrantissima. Afr J Pharm Pharmacol. 2013;7(32):2282–90.
Acknowledgements
The authors wish to thank Yardena Abudi (TAU) and Miriam Rindner (ARO) for their technical assistance.
Funding
This research was supported by research grant no. IS-4473-11 from BARD, the United States – Israel Binational Agricultural Research and Development Fund. This is publication 784/17 from the Agricultural Research Organization.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors’ contributions
AE carried out the study design, literature search and manuscript preparation. AT carried out the cell culture and biochemical experiments. RO collected and supplied the plants. YK isolated TTF from the plants and participated in drafting the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not Applicable
Ethics approval and consent to participate
Not Applicable
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Telerman, A., Ofir, R., Kashman, Y. et al. 3,5,4′-trihydroxy-6,7,3′-trimethoxyflavone protects against beta amyloid-induced neurotoxicity through antioxidative activity and interference with cell signaling. BMC Complement Altern Med 17, 332 (2017). https://doi.org/10.1186/s12906-017-1840-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-017-1840-y