
Abstract
Purpose
Organochlorine pesticides (OCPs) like lindane and DDT have been used extensively after World War II until the 1990s. Still, residues of these pesticides can be found in agricultural soils all over the world, especially in developing countries. Often, they occur in extensive areas and elevated concentrations so that food safety is jeopardized. Hence, simple, cheap, and fast analytical methods are needed for a straight-forward assessment of risks. A miniaturized solid–liquid extraction combined with solid-phase microextraction (SPME) based on a proven ISO method is presented.
Methods
The performance of the method is evaluated by extracting three different soils which were spiked with HCH and DDT congeners, and trifluralin, and aged for 35 days. The results are compared with those of a modified quick, easy, cheap, efficient, rugged, and safe (QuEChERS) method. For further validation, both methods are applied to three environmental soil samples.
Results
Validation results show limits of detection and quantification as well as recovery rates in good agreement with standard requirements. The new method was found to be quicker than QuEChERS, which requires time-consuming preparation of reagents.
Conclusion
Merits include low time and sample volume requirements (0.5 g) and the possibility to extract many samples simultaneously, which allows the screening of large sample sizes to determine the pollution status of whole landscape regions. However, access to an automated SPME apparatus is assumed. The authors can recommend this method as a cheap and fast alternative where SPME is available.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Soils are the foundation for human life. Among many services, they hold water and nutrients, are habitat for myriads of organisms, are the platform onto which structures are built, and most importantly, soils are the compartment where human food is grown. As such, keeping them in good condition is of utmost importance. All over the world, agricultural production systems are forced to develop in a more sustainable direction: simultaneously securing health and food safety for a growing population, minimizing detrimental environmental effects, preserving natural resources, and ensuring a sufficient yield and income especially in structurally weak, rural areas (subsistence farming) are only some of the associated challenges. However, soils are threatened by a multitude of factors, one of which is contamination with xenobiotics. To secure high crop yields, large quantities of pesticides are applied in agriculture each year, a great amount of which remains in the soil for a long time (Silva et al. 2019). While modern pesticides are required to be readily biodegradable and to minimize their impact on non-target organisms, historically applied compounds did not have these prerequisites. A recognized group of legacy compounds are organochlorine pesticides (OCPs), of which DDT (dichlorodiphenyltrichloroethane) and lindane (γ-hexachlorocyclohexane, γ-HCH) are well-known representatives. They were produced and used excessively after World War II until their toxicity, bioaccumulation, and persistence came to attention and led to various national restrictions and subsequent international ban on account of the Stockholm Convention 2001 (United Nations 2001). Yet, they are still found in occasionally large concentrations in agricultural soils all over the world, e.g., in Europe (Camenzuli et al. 2016), in states of the former Soviet Union (Aliyeva et al. 2012; Sharov et al. 2016; Tepanosyan et al. 2020), Africa (Olisah et al. 2020), or China (Li et al. 2014).
During HCH production, five main isomers are formed: 55–80% α-HCH, 5–14% β-HCH, 8–15% γ-HCH, 2–16% δ-HCH, and, 3–5% ε-HCH (Kutz et al. 1991). Of these, only γ-HCH has the desired insecticidal property, leading to approx. 85% of unwanted by-products. This technical mixture was either applied as is, or after purification to a formulation with a γ-HCH content of > 90% known as lindane. Apart from the contamination of agricultural land through intended application, many sites around the world were contaminated by the mainly uncontrolled dumping of the by-products (Vijgen et al. 2011).
During production of DDT, the two main congeners 4,4′-DDT and 2,4′-DDT make up 65–80% and 15–20% of the technical mixture, respectively (Haller et al. 1945). Only the first has insecticidal properties. When degraded in the soil, congeners retain their 2,4′ or 4,4′ conformation (Ricking and Schwarzbauer 2012). Under aerobic conditions, the primary transformation product (TP) of DDT is DDE (dichlorodiphenyldichloroethylene), and when anaerobic conditions prevail, it is DDD (dichlorodiphenyldichloroethane). In a study by Silva et al. (2019), in which the authors surveyed agricultural soils of 11 member states of the European Union, 4,4′-DDE was the fourth most found pesticide with median and maximum concentrations of 20 and 310 µg·kg−1, respectively.
Both analyte groups are stable, lipophilic, and semi-volatile, and, as such, are distributed widely from their points of origin by water, air, and soil (Garrison et al. 2014; Al Mahmud et al. 2015; Chen et al. 2017; Nežiková et al. 2019), leading to further contaminations of, e.g., ground and surface waters. They can enter the food chain, which leads to their bioaccumulation in fat cells of several types of organisms (Kutz et al. 1991; Nolan et al. 2012) also posing a threat to human health. From fat tissue they can be mobilized into the blood stream and further into breast milk among others (Longnecker et al. 1997; Smith 1999; Stuetz 2006).
HCH isomers as well as DDT and its TPs have been classified as carcinogenic and endocrine disruptors with genotoxic, mutagenic, and teratogenic effects (Kojima et al. 2004; Muñiz et al. 2017; Vandenberg et al. 2020). The latter is also assumed to be linked to obesity-related diseases like type 2 diabetes, and Alzheimer’s disease (Lee et al. 2011; Richardson et al. 2014; Loomis et al. 2015; Evangelou et al. 2016; Elmore and La Merrill 2019).
This endangers food supply, as crops grown on these soils can be contaminated in turn (Lunney et al. 2004; Namiki et al. 2013). Apart from the challenges mentioned above, this might also jeopardize a country’s export of foodstuff, if the contamination exceeds international regulations. Particularly affected groups are subsistence farmers, who rely solely on food grown on their own land to feed themselves. Quite often, this coincides with a lack of knowledge on correct and safe handling, storage, application or disposal of pesticides or even the general hazards associated with these substances (Oesterlund et al. 2014; Jallow et al. 2017; Sapbamrer 2018; Ndayambaje et al. 2019; Mubushar et al. 2019; Deng et al. 2019; Olisah et al. 2020).
In 2015, the nations of the UN defined and adopted 17 goals as calls to action for a more sustainable development on economic, social, and ecological scale in all member states (SDG, Social Development Goal; United Nations 2015). In order to support developing countries to screen and monitor large areas to determine pollution status (SDGs 2, 3, 6 and 15), simple, time, and cost-efficient methods are needed. Better yet, if these methods can be implemented in the framework of capacity building measures (SDG 17).
Generally proven methods to extract pesticides from environmental samples include solvent extraction, Soxhlet, ultrasound-assisted, microwave-assisted, or pressurized liquid extraction (Ayala-Cabrera et al. 2021). Nowadays, new developments in extraction and pre-extraction adhere to the principals of green chemistry and are therefore often miniaturized methods with little solvent consumption and small sample volumes (Ali et al. 2019; Prosen 2019). This includes QuEChERS (quick, easy, cheap, efficient, rugged, and safe; Anastassiades et al. 2003) extraction, originally intended for extraction of pesticide residues in food that has been adapted to other matrices, even soil (Vera et al. 2013). New methods on the basis of QuEChERS or dSPE (dispersive solid-phase extraction) continue to emerge (Eyring et al. 2021; Park et al. 2021). A lot of effort is directed into the development of new sorbents — e.g., on the basis of graphene (Li et al. 2021) or natural materials (Huang et al. 2020) — and new extraction materials like ionic liquids (Llaver et al. 2021) or deep eutectic solvents (Lee et al. 2020) among many others. Still, microextraction techniques both with solvent (e.g., liquid-phase microextraction, LPME; Prosen 2019; Rutkowska et al. 2019) and solventless (e.g., solid-phase microextraction, SPME; Liang et al. 2019; Naccarato and Tagarelli 2019) have been popular choices in extraction or clean-up of samples since their invention in the 1990s, and have seen new developments as well (Da Soares Silva Burato et al. 2020). Among the latter group, SPME is an extraction and preconcentration method (Arthur and Pawliszyn 1990) in which a reusable sorbent-coated fiber is exposed to the sample either directly (immersed) or in the sample’s headspace. During exposure, the fiber accumulates possible analytes which are subsequently thermally desorbed in the injector of a gas chromatograph, without the need of an additional clean-up step. Compared to conventional liquid injection where, usually, only a small portion of the sample extract is injected, everything the fiber extracted is injected with SPME. Especially when performed in headspace, co-extraction of interfering matrix compounds is reduced substantially and longevity of the fiber is increased (Zhang and Pawliszyn 1993; Zhang et al. 1994). SPME can also be used as a clean-up of extracts from solid samples if they have been extracted beforehand by a suitable solvent (Hernandez et al. 2000; Fidalgo-Used et al. 2003).
With the present study, a miniaturized solid–liquid extraction technique is validated (hereinafter referred to as MISOLEX) that combines a modified standard method (ISO 10382 2002) with SPME as simple clean-up and preconcentration to sensitively and selectively analyze pesticides in a large number of soil samples with little effort. Among the method’s merits are low solvent consumption, only one evaporation step, no laborious clean-up steps, and a sample requirement of only 0.5 g. Apart from SPME equipment and the requirement of internal standards (ISs) with appropriate physicochemical properties, it can be done with standard lab equipment and could be an affordable alternative. Larger sets of samples can be managed by this approach allowing for extensive area soil screening for OCPs to assess food security. With small adaptions, suitability of this method for further matrices (e.g., sediment or plant material) is conceivable. A preliminary version of this approach was used by Mukaj et al. (2016) on agricultural soils in Albania.
To prove the adequacy of MISOLEX, quality parameters such as recovery rate, limits of quantitation and detection (LOQ and LOD, respectively), and reproducibility were determined. As QuEChERS has long since been used to analyze soil samples for halogenated pesticides and other contaminants (Vera et al. 2013), the quality parameters were compared to a modified QuEChERS approach by Woldetsadik et al. (2021) that applies SPME as clean-up and preconcentration step as well. In addition, both methods were applied to analyze environmental samples from a contaminated site in Tamarissi municipality, Georgia. The results were used to further validate the performance of the two methods by demonstrating the applicability of the method to environmental samples.
2 Materials and methods
2.1 Chemicals
Organic solvents acetone, methanol, and acetonitrile (all gradient grade for HPLC) were purchased from VWR International (Radnor, PA, USA), petroleum ether (40–60 °C, p.a.) was purchased from Merck GmbH & Co. KG (Darmstadt, Germany), and n-pentane was purchased from Carl Roth GmbH & Co. KG (Karlsruhe, Germany). Standards (purity) of trifluralin (TriF; 99.6%) 2,4′-dichlorodiphenyldichloroethane (DDD, 97.5%), 2,4′-dichlorodiphenyldichloroethylene (DDE, 99%), 2,4′-DDT (99.5%), 4,4′-DDD (99.5%), 4,4′-DDE (98%), 4,4′-DDT (99.5%), 13C-2,4′-DDT (100%), γ-HCH (98.6%), and δ-HCH-D6 (98%) were purchased from Dr. Ehrenstorfer GmbH (Augsburg, Germany). α-HCH (≥ 98%) and δ-HCH (≥ 98%) were purchased from Sigma-Aldrich (St. Louis, MO, USA). β-HCH (99.5%) was obtained from Institute of Industrial Organic Chemistry (Warsaw, Poland). TriF-D14 (98%), 4,4′-DDD-D8 (99.7%), 4,4′-DDE-D8 (99.4%), and α-HCH-D6 (99.2%) were purchased from CDN Isotopes (Pointe Claire, Canada). 13C-4,4′-DDT (99%) was purchased from Cambridge Isotope Laboratories Inc. (Andover, MA, USA). Purity was considered when preparing stock solutions of standards. QuEChERS extraction salts magnesium sulfate (MgSO4) and sodium citrate dibasic sesquihydrate (≥ 99%) were purchased from VWR International and Sigma-Aldrich, respectively. Tri-sodium citrate dihydrate (≥ 99.5%) was obtained from Th. Geyer GmbH & Co. KG (Renningen, Germany) and primary-secondary amine (PSA) from Supelco (Bellefonte, PA, USA). Sodium chloride (NaCl, ≥ 99.5%) for preparation of SPME salt solution was purchased from Carl Roth GmbH & Co. KG (Karlsruhe, Germany). All salts were of analytical grade or better. Ultra-pure water was produced with Milli-Q A10 water purification system (Merck KGaA).
2.2 Preparation of spiked soil samples
Three soil samples were chosen according to their soil organic carbon (SOC) content, to cover representative SOC contents for arable soils in temperate climates: 1.25, 2.27, and 2.74%. An overview of these samples’ features is given in Table 1, while the full characterization is available in the Online Resource (Table S1). These samples were spiked with DDX (2,4′-DDD, 2,4′-DDE, 2,4′-DDT, 4,4′-DDD, 4,4′-DDE, 4,4′-DDT), HCHs (α-, β-, γ-, and δ-HCH), and TriF to a concentration of 50 µg·kg−1 per analyte. For that, n-pentane was added to 120 g of each soil until a supernatant of ca. 1 cm occurred, and standard mixes containing DDX, HCHs, and TriF were injected into the supernatant. After thorough stirring, samples were placed on a horizontal shaker (Swip KS-10, Edmund Bühler GmbH, Bodelshausen, Germany) at 200 rpm and shaken until the solvent had evaporated completely. Then, samples were stored at 8 °C in a refrigerator for 35 days, to simulate aging for generation of field-like samples as suggested by Škulcová et al. (2017).
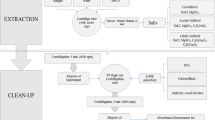
2.3 QuEChERS extraction
As comparative method, the QuEChERS extraction for plant material as described in Woldetsadik et al. (2021) was used. Extraction was done in quintuplet. In brief, 1 g of air-dried (30 °C, 24 h), homogenized sample was weighed into a 50 mL glass centrifuge tube, to which 10 mL of ultrapure water was added and vortexed for 10 s. After letting the samples rehydrate for 10 min, 15 mL acetonitrile was added and samples were shaken on a horizontal shaker for 15 min at 200 rpm (Swip KS-10, Edmund Bühler GmbH). Thereafter, 4 g MgSO4, 1 g NaCl, 1 g sodium citrate (tribasic) dihydrate, and 0.5 g sodium citrate (dibasic) sesquihydrate were added and vortexed for another 10 s. Then, samples were centrifuged for 10 min at 1,000 rpm (207.2 g; Rotanta 460 R, Hettich AG, Bäch, Switzerland). Afterwards, 8 mL of supernatant was transferred into a centrifuge tube containing 1.2 g MgSO4 and 0.2 g PSA. It was again vortexed for 10 s and centrifuged at 2,500 rpm (1,295 g) for 10 min. Of the supernatant, a 4 mL aliquot was transferred into a 20 mL brown glass head space vial; 2 µL of IS mix (see Table S2) was added and vortexed. Under a gentle stream of nitrogen at ambient temperature, the extract was evaporated to dryness and immediately resolubilized with 100 µL of methanol after which it was vortexed for 5 s, and 10 mL of salt solution (200 g NaCl in 1 L ultrapure water) was added. The glass vial was firmly closed with a septum screw cap for subsequent measurement by HS–SPME–GC–MS.
2.4 Miniaturized solid–liquid extraction (MISOLEX)
Extraction was done in quintuplet. 0.5 g of air-dried (30 °C, 24 h), homogenized soil sample was weighed in a 20 mL clear glass head space vial. 5 mL acetone and 5 mL petroleum ether were added and the vial was closed tightly with a screw cap. The sample was shaken on a horizontal shaker for 30 min at 200 rpm (Swip KS-10, Edmund Bühler GmbH) and then centrifuged for 10 min at 1000 rpm (207.2 g; Rotanta 460 R, Hettich AG). The supernatant was transferred into a 20 mL brown glass head space vial. Another 5 mL of petroleum ether was added to the sample, and the process was repeated. The supernatant was added to the one taken before, resulting in approx. 12 mL of extract. An aliquot of 10 mL was transferred to a fresh 20 mL brown glass head space vial, and 2 µL of an IS mix as well as 200 µL of acetonitrile acting as keeper was added. The extract was evaporated to keeper under a gentle stream of nitrogen at ambient temperature. Immediately thereafter, 10 mL of salt solution (200 g NaCl in 1 L ultrapure water) was added. The glass vial was firmly closed with a septum screw cap for subsequent measurement by HS–SPME–GC–MS.
2.5 Note on environmental samples
Even though only 1 g or 0.5 g of sample material is needed for QuEChERS and MISOLEX, respectively, standard protocols concerning representative sampling in the field do apply. That is, it is not correct to assume that only 0.5 g is supposed to be collected from a soil intended as representative sample. Rather, we recommend to collect adequately large (composite) samples to depict the in situ situation, to homogenize them sufficiently and only then to take the subsample of 1 g or 0.5 g intended for extraction from it. Although a larger sample amount has to be taken in the field, the small sample amount intended for pesticide extraction is still beneficial because the remaining soil sample can be used for further characterization (e.g., SOC, particle size analysis, pH).
2.6 Environmental sampling
The study area (108 m2) from which environmental samples were taken was located near Tamarissi municipality, Georgia (N41.444311° E44.76041°) next to an abandoned, ruinous pesticide storage house. Three composite samples (A, B, and C) were collected in 2018, each consisting of five 10-cm cores, sampled with a soil corer (inner diameter 20 mm) in a subplot of 4 m2. Detailed soil characteristics for these samples are available in the Online Resource (Table S1). The soil in the study area can be characterized as Vertic Kastanozem according to the World Reference Base for Soil Resources (IUSS Working Group WRB 2015). After transport to Germany, samples were air-dried (30 °C) for 24 h, homogenized, and sieved (2 mm) to gain the fine earth fraction. Samples were stored at −30 °C until extraction.
2.7 SPME and GC–MS analysis
OCP analysis in soil samples was carried out with a Trace 1310 gas chromatograph (Thermo Fisher Scientific, San Jose, CA, USA), a CombiPAL autosampler (CTC Analytics AG, Zwingen, Switzerland) equipped with a SPME fiber assembly, and an ISQ 7000 mass spectrometer (Thermo Fisher Scientific). For all measurements, a SPME fiber coated with PDMS/DVB (65 µm StableFlex fiber) was used (Sigma-Aldrich, St. Louis, MO, USA). SPME of prepared samples started with a heat-up phase of 5 min to 80 °C in the agitator, followed by headspace extraction at the same temperature for 60 min. After extraction, the fiber was thermally desorbed in splitless mode in the GC injector for 3 min at 260 °C, after which it switched back to a split flow of 30 mL·min−1. At the start and end of each SPME sample cycle, the fiber was desorbed in a needle heater for 7.5 min at 270 °C to prevent potential carry-over of analytes between samples. Chromatographic separation was conducted on a fused silica capillary column (TG-XLBMS: 60 m, 0.25 mm inner diameter, 0.25 μm coating thickness; Thermo Fisher Scientific). Helium (≥ 99.999%, Praxair Inc., Danbury, CT, USA) was used as carrier gas at a constant flow of 1.0 mL·min−1. The initial oven temperature was set to 90 °C and held for 3 min. The temperature was ramped to 150 °C at a rate of 15 °C·min−1. Then, it was ramped to 280 °C at a rate of 5 °C·min−1 and held for 3 min. Quantification was done in selected ion monitoring (SIM) mode based on one target and one qualifier ion. The list of utilized ions and retention times (RTs) is available in the Online Resource (Table S3). The peak areas of analytes in sediment samples were corrected with their respective ISs (Online Resource, Table S2). The according concentration was determined by interpolation of the relative peak areas for each pesticide to standard peak areas of the calibration curve (compare section “Method Validation”).
2.8 Method validation
Linearity, precision, and recovery rate were evaluated using a seven-level calibration curve at working solution concentrations of 0.01, 0.05, 0.1, 0.5, 1.0, 1.5, and 2.0 µg·L−1 for DDX and TriF, and 0.1, 0.5, 1.0, 1.5, 2.0, 2.5, and 3.0 µg·L−1 for HCHs. Calibrations were extended with concentrations of 12.5 and 25 µg·L−1 when analyzing environmental samples to assess linearity in very high concentration ranges. Recoveries of the analytes were calculated per soil as percentage of the expected concentration of 50 µg·kg−1. For determination of LODs and LOQs, the concentration result of each analyte in each (matrix) sample was divided by the corresponding signal to noise ratio of its peak as approximation to noise present at the analyte’s RT. The resulting noise concentrations were multiplied by 3 and 10 to obtain LOD and LOQ, respectively.
Both applied methods make use of a large number of isotopically labeled ISs. For four analytes (2,4′-DDD, 2,4′-DDE, β-HCH, and γ-HCH), instead of isotopologues the IS of a similar congener (4,4′-DDD-D8, 4,4′-DDE-D8, δ-HCH-D6, and α-HCH-D6, respectively) was used. Incidentally, these four analytes demonstrated implausibly high recovery rates. As the IS correction was deemed the cause for this overestimation, an alternative approach for this step was applied. In principle, the RT of an analyte was used as indicator for its physicochemical properties. The closer the RTs of two analytes (or an analyte and IS) are, the closer these two compounds may also be in terms of their physicochemical properties. Because of this assumption, correction factors for each of the four mentioned analytes were calculated proportionally between the nearest two ISs that surrounded each of them. In the case of 2,4′-DDD and 2,4′-DDE, this meant using other ISs as before (4,4′-DDE-D8 and 13C-2,4′-DDT for the first, and δ-HCH-D6 and 4,4′-DDE-D8 for the latter). Further explanations can be found in the Online Resource (Table S3 and Figure S1).
2.9 Statistics and figures
Statistical calculations to check assumptions and determine likeliness between methods were carried out with the computational software R, version 4.0.4 (R Core Team 2020). Shapiro–Wilk test was used to test for assumption of normality. Statistical significance was set at p < 0.05 level. Single pairwise comparisons were done with Welch’s t-test or ANOVA-type rank test by Brunner and Munzel (2013), if assumptions were not met, as it does require neither normal distribution nor homoscedasticity of data. Multiple pairwise comparisons to test for significant difference between three soils were computed with t-tests with Holm correction to correct for inflated familywise error rates. In those cases, where the assumptions were violated, the robust trimmed means bootstrap post hoc test (mcppb20) described by Wilcox (2016) was applied, which is available in the package WRS2 (Mair and Wilcox 2020). Figures were created with OriginPro 2020 (OriginLabs Corp., Northampton, MA, USA).
3 Results
For MISOLEX, LODs and LOQs of DDX congeners were 0.04–0.32 and 0.12–1.05 µg·kg−1, respectively. LOD of TriF was 0.005 µg·kg−1, and LODs of HCHs ranged from 0.27 to 1.16 µg·kg−1. LOQs were 0.02 and between 0.90 and 3.85 µg·kg−1, respectively. Recovery rates were between 65.8 and 172.6% for most DDX, and between 70.2 and 126.1% for TriF and most HCH isomers (Online Resource Fig. S2 and Table S4). Here, γ-HCH and 2,4′-DDD were overestimated with values up to 180.9% and 167.3%, respectively. The average recovery rate for the whole method was 110%. Repeatability expressed as relative standard deviation (RSD) was 23.7%. On a per soil basis, it ranged from 1.3 to 22.4%, and between 6.7 and 20.6% if soils were taken together. γ-HCH, again, demonstrated elevated values between 12.9 and 22.4%.
For the modified QuEChERS method, LODs and LOQs of DDX congeners were between 0.03 and 0.47 µg·kg−1, and between 0.10 and 1.57 µg·kg−1, respectively (Table 2). For TriF and HCHs, LODs were 0.001 and between 0.31 and 1.48 µg·kg−1, respectively, and LOQs were 0.004 and between 1.05 and 4.93 µg·kg−1, respectively. Recovery rates were satisfactory, ranging from 67.8 to 127.0% for most analytes (Online Resource Fig. S2 and Table S5). The rates for 2,4′-DDE and δ-HCH were above 120%, with values up to 169.3% and 140.7%, respectively. The overall average for all analytes was 102.4%. Repeatability (RSD) was 21.8% for the whole method. Separated into analytes, it ranged between 1.2 and 9.4% on a per soil basis and between 3.6 and 8.8% if calculated for all three soils together.
On average, the interpolated IS correction resulted in the preferred range of 80 to 120% for all analytes of both methods (Fig. 1 and Online Resource Tables S6 and S7), the exception being δ-HCH and 2,4′-DDD in the QuEChERS method. The former was not significantly altered and still remained mostly outside of range whereas the latter was slightly worsened. The most drastic effects in terms of recovery rate adjustment (all significant) were shown by 2,4′-DDE of the QuEChERS method and by γ-HCH, β-HCH, and 2,4’-DDD of the MISOLEX method. In terms of repeatability, the RSD of γ-HCH was reduced from 20.6 to 12.8%, while that of 2,4′-DDD was increased from 11.2 to 40.9%. Figures showing means and standard deviations (SDs) of the results discriminated by soil are included in the Online Resource (Figs. S2 and S3).

Boxplots of recovery rates of 11 different analytes in three spiked soils for the two examined methods with five replicates per soil and method (n = 15 per boxplot). Order follows elution sequence. γ-HCH, 2,4′-DDE, and 2,4′-DDD are derived from interpolated internal standard correction. Gray area depicts the targeted recovery rate range between 80 and 120%. Asterisks denote statistically significant difference between the two methods (p < 0.05). Figures discriminating between the three soils are available in the Online Resource (Figs. S2 and S3)
Comparing the results of regularly and interpolated IS-corrected environmental samples shows a very similar pattern (Fig. 2). Interpolation results in considerable reduction in concentration (mean of all three samples) of γ-HCH (−30%), 2,4′-DDE (−60%), and 2,4′-DDD (−74%) for MISOLEX as well as QuEChERS (−39%, −30%, and −40%, respectively). Consequently, the three mentioned analytes were corrected by the interpolation approach in the environmental samples.

Means of concentrations of 11 different analytes determined in three environmental soil samples A, B, and C from Tamarissi municipality, Georgia (n = 5 per bar). Note the different y-axes. Error bars depict standard deviation. Asterisks denote statistically significant difference between the two methods (p < 0.05)
Repeatability of the two methods is assessed by the RSD of the five replicates: averaged across all analytes, RSDs for MISOLEX extraction were 23%, 19%, and 16% for samples A, B, and C, respectively. For QuEChERS, averaged RSDs were 11%, 13%, and 15%, respectively.
Both methods yielded similar results in the same orders of magnitude on a per analyte basis but with significant differences in some cases. TriF was not detected in any sample, regardless of the method. In general, QuEChERS seemed to yield higher values for HCH isomers.
In samples A and B, values of HCHs were higher than those of DDX by at least one order of magnitude. In sample C, differences were not as pronounced between compound groups. Concentrations in the three samples ranged from 0.33 to 849.0 µg·kg−1, or from 0.39 to 1,006.9 µg·kg−1 if analyzed by MISOLEX or QuEChERS, respectively. For detailed concentration values, the reader is referred to the Online Resource (Tables S8 to S11).
4 Discussion
The miniaturized method showed good LODs, LOQs, recovery rates, and repeatability for the three tested soils, with only few exceptions. LOQs were far below the limit of 50 µg·kg−1 recommended by the European Commission (2021) for pesticide analyses in soil. Means of recovery rates and RSDs were mostly between 80 and 120% and below 20%, respectively. The parameters are comparable with previous results reported in other studies for samples from, e.g., a SOC rich garden soil and a clay rich soil from Spain (LOD 0.15–2.2 µg·kg−1, recovery rates 62–92%, SD 0.1–3.4 percentage points (pp); Pinto et al. 2010), two Portuguese strawberry farm soils (LOD 1.7–7.6 µg·kg−1, recovery rates 70–151%, relative SD 1–14 pp; Fernandes et al. 2013), field soils in China (LOD 0.1–0.8 µg·kg−1, recovery rates 80.6–118.3%, SD 2.4–8.4 pp; Ma et al. 2020), mineral and peat soils from vegetable farms in Malaysia (LOD 1 µg·kg−1, LOQ 3.33 µg·kg−1, recovery rates 82–104%; Chai et al. 2013), and an agricultural soil from Gran Canaria (LOD 0.024–6.25 µg·kg−1, LOQ 0.5–20 µg·kg−1, recovery rates 63.7–122.8%; Acosta-Dacal et al. 2021). In fact, the low LODs and LOQs make the presented MISOLEX method not only applicable in screening cases, but constate suitability for use in trace analyses as well.
Differences of recovery rates between the three soils — while being significant in several cases — were mostly relatively small, attesting good reproducibility for each separate case. Preliminary experiments with soils of lower and higher SOC content (between 0.13 and 5.86%) showed similar reproducibility, although these results were not further validated and are therefore not considered here (Online Resource Fig. S4).
The performance of MISOLEX is comparable to that of the modified QuEChERS method, both in terms of sensitivity and recovery. In terms of quickness, however, MISOLEX was found to be faster, as there was no necessity to prepare salt mixtures and subsequently weigh them into each vessel separately. In general, the variability of MISOLEX was higher than that of QuEChERS, both in spiked and environmental samples, which might be due to the lower sample volume that is extracted (0.5 g vs. 1 g).
The overestimation of γ-HCH, 2,4′-DDE and 2,4′-DDD could be alleviated with the interpolated IS correction for these analytes. The recovery rates of γ-HCH showed even less variability than before. In the case of 2,4′-DDD, however, the correction led to an increasing variability. This was less pronounced when regarding the results for each soil separately (Online Resource Fig. S3). The large standard deviation of 2,4′-DDD in Fig. 1 is mainly due to the large differences between the soils.
Recovery rates differed significantly, which was confirmed by the results of the environmental samples. Based on this, the authors can recommend to apply the interpolated IS correction to 2,4′-DDD, 2,4′-DDE, and especially γ-HCH when using MISOLEX and to 2,4′-DDE when using the modified QuEChERS method.
Additionally, several other interpolation approaches of an increasing range between the applied ISs were calculated that would even further reduce the number of ISs needed. But as these did not yield consistent results especially between the two different methods, and would therefore require further experiments, they are not discussed here. Still, this interpolation approach should serve as incentive for other researchers. It could be adapted and made suitable for other applications or ISs and by this help reduce effort and costs.
To the best of the authors’ knowledge, this approach has not been published before. Although the combination of two IS was reported as an option in an analytics software (Thermo Electron PlasmaLab) for ICP-MS (inductively coupled plasma mass spectrometry) as “interpolation technique between masses when multiple ISs” a detailed description of this combination was not given (Niemelä et al. 2005). In the recent past, other studies in analytics of organic contaminants have rather focused on determining the most suitable IS for larger groups of analytes in multiresidue methods. This was done on the basis of, e.g., shared variation, similar chemical properties, and matrix effect (Ueno et al. 2004; Cervera et al. 2010; Tsuchiyama et al. 2017).
ISs are intended to parallel as much as possible the physico-chemical properties of the analytes they are supposed to correct (Poerschmann et al. 1997; Stokvis et al. 2005). Ideally, there would be one IS for each analyte and this — in case of mass spectrometric detection — would be an isotope-labeled version of its analyte counterpart. Such standards are usually quite expensive or even not available at all, which was one reason why interpolation of ISs was examined here. The other fact was the overestimation of those analytes that did not have a very close IS counterpart. As the RT is a function of the substance’s properties, it serves as an indication of similar behavior. Therefore, it could be assumed that greater distances in RT between two substances imply less similarity and less similar behavior. If this applies to the pair of a substance and its IS, the latter is not as representative of losses of the former that may occur during execution of an analysis, possibly resulting in overestimation or underestimation of the substance when corrected. In terms of losses, the most critical part in the methods presented here is the evaporation step, for which vapor pressure and lipophilicity are very important: two properties that are of high influence for RT as well. Another important factor could be the presence of organic residues in the extract, which could serve as sorbent for analytes and ISs that prevents their evaporation or drift with the nitrogen stream when the supernatant level decreases. This is corroborated by results from Woldetsadik et al. (2021) who used the modified QuEChERS method for plant material, where overestimation did not occur. This factor would be very much dependent on hydrophobicity.
Compared to the classic ISO method, MISOLEX features several advantages. The standard method relies on a great number of different steps that increase labor and necessary time per sample, and with that the risks for errors and losses of analyte. As well, the required amount of solvent per sample (ca. 200 mL compared to ca. 15 mL) is by far higher than with MISOLEX. Additionally, each extract has to be washed with water to remove acetone, resulting in 1 L of contaminated washing water per sample. The relatively high sample amount of 20 g compared to 0.5 g renders it unsuitable for all cases, where sample volume is scarce or is impractical to collect, e.g., when a large number of samples need to be shipped. In the ISO method, apart from the washing step, multiple evaporations and transfers are necessary. Process standards that are added before these critical steps could serve as indicator if analyte losses occurred. With the utilization of mere injection standards as it is required in the ISO method, however, only deviations of measurement performance are considered. Taken together, the features of MISOLEX are in accordance with several principles of green analytical chemistry (Gałuszka et al. 2013), e.g., to generate as little waste as possible, to avoid derivatization, to analyze multiple parameters or analytes at once, and to automate and miniaturize.
But MISOLEX is not without disadvantages: The most prominent is the limitation that each extract can only be measured once, as the SPME alters the concentration of analytes in a sample. If a sample is to be analyzed in, e.g., a triplet, there are two options. (1) It could be solvent-extracted the same number of times, at the cost of the maximum number of samples that could be simultaneously extracted. Or (2) the method could be upscaled with larger vessels that can contain thrice as much sample as well as solvent to generate the necessary extract volume for three aliquots in one step. Of course, if analyte concentrations are sufficiently high, the extract could be divided in as many aliquots as adequate before evaporation and SPME extraction without upscaling, also allowing for retention samples. Another point which is just as much an advantage as it is a disadvantage is the use of a large number of isotope-labeled standards. While they greatly enhance the certainty of recovery rates of the analytes, they are also quite costly. They could be exchanged for alternative, unlabeled, and thus cheaper standards, whose suitability needs to be validated beforehand. When using such a low sample amount as presented here, particular care has to be given to the homogenization of samples. The main sample from which the 0.5 g is taken has to be of adequate size and be homogenized sufficiently beforehand to ensure representative results for depiction of a real pollution situation in situ. And finally, as is the case with every new system, the implementation of SPME into existing routines initially requires additional method development after installment. It should be noted, however, that it is possible to use SPME manually if no autosampler is available, but for that, great care to timing needs to be exercised. The implementation of SPME in general enables the adoption of certain other standard methods for analysis of, e.g., water samples (ISO 27108 2010; ISO 17943 2016). It is of course possible to adapt this method to include more OCPs like aldrin, dieldrin, or endosulfan, but also pollutants in general like polychlorinated biphenyls (PCBs) or polycyclic aromatic hydrocarbons (PAHs).
5 Conclusion
The presented miniaturized method allows to quickly and simply determine OCPs in soil samples. With its low sample amount and solvent requirements, it fulfills several principles of green analytical chemistry and is well suited for large area soil screenings or even trace amount analytics. It applies SPME and a single-quadrupole GC–MS, the latter of which is considered standard in terms of mass spectrometry. With more sophisticated instrumentation, e.g., a triple-quadrupole or high-resolution MS, the method’s performance could likely be improved. SPME adds the advantage of a fully automized clean-up procedure. The authors are aware that the appropriate equipment is not available in every laboratory. In such cases, SPME could be operated manually, which would save instrument costs but at the same time remove the advantage of automation.
In our laboratory, the extraction of 40 samples per day was very well manageable. So, for 80 samples, including evaporation and subsequent measurement with calibrations, it took 7 days in total. With more samples to be measured, the next set could be extracted during evaporation and measurement, so that a continuous routine with considerable throughput of samples would be established.
The reproducibility of the MISOLEX method in this case leaves room for improvement, especially in comparison with the very well performing modified QuEChERS method, but is more than sufficient for screening purposes. This could be demonstrated with the application on environmental samples that cover a broad concentration range. MISOLEX might be improved by higher sample amounts, e.g., 1 g instead of 0.5 g, to decrease the impact of the samples’ homogeneity, but this requires further experiments.
In addition, a first test of a new approach to interpolate different ISs was introduced. It uses the analytes’ RTs as indication for likeliness of physical properties. In the present study, it performed well for two out of four analytes and improved their recovery rates. Future studies should evaluate additional samples with differing properties, different combinations of ISs between which interpolation is applied, and should expand the method to more substance groups.
The described approach of a highly efficient analytical method meets the urgent need to further identify the widespread contamination of soils and sediments with OCPs, especially in developing countries.
Availability of data and material
Detailed data together with additional information and figures is supplied alongside the text in the Online Resource on the article’s webpage.
Code availability
Not applicable.
References
Acosta-Dacal A, Rial-Berriel C, Díaz-Díaz R, Del Bernal-Suárez MM, Luzardo OP (2021) Optimization and validation of a QuEChERS-based method for the simultaneous environmental monitoring of 218 pesticide residues in clay loam soil. Sci Total Environ 753:142015. https://doi.org/10.1016/j.scitotenv.2020.142015
Al Mahmud MNU, Khalil F, Rahman MM, Mamun MIR, Shoeb M, Abd El-Aty AM, Park J-H, Shin H-C, Nahar N, Shim J-H (2015) Analysis of DDT and its metabolites in soil and water samples obtained in the vicinity of a closed-down factory in Bangladesh using various extraction methods. Environ Monit Assess 187:743. https://doi.org/10.1007/s10661-015-4965-9
Ali I, Suhail M, Alharbi OML, Hussain I (2019) Advances in sample preparation in chromatography for organic environmental pollutants analyses. J Liq Chromatogr Relat Technol 42:137–160. https://doi.org/10.1080/10826076.2019.1579739
Aliyeva G, Kurkova R, Hovorkova I, Klánová J, Halsall C (2012) Organochlorine pesticides and polychlorinated biphenyls in air and soil across Azerbaijan. Environ Sci Pollut Res Int 19:1953–1962. https://doi.org/10.1007/s11356-012-0944-7
Anastassiades M, Lehotay SJ, Štajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86:412–431. https://doi.org/10.1093/jaoac/86.2.412
Arthur CL, Pawliszyn J (1990) Solid phase microextraction with thermal desorption using fused silica optical fibers. Anal Chem 62:2145–2148. https://doi.org/10.1021/ac00218a019
Ayala-Cabrera JF, Santos FJ, Moyano E (2021) Recent advances in analytical methodologies based on mass spectrometry for the environmental analysis of halogenated organic contaminants. Trends Environ Anal Chem 30:e00122. https://doi.org/10.1016/j.teac.2021.e00122
Brunner E, Munzel U (2013) Nichtparametrische Datenanalyse. Unverbundene Stichproben. (Nonparametric data analysis. Unpaired samples). Springer, Berlin, Heidelberg
Burato DSS, J, Vargas Medina DA, Toffoli AL de, Vasconcelos Soares Maciel E, Mauro Lanças F (2020) Recent advances and trends in miniaturized sample preparation techniques. J Sep Sci 43:202–225. https://doi.org/10.1002/jssc.201900776
Camenzuli L, Scheringer M, Hungerbühler K (2016) Local organochlorine pesticide concentrations in soil put into a global perspective. Environ Pollut 217:11–18. https://doi.org/10.1016/j.envpol.2015.08.028
Cervera MI, Medina C, Portolés T, Pitarch E, Beltrán J, Serrahima E, Pineda L, Muñoz G, Centrich F, Hernández F (2010) Multi-residue determination of 130 multiclass pesticides in fruits and vegetables by gas chromatography coupled to triple quadrupole tandem mass spectrometry. Anal Bioanal Chem 397:2873–2891. https://doi.org/10.1007/s00216-010-3597-8
Chai L-K, Elie F, Jinang C (2013) Determination of 24 pesticides residues in mineral and peat soils by modified QuEChERS method and gas chromatography. Int J Environ Anal Chem 94:519–530. https://doi.org/10.1080/03067319.2013.871713
Chen L, Feng Q, He Q, Huang Y, Zhang Y, Jiang G, Zhao W, Gao B, Lin K, Xu Z (2017) Sources, atmospheric transport and deposition mechanism of organochlorine pesticides in soils of the Tibetan Plateau. Sci Total Environ 577:405–412. https://doi.org/10.1016/j.scitotenv.2016.10.227
Deng Y, Dai H, Zeng M, Guan L, Luo X, Zhang C, Tian J, Zhang J, Li Y, Xi Q, Zhao M, Jiang M, Zhao L (2019) Knowledge and behavior regarding pesticide use: a survey among caregivers of children aged 1–6 years from rural China. Environ Sci Pollut Res Int 26:23037–23043. https://doi.org/10.1007/s11356-019-05560-w
Elmore SE, La Merrill MA (2019) Oxidative phosphorylation impairment by DDT and DDE. Front Endocrinol (lausanne) 10:122. https://doi.org/10.3389/fendo.2019.00122
European Commission (2021) SANTE/2020/12830, Rev. 1: Guidance Document on Pesticide Analytical Methods for Risk Assessment and Post-approval Control and Monitoring Purposes. European Comm: Directorate Gen Health and Consu Protection. https://ec.europa.eu/food/system/files/2021-02/pesticides_mrl_guidelines_2020-12830.pdf. Accessed 15 September 2021
Evangelou E, Ntritsos G, Chondrogiorgi M, Kavvoura FK, Hernández AF, Ntzani EE, Tzoulaki I (2016) Exposure to pesticides and diabetes: a systematic review and meta-analysis. Environ Int 91:60–68. https://doi.org/10.1016/j.envint.2016.02.013
Eyring PRMP, Herrmann SS, Poulsen ME (2021) Multiresidue analysis of 184 pesticides in high-fat fish feed using a new generic extraction method coupled with gas and liquid chromatography-tandem mass spectrometry Appl Biol Chem 64 https://doi.org/10.1186/s13765-021-00610-9
Fernandes VC, Domingues VF, Mateus N, Delerue-Matos C (2013) Multiresidue pesticides analysis in soils using modified QuEChERS with disposable pipette extraction and dispersive solid-phase extraction. J Sep Sci 36:376–382. https://doi.org/10.1002/jssc.201200673
Fidalgo-Used N, Centineo G, Blanco-González E, Sanz-Medel A (2003) Solid-phase microextraction as a clean-up and preconcentration procedure for organochlorine pesticides determination in fish tissue by gas chromatography with electron capture detection. J Chrom A 1017:35–44. https://doi.org/10.1016/S0021-9673(03)01321-9
Gałuszka A, Migaszewski Z, Namieśnik J (2013) The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. TrAC Trends Anal Chem 50:78–84. https://doi.org/10.1016/j.trac.2013.04.010
Garrison AW, Cyterski M, Roberts KD, Burdette D, Williamson J, Avants JK (2014) Occurrences and fate of DDT principal isomers/metabolites, DDA, and o, p’-DDD enantiomers in fish, sediment and water at a DDT-impacted Superfund site. Environ Pollut 194:224–234. https://doi.org/10.1016/j.envpol.2014.07.025
Haller HL, Bartlett PD, Drake NL, Newman MS, Cristol SJ, Eaker CM, Hayes RA, Kilmer GW, Magerlein B, Mueller GP, Schneider A, Wheatley W (1945) The chemical composition of technical DDT 1. J Am Chem Soc 67:1591–1602. https://doi.org/10.1021/ja01225a058
Hernandez F, Beltran J, Lopez FJ, Gaspar JV (2000) Use of solid-phase microextraction for the quantitative determination of herbicides in soil and water samples. Anal Chem 72:2313–2322. https://doi.org/10.1021/ac991115s
Huang H, Li J, Zhang Y, Chen W, Ding Y, Chen W, Qi S (2020) How persistent are POPs in remote areas? A case study of DDT degradation in the Qinghai-Tibet Plateau. China Environ Pollut 263:114574. https://doi.org/10.1016/j.envpol.2020.114574
IUSS Working Group WRB (2015) World reference base for soil resources 2014, update 2015 International soil classification system for naming soils and creating legends for soil maps (World soil resources reports, 106). FAO, Rome
ISO 10382 (2002) Soil quality — determination of organochlorine pesticides and polychlorinated biphenyls — gas-chromatographic method with electron capture detection
ISO 17943 (2016) Water quality — determination of volatile organic compounds in water — method using headspace solid-phase micro-extraction (HS-SPME) followed by gas chromatography-mass spectrometry (GC-MS)
ISO 27108, (2010) Water quality — determination of selected plant treatment agents and biocide products — method using solid-phase microextraction (SPME) followed by gas chromatography-mass spectrometry (GC-MS)
Jallow MFA, Awadh DG, Albaho MS, Devi VY, Thomas BM (2017) Pesticide knowledge and safety practices among farm workers in Kuwait: results of a survey Int J Environ Res Public Health 14 https://doi.org/10.3390/ijerph14040340
Kojima H, Katsura E, Takeuchi S, Niiyama K, Kobayashi K (2004) Screening for estrogen and androgen receptor activities in 200 pesticides by in vitro reporter gene assays using Chinese hamster ovary cells. Environ Health Perspect 112:524–531. https://doi.org/10.1289/ehp.6649
Kutz FW, Wood PH, Bottimore DP (1991) Organochlorine pesticides and polychlorinated biphenyls in human adipose tissue. Rev Environ Contam Toxicol 120:1–82. https://doi.org/10.1007/978-1-4612-3080-9_1
Lee D-H, Steffes MW, Sjödin A, Jones RS, Needham LL, Jacobs DR (2011) Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS One 6:e15977. https://doi.org/10.1371/journal.pone.0015977
Lee J, Kim H, Kang S, Baik N, Hwang I, Chung DS (2020) Applications of deep eutectic solvents to quantitative analyses of pharmaceuticals and pesticides in various matrices: a brief review. Arch Pharm Res 43:900–919. https://doi.org/10.1007/s12272-020-01266-7
Li H, Zeng EY, You J (2014) Mitigating pesticide pollution in China requires law enforcement, farmer training, and technological innovation. Environ Toxicol Chem 33:963–971. https://doi.org/10.1002/etc.2549
Li Y, Lan S, Zhu T (2021) Recent advances of graphene-based sorptive materials in extraction: a review. TrAC Trends Anal Chem 142:116319. https://doi.org/10.1016/j.trac.2021.116319
Liang D, Liu W, Raza R, Bai Y, Liu H (2019) Applications of solid-phase micro-extraction with mass spectrometry in pesticide analysis. J Sep Sci 42:330–341. https://doi.org/10.1002/jssc.201800804
Llaver M, Oviedo MN, Fiorentini EF, Quintas PY, Wuilloud RG (2021) Analytical developments and applications of ionic liquids for environmental studies. Trends Environ Anal Chem 31:e00131. https://doi.org/10.1016/j.teac.2021.e00131
Longnecker MP, Rogan WJ, Lucier G (1997) The human health effects of DDT (dichlorodiphenyltrichloroethane) and PCBS (polychlorinated biphenyls) and an overview of organochlorines in public health. Annu Rev Public Health 18:211–244. https://doi.org/10.1146/annurev.publhealth.18.1.211
Loomis D, Guyton K, Grosse Y, El Ghissasi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Mattock H, Straif K (2015) Carcinogenicity of lindane, DDT, and 2,4-dichlorophenoxyacetic acid. Lancet Oncol 16:891–892. https://doi.org/10.1016/S1470-2045(15)00081-9
Lunney AI, Zeeb BA, Reimer KJ (2004) Uptake of weathered DDT in vascular plants. Potential for Phytoremediation Environ Sci Technol 38:6147–6154. https://doi.org/10.1021/es030705b
Ndayambaje B, Amuguni H, Coffin-Schmitt J, Sibo N, Ntawubizi M, VanWormer E (2019) Pesticide application practices and knowledge among small-scale local rice growers and communities in Rwanda: a cross-sectional study Int J Environ Res Public Health 16 https://doi.org/10.3390/ijerph16234770
Ma T, Li S, Li Y, Li X, Luo Y (2020) Simultaneous determination and exposure assessment of six common pesticides in greenhouses through modified QuEChERS and gas chromatography–mass spectrometry. Stoch Environ Res Risk Assess 34:1967–1982. https://doi.org/10.1007/s00477-020-01844-7
Mair P, Wilcox R (2020) Robust statistical methods in R using the WRS2 package. Behav Res Methods 52:464–488. https://doi.org/10.3758/s13428-019-01246-w
Mubushar M, Aldosari FO, Baig MB, Alotaibi BM, Khan AQ (2019) Assessment of farmers on their knowledge regarding pesticide usage and biosafety. Saudi J Biol Sci 26:1903–1910. https://doi.org/10.1016/j.sjbs.2019.03.001
Mukaj M, Mai S, Cara M, Düring R-A (2016) Status of soil organic matter and levels of DDT residues in some agricultural soils in Albania. Glob J Agric Res 4:39–47
Muñiz S, Gonzalvo P, Valdehita A, Molina-Molina JM, Navas JM, Olea N, Fernández-Cascán J, Navarro E (2017) Ecotoxicological assessment of soils polluted with chemical waste from lindane production: use of bacterial communities and earthworms as bioremediation tools. Ecotoxicol Environ Saf 145:539–548. https://doi.org/10.1016/j.ecoenv.2017.07.070
Naccarato T (2019) Recent applications and newly developed strategies of solid-phase microextraction in contaminant analysis: through the environment to humans. Separations 6:54. https://doi.org/10.3390/separations6040054
Namiki S, Otani T, Seike N (2013) Fate and plant uptake of persistent organic pollutants in soil. Soil Sci Plant Nutrition 59:669–679. https://doi.org/10.1080/00380768.2013.813833
Nežiková B, Degrendele C, Čupr P, Hohenblum P, Moche W, Prokeš R, Vaňková L, Kukučka P, Martiník J, Audy O, Přibylová P, Holoubek I, Weiss P, Klánová J, Lammel G (2019) Bulk atmospheric deposition of persistent organic pollutants and polycyclic aromatic hydrocarbons in Central Europe. Environ Sci Pollut Res Int 26:23429–23441. https://doi.org/10.1007/s11356-019-05464-9
Niemelä M, Kola H, Perämäki P, Piispanen J, Poikolainen J (2005) Comparison of microwave-assisted digestion methods and selection of internal standards for the determination of Rh, Pd and Pt in dust samples by ICP-MS. Microchim Acta 150:211–217. https://doi.org/10.1007/s00604-005-0356-1
Nolan K, Kamrath J, Levitt J (2012) Lindane toxicity: a comprehensive review of the medical literature. Pediatr Dermatol 29:141–146. https://doi.org/10.1111/j.1525-1470.2011.01519.x
Oesterlund AH, Thomsen JF, Sekimpi DK, Maziina J, Racheal A, Jørs E (2014) Pesticide knowledge, practice and attitude and how it affects the health of small-scale farmers in Uganda: a cross-sectional study. Afr Health Sci 14:420–433. https://doi.org/10.4314/ahs.v14i2.19
Olisah C, Okoh OO, Okoh AI (2020) Occurrence of organochlorine pesticide residues in biological and environmental matrices in Africa: a two-decade review. Heliyon 6:e03518. https://doi.org/10.1016/j.heliyon.2020.e03518
Park E, Lee J, Lee J, Lee J, Lee HS, Shin Y, Kim J-H (2021) Method for the simultaneous analysis of 300 pesticide residues in hair by LC-MS/MS and GC-MS/MS, and its application to biomonitoring of agricultural workers. Chemosphere 277:130215. https://doi.org/10.1016/j.chemosphere.2021.130215
Pinto CG, Laespada MEF, Martín SH, Ferreira AMC, Pavón JLP, Cordero BM (2010) Simplified QuEChERS approach for the extraction of chlorinated compounds from soil samples. Talanta 81:385–391. https://doi.org/10.1016/j.talanta.2009.12.013
Poerschmann J, Zhang Z, Kopinke F-D, Pawliszyn J (1997) Solid phase microextraction for determining the distribution of chemicals in aqueous matrices. Anal Chem 69:597–600. https://doi.org/10.1021/ac9609788
Prosen H (2019) Applications of hollow-fiber and related microextraction techniques for the determination of pesticides in environmental and food samples—a mini review. Separations 6:57. https://doi.org/10.3390/separations6040057
R Core Team (2020) R: a language and environment for statistical computing. Version 4.0.4, Vienna, Austria. R Foundation for Stats Comp. https://www.R-project.org/
Richardson JR, Roy A, Shalat SL, von Stein RT, Hossain MM, Buckley B, Gearing M, Levey AI, German DC (2014) Elevated serum pesticide levels and risk for Alzheimer disease. JAMA Neurol 71:284–290. https://doi.org/10.1001/jamaneurol.2013.6030
Ricking M, Schwarzbauer J (2012) DDT isomers and metabolites in the environment: an overview. Environ Chem Lett 10:317–323. https://doi.org/10.1007/s10311-012-0358-2
Rutkowska M, Płotka-Wasylka J, Sajid M, Andruch V (2019) Liquid–phase microextraction: a review of reviews. Microchem J 149:103989. https://doi.org/10.1016/j.microc.2019.103989
Sapbamrer R (2018) Pesticide use, poisoning, and knowledge and unsafe occupational practices in Thailand. New Solut 28:283–302. https://doi.org/10.1177/1048291118759311
Sharov P, Dowling R, Gogishvili M, Jones B, Caravanos J, McCartor A, Kashdan Z, Fuller R (2016) The prevalence of toxic hotspots in former Soviet countries. Environ Pollut 211:346–353. https://doi.org/10.1016/j.envpol.2016.01.019
Silva V, Mol HGJ, Zomer P, Tienstra M, Ritsema CJ, Geissen V (2019) Pesticide residues in European agricultural soils - a hidden reality unfolded. Sci Total Environ 653:1532–1545. https://doi.org/10.1016/j.scitotenv.2018.10.441
Škulcová L, Hale SE, Hofman J, Bielská L (2017) Laboratory versus field soil aging: Impact on DDE bioavailability and sorption. Chemosphere 186:235–242. https://doi.org/10.1016/j.chemosphere.2017.07.159
Smith D (1999) Worldwide trends in DDT levels in human breast milk. Int J Epidemiol 28:179–188. https://doi.org/10.1093/ije/28.2.179
Stokvis E, Rosing H, Beijnen JH (2005) Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: necessity or not? Rapid Commun Mass Spectrom 19:401–407. https://doi.org/10.1002/rcm.1790
Stuetz W (2006) Global surveillance of DDT and DDE levels in human tissues. Int J Occup Med Environ Health 19:83. https://doi.org/10.2478/v10001-006-0009-6
Tepanosyan G, Sahakyan L, Belyaeva O, Beglaryan M, Pipoyan D, Hovhannisyan A, Saghatelyan A (2020) Studying DDTs in agricultural soils of selected rural communities of Armenia. Acta Geochim 39:487–496. https://doi.org/10.1007/s11631-019-00376-4
Tsuchiyama T, Katsuhara M, Nakajima M (2017) Compensation of matrix effects in gas chromatography-mass spectrometry analysis of pesticides using a combination of matrix matching and multiple isotopically labeled internal standards. J Chromatogr A 1524:233–245. https://doi.org/10.1016/j.chroma.2017.09.072
Ueno E, Oshima H, Saito I, Matsumoto H, Yoshimura Y, Nakazawa H (2004) Multiresidue Analysis of Pesticides in Vegetables and Fruits by Gas Chromatography/Mass Spectrometry after Gel Permeation Chromatography and Graphitized Carbon Column Cleanup. J AOAC Int 87:1003–1015. https://doi.org/10.1093/jaoac/87.4.1003
United Nations (2001–05–22) Stockholm convention on persistent organic pollutants. No. 40214. Treaty Series 2256:119–403. https://treaties.un.org/Pages/ViewDetails.aspx?src=TREATY&mtdsg_no=XXVII-15&chapter=27. Accessed 24 February 2021
United Nations (2015) Transforming our World: The 2030 Agenda for Sustainable Development. A/RES/70/1. https://sdgs.un.org/2030agenda. Accessed 01 February 2021
Vandenberg LN, Najmi A, Mogus JP (2020) Agrochemicals with estrogenic endocrine disrupting properties: Lessons Learned? Mol Cell Endocrinol 518:110860. https://doi.org/10.1016/j.mce.2020.110860
Vera J, Correia-Sá L, Paíga P, Bragança I, Fernandes VC, Domingues VF, Delerue-Matos C (2013) QuEChERS and soil analysis An Overview Sample Prep 1 https://doi.org/10.2478/sampre-2013-0006
Vijgen J, Abhilash PC, Li YF, Lal R, Forter M, Torres J, Singh N, Yunus M, Tian C, Schäffer A, Weber R (2011) Hexachlorocyclohexane (HCH) as new Stockholm Convention POPs—a global perspective on the management of Lindane and its waste isomers. Environ Sci Pollut Res Int 18:152–162. https://doi.org/10.1007/s11356-010-0417-9
Wilcox RR (2016) Introduction to robust estimation and hypothesis testing. Academic Press, Amsterdam
Woldetsadik D, Simon MP, Knuth D, Hailu H, Gebresilassie A, Dejen A, Düring R-A (2021) Exposure to DDT and HCH congeners and associated potential health risks through khat (Catha edulis) consumption among adults in South Wollo, Ethiopia. Environ Geochem Health. https://doi.org/10.1007/s10653-021-00846-w
Zhang Z, Pawliszyn J (1993) Headspace solid-phase microextraction. Anal Chem 65:1843–1852. https://doi.org/10.1021/ac00062a008
Zhang Z, Yang MJ, Pawliszyn J (1994) Solid-phase microextraction. A solvent-free alternative for sample preparation. Anal Chem 66:844A-853A. https://doi.org/10.1021/ac00089a001
Funding
This work was conducted in the framework of project POPcont (Environmental Health Risks of POP contaminated Soils in the South-Caucasus Region: Monitoring and Mitigation), funded by the German Federal Ministry of Education and Research (BMBF) (funding code: 01DK17030A). Open Access funding was enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: Rolf-Alexander Düring; methodology: Marcel Pierre Simon, Dennis Knuth, Leonard Böhm, Katrin Wiltschka, Marlene Schatz, Rolf-Alexander Düring; software: not applicable; validation: Marcel Pierre Simon, Dennis Knuth, Leonard Böhm, Katrin Wiltschka, Marlene Schatz, Rolf-Alexander Düring; formal analysis: Marcel Pierre Simon, Dennis Knuth, Katrin Wiltschka; investigation: Marcel Pierre Simon, Dennis Knuth, Leonard Böhm, Katrin Wiltschka, Marlene Schatz, Rolf-Alexander Düring; resources: Rolf-Alexander Düring; data curation: Marcel Pierre Simon; writing — original draft: Marcel Pierre Simon; writing — review and editing: Marcel Pierre Simon, Dennis Knuth, Leonard Böhm, Katrin Wiltschka, Marlene Schatz, Rolf-Alexander Düring; visualization: Marcel Pierre Simon; supervision: Rolf-Alexander Düring; project administration: Rolf-Alexander Düring; funding acquisition: Rolf-Alexander Düring.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Responsible Editor: Yanzheng Gao
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Simon, M.P., Knuth, D., Böhm, L. et al. A miniaturized method for fast, simple, and sensitive pesticide analysis in soils. J Soils Sediments 22, 496–508 (2022). https://doi.org/10.1007/s11368-021-03080-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-021-03080-0