
Abstract
Radiolabeled amino acids, their derivatives and peptides have a broad scope of application and can be used as receptor ligands, as well as enzyme substrates for many different diseases as radiopharmaceutical tracers. Over the past few decades, the application of molecular imaging techniques such as positron emission tomography (PET) has gained considerable importance and significance in diagnosis in today’s advanced health care. Next to that, the availability of cyclotrons and state-of-the-art radiochemistry facilities has progressed the production of imaging agents enabling the preparation of many versatile PET radiotracers. Due to many favorable characteristics of radiolabeled amino acids and peptides, they can be used for tumor staging and monitoring the progress of therapy success, while aromatic amino acids can be employed as PET tracer to study neurological disorders. This review provides a comprehensive overview of radiosynthetic and enzymatic approaches towards carbon-11 amino acids, their analogues and peptides, with focus on stereoselective reactions, and reflects upon their clinical application.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the past few decades, the application of molecular imaging techniques has become widely available for all fields of today’s health care. Especially, molecular imaging techniques using tracers for single photon emission computed tomography (SPECT) and positron emission tomography (PET) have gained great attention for the study of disease biology in vivo due to their sensitivity and specificity. Next to that, the availability of cyclotrons, coupled with a great variety of radiolabeling methods and automated synthesis modules for the production of PET radiopharmaceuticals, has enabled the preparation of the most frequently used PET radiotracer 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) as well as a large variety of other PET tracers like receptor ligands or enzyme substrates. Among all classes of PET tracers, radiolabeled amino acids (AAs), their derivatives and peptides have a broad scope of clinical applications [1].
Natural, L-AAs play a crucial role in virtually all biologic processes, e.g., protein synthesis, cell signaling, precursors for biomolecular transmission, and as neurotransmitters [2, 3]. Their constant and dynamic degradation and recirculation results in metabolic intermediates, such as ureas, fatty acids, or glucose. Furthermore, AAs are used as metabolic fuel or reused by other tissues or cells. It is known that AAs can diffuse into cells; however, the main transport of AAs into cells occurs through specific membrane-associated proteins, so-called AA-transporters [4]. Due to the imperative role of generally existing or tissue-specific AA-transport systems and their consumption in many cellular processes, radiolabeled AAs have become an appealing class of compounds to be used as PET tracer to study cancer biology, considering that malignant tissue often relies on increased AA uptake [2, 5]. Next to cancer imaging with radiolabeled AAs, aromatic AAs can be employed as PET tracer to study neurological disorders. Additionally, the use of AAs as PET tracers often result in high contrast images which are not distorted by tissue inflammation, as is known for glucose-based PET tracer [18F]FDG. The brain, heart, and inflamed areas use glucose as nutrient; therefore, these regions are difficult to distinguish from metastasis in [18F]FDG PET studies [2, 6, 7]. The radionuclides used for PET to label natural and unnatural AAs include C-11 (t1/2 20.4 min), N-13 (t1/2 10.0 min), F-18 (t1/2 1.8 h), and I-124 (t1/2 4.2 days) [3].
Peptides, typically consisting of less than 50 AAs, are another important class of compounds to be used as PET tracer, and have gained increasing importance as imaging agents. Furthermore, they generally have a high affinity and specificity for their cellular target, which often occur in nanomolar concentrations, in diseased tissue [8, 9]. Due to many favorable properties for molecular imaging, such as advantageous pharmacokinetics and specific tumor targeting characteristics, including the overexpression of peptide receptors on the tumor cells, radiolabeled peptides have an important impact not only in PET imaging of cancer, but also in targeted radionuclide therapy [10]. Peptide-based radiopharmaceuticals have been successfully employed for imaging of somatostatin (SST) and bombesin or cholecystokinin/gastrin receptor overexpressing tumors, all common human cancers [10,11,12,13,14].
Contemporary radiolabeling methods to label peptides with radiometals such as Ga-68 (t1/2 1.1 h), Tc-99m (t1/2 6.0 h), Cu-64 (t1/2 12.7 h), In-111 (t1/2 67.2 h), and Lu-177 (t1/2 160.8 h) require chemical modifications introducing a bulky bifunctional chelators on the peptide in order to incorporate a radiometal. For non-metal isotope F-18 (t1/2 1.8 h), as well as C-11 (t1/2 20.4 min), bifunctional prosthetic groups are installed on the peptide allowing conjugation to the biologically relevant peptide [15,16,17,18,19]. Present peptide radiolabeling techniques require structural modification of the lead structure, consequently influencing the pharmacokinetics, receptor affinity, and/or selectivity, usually in a negative manner [12, 16]. Ideally, a radionuclide should be added directly into a lead peptide structure, preserving an unmodified, so-called “native” peptide without changing its chemical structure and thus its biological behavior. Direct labeling of small peptides has become increasingly popular; however, site-specific labeling without disrupting key target interactions remains a challenge.
The presence of carbon in organic molecules makes the PET isotope C-11 (99 % β+, 0.96 MeV) [20] an attractive and valuable radionuclide for PET tracer development and thus also for the labeling of AAs, their derivatives and peptides. Isotopologue labeling without structural modifications conveniently includes C-11 as a normal constituent in well-characterized molecules of known biological properties [6, 21,22,23]. Currently, there is a great toolbox available for labeling AAs with C-11, which includes viable methods using two major C-11 precursors produced directly by cyclotrons: [11C]carbon dioxide ([11C]CO2) and [11C]methane ([11C]CH4), and their transformed products, like [11C]methyl iodide ([11C]CH3I), [11C]methyl triflate ([11C]CH3OTf), [11C]cyanide, or other 11C-alkylating agents [23]. The half-life of C-11 is, with the current radiolabeling techniques, long enough to perform multi-step synthesis and biological PET tracer analysis [24]. Nevertheless, carbon-11-labeled radiotracers necessitate production at onsite radiochemistry facilities which includes a cyclotron. Besides synthetic challenges, the clinical advantage is predominant allowing multi-tracer administrations in patients in a single day. Moreover, C-11-labeled radiotracers often provide a better match of physical and in vivo peptide half-live compared to long-lived radiometals.
The merits of radiosynthetic methods enabling to label a variety of [11C]AA and their derivatives, and their value as PET tracer, is well-recognized, exemplified by L-[11C]methionine ([11C]MET), which is studied extensively for tumor imaging. On the other hand, C-11-labeled small peptides have received less attention until recently, where multiple independent papers described different methods for C-11 labeling of peptides, though clinical translation has thus far been inadequately examined [10, 15, 19]. With current achievements, the scope of C-11 labeling of peptides ranges from small proteins that have been successfully labeled with C-11 and used for applications such as blood flow permeability studies, as well as C-11-labeled peptides for inflammation and tumor imaging [25,26,27,28].
To the best of our knowledge, we describe all currently available synthesis strategies for the radiosynthesis of [11C]AAs, their most common derivatives and peptides, with an emphasis on stereoselective synthesis. Besides production options for these classes of radiopharmaceuticals, we will also discuss the clinical application and value of [11C]AA, and we will identify remaining challenges in radiosynthesis procedures for widespread adoption of these PET tracers.
Carbon-11 Amino Acid Labeling
As depicted in Fig. 1, despite the fact that theoretically all AAs can be labeled with C-11, the numerous attempts to radiolabel these molecules have shown that there is not one preferred strategy. All described methodologies to obtain radiolabeled AAs share both advantages and disadvantages. Synthesis strategies to obtain C-11-labeled AAs have to consider the labeling position and chirality in the AA structure, as well as the metabolic stability for intended biological applications. All these have resulted in the development of distinct strategies to radiolabel [11C]AAs for imaging purposes.

Short historical overview of general synthetic routes to yield carbon-11-labeled alanine and phenylalanine in two possible positions.
Carbon-11 Amino Acid Labeling of [1-11C]Carboxyl-Function
Bucherer-Strecker Synthesis
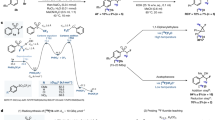
The Strecker reaction, and its modified Bucherer-Strecker hydantoin synthesis, is one of the earliest one-pot and atom economic multi-component reactions for AA synthesis in general. The simplicity of the synthesis—simple mixing of the reagents acetaldehyde, ammonia (or amines) and hydrogen cyanide resulting in an α-aminonitrile adduct and its subsequent hydrolysis—has attracted radiochemists to synthesize [11C]AAs using [11C]carbonyls or [11C]cyanide. The first approaches described the use of [11C]hydrogen cyanide ([11C]HCN) to yield the corresponding [11C]aminonitrile [29], whereas the second approach employed [11C]CO2 which reacted with α-lithiated precursor [30]. In both cases, subsequent hydrolysis affords the corresponding AA as exemplified by Hayes et al. [1-11C]Aminocyclopentanecarboxylic acid (ACPC) was first obtained by a two-step high temperature reaction of cylcopentanone with [11C]HCN via a [11C]hydantoin anionic salt [29]. Unfortunately, when this methodology was developed, most procedures to obtain C-11-labeled AAs required addition of “carrier-added” cyanide to the reaction mixture which resulted in diminished molar activity (AM) of the final radiopharmaceutical. In the following years after the first publications, the Bucherer-Strecker radiolabeling method was further optimized, providing successful radiosynthesis of [11C]valine [31], [11C]tryptophan [32], [11C]leucine [32], and α-[11C]methylvaline [32] within 1 h including hydrolysis, final tracer purification, and formulation. Subsequently, advances in technology generated other modified routes, for example the “no-carrier-added” synthesis by Iwata et al., furnishing good yields for [11C]phenylalanine, [11C]valine, and [11C]leucine. Here, the starting material amino sulfonate reacted with on-line produced [11C]HCN [33]. Due to the high amount of precursor, around 400 mg (~ 2 mmol) of amino sulfonate, the application of this method might not be preferred, however could be realized using aldehydes and unhindered ketones, but not sterically hindered ketones (e.g., for the synthesis of α-[11C]methylphenylglycine). However, modifications by Studenov et al. in 2003 dismissed “carrier-added” cyanide and high amounts of amino sulfonates (only 4 mg, 20 μmol, needed). The synthesis of [11C]phenylalanine and [11C]tyrosine was performed at a slightly elevated temperature and after acidic hydrolysis AAs were obtained molar activities as high as 74–111 GBq μmol−1, as depicted in Fig. 2a [34].

Non-stereoselective (a) and stereoselective (b) modified Bucherer-Strecker synthesis.
Due to the general increased knowledge on radiochemical methods, this strategy for C-11-labeled AA synthesis gradually attracted more attention and finally resulted in an alternative radiosynthesis, the so-called Zelinski-Stadnikoff variation. Prenant et al. described the unique labeling of an AA in the 2-position of the AA using the C-11 reagent [11C]acetone, obtained from [11C]CO2 after reaction with methyllitihium (CH3Li) [35]. An advantage of the Zelinski-Stadnikoff compared to the Bucherer reaction is the one-pot procedure in aqueous media. Utilizing this method, the labeling of α-[2-11C]aminoisobutyric acid was described in a radiochemical yield (RCY) of 4 % with AM of 16–20 GBq μmol−1 in approximately 50 min. Inconveniently, side reactions have occurred and two known by-products were reported: [11C]lactic acid for incomplete conversion and/or [11C]tert-butanol for an excessive concentration of CH3Li.
Recently, an automated synthesis of no-carrier added [11C]carbonyl-labeled AAs using a modified Strecker synthesis has been reported. To overcome stated limitations such as “carrier” cyanide, the need for intermediate purification and reactions conducted in sealed tubes (which are mostly incompatible for automation due to high temperature and pressures required), Xing et al. implemented an alternative design to label the carbonyl moiety by forming an [11C]amino nitrile with [11C]NaCN. A fully automated synthesis for [11C]sarcosine has been achieved in a non-decay corrected (n.d.c.) 1 % RCY, calculated from [11C]HCN, and radiochemical purity (RCP) of > 90 % with AM 55.5 GBq mmol−1 in 40 min, sufficient for preliminary imaging. Furthermore, application of this method towards other C-11 labeled AAs was achieved with variable success for methionine (5 % RCY; n.d.c. based on [11C]HCN), glycine (14 % RCY), and N-phenylglycine (4 % radiochemical conversion (RCC)Footnote 1) [36].
Nonetheless, very recently, Hienzsch et al. have focused on developing a simple and efficient synthesis of enantiomerically pure [11C]AA using a microfluidics platform and extended the use of an asymmetric version of the Strecker synthesis. As depicted in Fig. 2b, [11C]HCN is bubbled through a sulfinylimine precursor (1–3 mg, 4–20 μmol) in 1,4-dioxane/methanol in the HCN chamber of a microfluidic chip. Next, hydrolysis occurs in a separate chamber at elevated temperature, followed by a purification using a cation exchange cartridge and enzymatic enrichment by the D-α-amino acid oxidase (D-AAO) to exclusively obtain L-AA (enantiomeric excess (ee) of 100 %) [37].
In summary, all described variations of the Strecker synthesis lead to a racemic mixture of C-11-labeled AAs where further enzymatic resolution to obtain enantiopure product is necessary. Time-consuming and low-yielding chiral purification to obtain a single stereoisomer, has, to date, hampered the application of Bucherer-Strecker synthesis or its variations as usually only one enantiomer displays favorable biological activity.
[1-11C]Carboxylations Using [11C]Carbon Dioxide
[11C]CO2, mostly the first C-11-labeled reagent formed in a cyclotron target, is also a highly convenient reagent used to form C-11-labeled AAs. A route affording the [1-11C]carboxyl-labeled position, based on carboxylation of α-lithioisocyanides with [11C]CO2, was for the first time reported in 1976 by Vaalburg et al. [26]. Application of the direct carboxylation requires the activation of the α-carbon, achieved by the introduction of the isocyanide functionality on the N-terminus. At the same time, abstraction of the proton on the α-carbon with n-butyl lithium (n-BuLi) followed by hydrolysis leads to the desired amine. Using this route, as described in Fig. 3a, a vast number of racemic [11C]AAs have been described in literature, namely phenylalanine [26], ornithine and lysine [38], tyrosine [28, 30], proline [39] and phenylglycine [26], and achiral glycine [25, 26]. The [1-11C]proline has been reported in 12–18 % RCY with a RCP of > 95 % in total synthesis time of 45 min [39].

[1-11C]carboxylation reactions with activated precursors.
As mentioned, direct use of [11C]CO2 results in shorter synthesis time which improves the non-decay corrected RCY. This was achieved by using lower precursor quantities, performing the synthesis in a “no-carrier-added” manner, and use of stronger base (e.g., sec-butyl lithium) to lithiate the precursor. Furthermore, essential technical details, such as stirring, temperature control, and successive addition of chemicals, had to be fine-tuned as well. By adjusting the conditions, Bolster et al. successfully synthesiezed [1-11C]tyrosine [40], 3,4-[11C]dihydroxyphenylalanine ([11C]DOPA) [27] and [1-11C]MET [41, 42] with RCP > 94 % within 35 min, followed by chiral resolution to obtain the desired enantiomer as PET tracer in RCY of 10–15 %.
[1-11C]Carboxylation of N-(α-Lithioalkyl)Oxazolidinones
There has been continuous interest in the refinement of the [11C]CO2-fixation method towards chiral syntheses in order to overcome the challenges of time-consuming and low-yielding chiral separation. Driven by the success of C-11-labeled methionine, firstly C-11 carboxylation using the N-(α-lithioalkyl)oxazolidinone reagents have been thoroughly explored to obtain the L-enantiomer of [11C]MET [43]. This procedure successfully yielded L-[11C]MET in excellent ee of 95 % and RCY of 85 %, with short synthesis times of 35–40 min [44], and is now used for PET imaging studies. Using this approach, the general starting material, N-(α-lithioalkyl)oxazolidinon, was transformed by a tin-lithium exchange to the corresponding N-(α-stannylalkyl)oxazolidinon, which equilibrated rapidly to one diastereomer. This intermediate was further carboxylated using [11C]CO2 to the desired and diastereopure (α-carboxyalkyl)oxazolidinon, presented in Fig. 3b. Finally, the chiral auxiliary was removed by a Birch-type reduction to yield the unprotected and entantiomerically pure C-11-labeled AA, which was purified by ion exchange chromatography. In view of the good results, this method has been extended to C-11-labeled L-leucine, L-alanine, and L-homocysteine, accomplished with an overall yield of 13–33 % with 92–95 % ee [45].
To conclude, oxazolidinone moiety precursors necessitate longer times for organic synthesis, but the radiosynthesis itself can be achieved in beneficial shorter time frames and more importantly in high stereoselective manner. The optimization for the routine preparation of labeled L-[11C]MET has been initiated, however to the best of our knowledge has not been published [45].
Racemic and Asymmetric Syntheses to Label Amino Acids in the Side Chain
With variable success, many [11C]AA isotopologues have been synthesized with the label in the carboxylic position. Alternatively, the carbon-11 label can also be positioned in the side chain of the AA, which might be beneficial to detect different metabolism of the investigated AA. Therefore, alternative synthetic approaches for labeling the 3-position, or β, have been developed. An early approach towards novel 11C-C bond formation in the backbone of the molecule, utilized SN2 alkylations of an activated methylene group, the glycine Schiff base, by alkylation with C-11-labeled aliphatic or aromatic alkyl halides. The starting Schiff base precursor, N-(diphenylmethylene) glycine tert-butyl ester, is rather straightforward to synthesize and is even commercially available, and the α-carbon can be deprotonated easily under basic conditions. The first encouraging reports on racemic labeled [11C]AA by research groups in Uppsala, Sweden [46], and St. Louis, USA [47] date back to the early 1990s. These initial attempts made use of achiral approaches and the AAs formed required enantiomer separation, either by chromatographic or enzymatic methods, resulting in longer synthesis times and low radiochemical yields. To circumvent this problem, asymmetric alkylation methodologies have attracted radiochemists aiming to prepare the desired enantiomer in a highly stereoselective manner and with good RCY. Reliable methods to synthesize enantiomerically pure [11C]AAs have been accomplished with a variety of available Schiff bases and C-11 alkylating agents, as discussed further on in this section [6, 48, 49].
Racemic and Asymmetric Synthesis with Schiff Base Precursor: N-(Diphenylmethylene) Glycine Tert-Butyl Ester
The alkylation of the achiral Schiff base glycine precursor was first reported for the radiosynthesis of [11C]phenylalanine by Kilbourn et al. [47]. Thereafter, this strategy was followed to radiolabel a wide range of other racemic [3-11C]AAs, namely alanine, 2-aminobutyric acid, norvaline, norleucine, and leucine [46]. All these reactions employed phase-transfer alkylation conditions using achiral catalyst tetra-butylammonium hydrogen sulfate and the achiral precursor, N-(diphenylmethylene) glycine tert-butyl ester, with different [11C]alkyl iodides. The racemic [11C]AAs of interest were obtained in a d.c. RCY of 10–50 % and RCP of 93–99 %. The reaction times for the C-11 alkylation were between 5 and 10 min and typical reaction temperature of 40–45 °C, though reaction conditions were always dependent on the reactivity of [11C]alkyl iodides. The final step to obtain the unprotected [11C]AA, removal of the protecting group of the alkylated intermediate, occurred in 5 min at 130 °C using strong acidic conditions (e.g., concentrated HCl). An inconvenience with this methodology, when first reported, was its non-stereoselectivity. Therefore, special attention was devoted to treatment of [3-11C]alanine and [3-11C]phenylalanine with the enzyme D-AAO to obtain enantiopure (99 % ee) product [46]. Though very successful, this enzymatic resolution was time-consuming and challenging to perform.
Overall, these studies have identified phase-transfer alkylation is reliable and a practical synthetic pathway to label [11C]AAs in the side chain [46]. The formidable challenge in this methodology still lies in performing a stereoselective alkylation on the α-carbon starting from an achiral precursor. Important parameters that apply to all radiopharmaceuticals are the convenience and reliability of the radiolabeling process, however limited by synthesis time and AM. To overcome these challenges, especially the RYCs for these particular C-11 alkylations, more reactive alkylating reagents have been explored. Employing the highly reactive [11C]CH3OTf on the same precursor, N-(diphenylmethylene) glycine tert-butyl ester, resulted in very rapid and efficient C-11 methylation in only 1 min at 60 °C. Acidic deprotection over 5 min at 170 °C finally yielded [1-11C]alanine where the RCY was increased to 70–90 % within 25–30 min [50].
Considering the utility of obtaining one enantiomer of [11C]AAs, this attractive alkylation approach has been brought to the next level by taking inspiration from the wealth of literature surrounding asymmetric phase-transfer catalysis [51, 52]. Recently, Windhorst et al. achieved rapid and highly enantioselective 11C-C bond formation of L- and D-[11C]alanine [53] and L- and D-[11C]phenylalanine [54], as illustrated in Fig. 4a. The commercially available glycine Schiff base was utilized, in the presence of quaternary ammonium salts as chiral phase-transfer catalysts (PTCs; Fig. 4b) to yield specifically L- or D-[11C]AAs. Reactions were performed in near quantitative RCC of the alkylation reactions, resulting in L-[11C]alanine and L-[11C]phenylalanine in excellent ee of > 90 %, desirable AM of > 50 and 85–135 GBq μmol−1, and sufficient d.c. RCY of 20 % and RCP > 95 %, respectively [53, 54].

Asymmetric phase-transfer alkylation reaction (a) with chiral phase-transfer catalysts (b).
Furthermore, Windhorst et al. aimed to extend the catalyst-controlled asymmetric induction to 1,4-Michael addition reactions with [11C]tert-butylacrylate and [11C]N-tritylacrylamide to yield [11C]glutamate and [11C]glutamine, respectively [55]. Despite the versatility of C-11 synthons, so far, the enantioselective addition reaction has been only moderately successful, which might be attributed to the Pd-ligand complex utilized in carbonylation reaction with [11C]CO.
Asymmetric Synthesis with Schiff Base Precursor: [(+)-2-Hydroxypinanyl-3-Idene]Glycine Ester
Alternative strategies to perform asymmetric alkylation reactions date back to 1979, where Långström and Stridsberg recognized the purification issues, and this consequently led to the development of chiral starting materials. First attempts employed a chiral precursor 8-phenylmethyl-2-isocyanoacetate and alkylation reagent [11C]CH3I furnishing partially resolved [3-11C]alanine obtained in an ee of 48 % with an excess of L-alanine in 50–60 min [56]. In due course, a similar chiral Schiff base precursor, [(+)-2-hydroxypinanyl-3-idene]glycine tert-butyl ester, afforded under anhydrous conditions the first chiral synthesis of L-[3-11C]alanine in a high ee of 89 %, determined by enantiomeric analysis using gas chromatography (GC), in 30–45 min (Fig. 5a). As a final step, a two-step hydrolysis of the protecting groups, the imine and tert-butyl ester group, was performed without racemization [57]. Encouraged by the benefits of a single enantiomer, especially the L-form, asymmetric C-11 labeling of AAs was further extended using different [11C]alkyl iodides towards L-2-amino-[3-11C]butyric acid, L-[3-11C]norvaline, and L-[3-11C]valine with a synthesis time of 50–55 min [58]. In addition to the previously employed Schiff base precursor, an additional chiral handle on the C-terminus (Fig. 5a, in green) was feasible, but unfortunately resulted in lower asymmetric induction affording ee ranging from 36 to 80 %, d.c. RCY of 40 %, and excellent RCP (> 98 %) in 20–25 min [59].

Asymmetric syntheses of various amino acids with different chiral starting material.
Nevertheless, these examples highlight the potential for asymmetric C-11 alkylation with chiral Schiff base precursors. However, a general observation was that larger [11C]alkyl iodides proceed slowly, compared to the aforementioned [11C]alanine synthesis. An acceleration of the reaction and considerable increase in radiochemical conversion (up to 90 %) was observed as a function of the addition of more the polar solvent 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone and its concentration, next to the addition of a strong base (BuLi) and increase of temperature. Nonetheless, the synthetic approach by Antoni et al. [58] afforded previously mentioned [11C]AAs with decent RCY and excellent enantiomeric purities (> 98 %) which, however, has been discontinued due to occasional precipitations of lithiated precursor, inaccuracy in the ee determinations, and lastly, more approachable strategies.
Asymmetric Synthesis with Schiff Base Precursor: (−)-8-Phenylmenthan-3-yl N-(Diphenylmethylene)Glycinate
Comparable to the procedure described in the previous paragraph, herein the C-terminal chiral auxiliary (Fig. 5b), appeared as a simple method with high reproducibility and minimized technical handling. The starting material (-)-8-phenylmenthan-3-yl N-(diphenylmethylene)glycinate was subjected to the achiral phase-transfer catalyst tetra-butyl ammonium hydroxide and [11C]CH3I or [11C]CH2PhI to furnish L-[3-11C]alanine and L-[3-11C]phenylalanine, respectively. Similarly to the previous approach, both [11C]AAs had reasonable RCY, 40 and 15 %, respectively, and excellent RCP > 98 %, however enantioselectivity was lower [60].
Asymmetric Synthesis with the Oppolzer Chiral Sultam-Derived Glycine
During the years, Schiff base precursors have been the spotlight of research in which their C- and N-terminal sides have been modified, aiming to improve the RCY and, more importantly, the ee of the desired [11C]AA product. A very successful chiral precursor proved to be the sultam-derived glycine synthon, developed by Oppolzer et al. [61]. Advantageously, Nagren et al. adopted and improved this methodology by reacting the precursor with n-BuLi at low temperature for 30 min, followed by [11C]CH3I, trapping at the same temperature, enabling a quantitative alkylation within 5 min, while the reaction temperature slowly reached room temperature. Subsequently, the alkylated intermediate was subjected to two-step hydrolysis yielding L-[3-11C]alanine in d.c. RCY 40–50 %, counted from [11C]CH3I, and 94 % ee in a total synthesis time of 50 min (Fig. 5c) [6, 49]. Since these very encouraging results, even the precursor has become commercially available which maintained great interest for chiral alkylation reactions to obtain [11C]AAs.
Asymmetric Synthesis of α-[11C]Methyl Tryptophan with [11C]Methyl Iodide
Considerable potential in the use of Schiff base precursors has also been displayed by the radiosynthesis of the unnatural α-[11C]methyl tryptophan. Here, the natural amino acid, transformed into a Schiff base, is used as a precursor which is on its turn was alkylated at the α-carbon. The scope and potential of these precursor has, however, only been explored to a limited extent. Previous successful syntheses of [1-11C]tryptophan were not desirable, as the radiolabel would be lost during metabolism. To circumvent this, Chaly et al. developed the tryptophan imine moiety precursor, allowing stereospecific “no-carrier added” C-11 methylation to obtain the alternative tracer of [11C]tryptophan. The stereospecific alkylation with [11C]CH3I was achieved in 30 min and resulted in a 20–25 % n.d.c. RCY and AM of 74 GBq μmol−1 (Fig. 5d). Intriguing, the authors proposed that the intermediate is “locked” in the position existing in the original AA, in this case L-tryptophan, resulting in product of the same chiral configuration as starting precursor, with no D-stereoisomer present [62].
Asymmetric Synthesis with Chiral Nickel Complexes
With respect to the alkylations of Schiff bases, the scope of avaliable methods for PET-labeling has been broadened by the chemistry of metallo-complex AA synthons for the asymmetric preparation of α-AA. The methodology has been introduced in organic and organometallic chemistry by Belokon in 1985 [63]. These chiral nickel complexes proved to be highly versatile and allowed a very high level of stereocontrolled C-11 alkylation reactions due to high acidity of the α-hydrogen. Also, F-18 analogues have been obtained via this route, though these will not be further discussed here [7, 64]. Consequently, the condensation of Ni(ІІ) with (S)-N-(2-benzoylphenyl)-1-benzylpyrrolidine-2-carboxamide (BPB) and glycine has become popular due to commercial availability of BPB, the chiral scaffold required for the stereoselective alkylation reaction and its reusage potential. The first radiosynthetic approach to obtain [11C]AAs was evaluated by Fasth et al. [65]. Accordingly, [11C]alanine (de of 80 %; RCY of 60 %) and [11C]phenyalanine (de of 90 %; RCY of 40 %) were synthesized, as well as [11C]tyrosine with an excellent de of 90 % and RCY of 12 % (Fig. 5e).
Furthermore, by adapting the metallo-complex of Ni(ІІ) with BPB, with either tyrosine or dopamine (green part in Fig. 5e), Popkov et al. was also able to synthesize unnatural α-[11C]methyl-tyrosine and α-[11C]methyl-DOPA with [11C]CH3I, giving slight excess of the LD-stereoselectivity with RCY of 7 and 9 %, respectively [7, 66]. Demonstrating the general applicability of this methodology, chiral Ni(II)-complexes were also explored with [11C]formaldehyde as alkylating reagent by Popkov et al. [67]. While the successful and reliable synthesis D-[11C]serine resulted in 50 % d.c. RCY, the diastereoselectivity achieved was only 80 %, indicating improvements in the high performance liquid chromatrography (HPLC) separation of intermediate diastereomers are needed [67].
Specific Carbon-11 Labeling in the Side Chain of Amino Acid
Condensation Reaction with 2-Aryl-5-Oxazolones
The merits of C-11 alkylation, in particular the C-11 methylation, as a choice for radiotracer production have been well-recognized. Schiff bases have been acknowledged as valuable precursors for stereoselective synthesis of various [11C]AAs by the formation of new 11C-C bond through alkylation. However, multiple attempts have been reported to apply other precursors as well, aiming to obtain a C-11-labeled AA under similar alkylation reactions [49]. For example, α-[11C]methyl-L-tryptophan, also described by Chaly et al. [61] in the “Asymmetric Synthesis of α-[11C]Methyl Tryptophan with [11C]Methyl Iodide” section was reported by Plenevaux et al. [68], where the N-terminal amine functionality was covalently attached to the indole precursor’s side chain to induce activation at the α-carbon as well as to protect the amine-functionality. The precursor was deprotonated with the base lithium diisopropylamide at low temperature to activate the α-carbon, followed by alkylation with [11C]CH3I in 5 min (Fig. 6a). Subsequently, deprotection of the labeled precursor was achieved by acidic hydrolysis to yield the alkylated product in 36 % d.c. RCY in 22 min with an exceptional ee > 97 % [68]. When the results of Plenevaux et al. are compared to the method described by Chaly et al. in the “Asymmetric Synthesis of α-[11C]Methyl Tryptophan with [11C]Methyl Iodide” section, this approach yields the product slightly faster in a higher RCY; however, the precursor synthesis for the Plenevaux’s approach is more demanding.

Miscellaneous C-11 alkylations with various starting material.
Asymmetric Synthesis of the 2-Tert-Butyl-3-Methyl-1,3-Imidazolidin-4-on with [11C]Alkyl Iodides
Another promising method that provides [11C]AAs in an almost enantiomerically pure form is the alkylation of imidazolidinone derivatives. These precursors form a highly stereoselective 11C-C bond with [11C]alkyl iodides on stabilized carbanions, as depicted in Fig. 6b. The imidazolidinone derivatives, (R)- or (S)-oc-2-tert-butyl-3-methyl-1,3-imidazolidin-4-one (BMI), is treated with lithium 2,2,6,6-tetramethylpiperidide in tetrahydrofuran at low temperature to initiate deprotonation on the α-carbon, followed by an C-11 alkylation and subsequent acidic hydrolization to furnish the desired [11C]AAs. Via this route, D- and L-[3-11C]alanine and D- and L-[3-11C]phenylalanine have been obtained within 50 min. Using this approach, the [11C]alanine synthesis was achieved in d.c. RCY of 75 % with an ee of exceptional 98 %. On the other hand, [11C]phenylalanine could only be obtained in a RCY of 30 %. This lower RCY might be explained by the lower electrophilicity of [11C]CH2PhI as reagent. Nevertheless, the ee of the product was 96 %, which is highly satisfactory. Additionally, this methodology has also been used to obtain [6-11C]lysine with (S)-Boc-BMI as precursor that was radiolabeled with [11C]4-iodobutyronitrile achieving an ee of 96 %, though these were only preliminary findings. In the same paper, attempts to obtain [1-11C]valine with 2-[11C]isopropyl iodide were described, but proved to be unsuccessful. Unfortunately, the application of this methodology is hampered by the commercially available (R)- or (S)-Boc-BMI precursors as their enantiomeric purities can vary a few percent dependent on the batch, as revealed by Fasth and Låndström [130, 67], which may influence the enantioselective synthesis of [11C]AAs.
S-Methylations on Homocysteine with [11C]Methyl Iodide
Among alkylations to form a new 11C-C bond, C-11 methylations on homocysteine residue are important as well, especially considering their versatility in peptide radiolabeling, discussed later. As early as 1976, Comar et al. recognized C-11 as a beneficial radioisotope for 11C-methylmethionine labeling of DL-homocysteine or L-homocysteine thiolactone with [11C]CH3I [69]. Continuous research strived to improve and simplify methods to obtain latest PET tracer resulting in a “no-carrier added” and rapid S-alkylation of L-homocysteine adsorbed on solid-supported Al2O3/KF with [11C]CH3I (Fig. 6c). The acidic S-H proton of the homocystein is easily removed by the strong base. Therefore, the final purification was simplified, as 90 % of the precursor remained adsorbed on the solid support and was eliminated by a simple filtration, providing a superb d.c. RCY of 94 % with no required preparative HPLC. Hence, this methodology provides important simplification, enabling fast production of L-[11C-methyl]methionine ready for injection within 10 min in excellent RCP of 99 % and acceptable AM of 37 ± 6 GBq μmol−1 [70].
Palladium-Mediated Coupling with Organo-Boron Precursor and [11C]Methyl Iodide
Another research advance which should enable easier access to [11C]AAs is the Pd-catalyzed coupling reaction between a boronic ester precursor and [11C]CH3I. In this reaction, a Suzuki coupling is employed between an unsaturated AA precursor subjected to cross-coupling reactions. The newly formed 11C-C bond can then be reduced to obtain a saturated alkyl side chain of the AA. The organo-boron precursor reacted within 5 min with [11C]CH3I in the presence of tris(dibenzylideneacetone)dipalladium(0), tri(o-tolyl)phosphine and base at elevated temperature, followed by mild hydrogenation and deprotection to furnish L-[1-11C]leucine. Ozaki’s radiosynthesis is illustrated in Fig. 6d [71]. By a similar method, unnatural S-α-[11C]methyl leucine was also synthesized, showing the suitability for syntheses of various 11C-methyl-labeled branched alkanes [71].
[11C]Cyanide Nucleophilic Reactions
Alternatively to the Bucherer-Strecker synthesis, described earlier in the “Carbon-11 Amino Acid Labeling of [1-11C]Carboxyl-Function (Bucherer-Strecker Synthesis)” section, the highly polarized soft base [11C]cyanide has been further explored to radiosynthesize [11C]AAs. This methodology was more recently published by Qu et al., where the nucleophilic substitution reaction with aliphatic halides furnished the enantiopure radiosynthesis of L-[5-11C]glutamine [70]. The radiosynthesis depicted in Fig. 7a was achieved by a reaction of [11C]KCN with protected enantiopure precursor 4-iodo-2-amino-butanoic ester in a three-step synthesis, and successive deprotection and hydrolysis yielding L-[5-11C]glutamine in RCY of 25 % with high purity, but unfortunately rather low AM of 1.85 GBq μmol−1.

Nucleophilic reactions with [11C]cyanide.
Aziridine Ring-Opening Reactions
The versatility of the [11C]cyanide reagent allows the facile functionalization and derivatization of many precursors where aziridines can serve as alternative electrophiles for development of different [11C]carbonyl AAs. Aziridine ring-opening reactions have been reported by Gillings et al. as a novel and fast methodology for the synthesis of the following [11C]AAs: [4-11C]aspartic acid, [4-11C]asparagine, and 2,3-diamino-[4-11C]butyric acid, all obtained in a d.c. RCY of 30–40 % within 30 min [72]. Racemic N-activated aziridine-2-carboxylates were synthesized in four steps, where in particular N-(tert-butoxycarbonyl)aziridine-2-isopropyl carboxylate underwent nucleophilic ring-opening reactions with [11C]tetra-butylammonium cyanide by heating in dimethylformamide (DMF), shown in Fig. 7b. The use of [11C]tetra-butylammonium cyanide with the tetra-butylammonium counter ion was crucial since half of the radioactivity remained unreacted when [11C]HCN in DMF was used. The low reactivity of [11C]HCN in DMF was attributed to insolubility of the reagent in the solvent. To establish a stereoselective synthesis using this methodology, the racemic starting material was resolved by chiral HPLC; however, racemization occurred mainly during the ring-opening reaction due to the acidity of the β-hydrogen. Up till now, despite several attempts, no successful chiral syntheses have been reported using this methodology [72].
Enzyme-Catalyzed Synthesis of Carbon-11 Amino Acids
The relative short half-life of C-11 urges fast and efficient synthetic methods for PET tracer production and therefore biocatalysis offers attractive solutions, such as stereoselective reactions or the possibility to omit protecting groups in the precursor. To date, enzymes have been used in a number of radiosyntheses to obtain C-11-labeled AAs and this approach resulted in exquisite selectivity and high turnover numbers, enabling rapid high-yielding reactions to obtain pure products under mild conditions. Despite the general challenge of enzymatic reaction kinetics and the speed of radiolabel incorporation to the molecule, the application of low enzyme concentrations in C-11-labeled radiotracers preparation has proven to be a rewarding strategy for 12 carbon-11-labeled AAs [49].
Thus far, two different types of enzymatic transformations can be distinguished, firstly the 11C-C bond formation and secondly the functional group transfer, exemplified by transaminiations or oxidations of amino groups to ketones [49].
The first example of a pioneering, well-performed enzyme application, where an enzyme served as a resolving agent, was the preparation of L-[3-11C]phenylalanine and L-[3-11C]alanine, exemplified in Fig. 8. Treatment of the corresponding racemic mixture by D-AAO (EC 1.4.3.3) in 10–20 min resulted in enantiomerically pure [11C]AAs, where ee of the product was determined by GC (N-trifluoroacetylalanine methyl ester) or LC ([3-11C]phenylalanine as free acid) [46]. Advantageously, this method uses immobilized enzymes which improved the previous challenges regarding the product purification and reduced the risk of product contamination. Next to that, immobilization allowed effortless recovery of the enzyme and enabled the possibility to perform several syntheses with the same enzyme [49].

Enzymatic synthesis of aromatic L-enriched [11C]AAs (carboxylic- (in red) or the methyl- (in blue) position).
Nowadays, the most described methodology remains the multi-step enzymatic [11C]AA synthesis, where initially a radiochemical reaction is combined with one or more enzymatic reactions. Employing this approach, the syntheses of four aromatic AAs were described: 3,4-dihydroxy-L-[11C]phenylalanine, known as [11C]DOPA, L-[11C]tyrosine, L-[11C]tryptophan, and [5-11C]hydroxytryptophan, as well as L-[11C]alanine achieved using [11C]CH3I or [11C]CN−. As illustrated in Fig. 8, the synthesis of all these [11C]AAs starts from [11C]alanine, labeled at the carboxylic acid position (in red) or the β-carbon position (in blue). After initial labeling, [11C]alanine is resolved using D-AAO/catalase and further converted to 11C-pyruvic acid by glutamic-pyruvic transaminase (GPT). Accordingly, subsequent one-pot 11C-C bond forming reaction between the aromatic precursor (indole or phenol) and 11C-pyruvate by β-tyrosinase (tyrosine phenol-lyase; EC 4.1.99.2) or tryptophanase (tryptophan indole-lyase; EC 4.1.99.1) yielded products in more than 99 % ee. After the addition of HCl, the enzymes precipitated and final crude AA was purified by prep HPLC. Synthesis of L-[11C]DOPA required also addition of ascorbic acid, in order to prevent possible oxidation [48, 73].
Next to the aromatic [11C]AAs, laudable achievements have been also made towards C-11 aliphatic AAs by a variety of combined 11C-C bond-forming chemo-enzymatic reactions published in the last three decades. As a first example, the synthesis of L-[4-11C]aspartic acid [74] and L-[4-11C]glutamic acid [75] by O-acetyl-L-serinase and O-Acetyl-L-homoserine was published in early 1980 [76], where further meticulous improvements by Antoni et al. in 2001 enabled their synthesis in 45–55 min [76]. Satisfactory d.c. RCY of 50–60 % and 60–70 %, respectively, and RCP > 95 % and an overall AM of 30 GBq μmol−1 starting from [11C]HCN were obtained. Additional success was the high enantiomeric purity, determined by derivatization with Marfay’s reagent, of 98 % ee for both aliphatic [11C]AAs.
Finally, chemo-enzymatic synthesis advances were achieved for L-[11C]methionine ([11C]MET) via11C-S bond formation. Based on a wide spectrum for substrate specificity of O-acetyl-L-homoserine and its success in the synthesis of γ-[11C]cyano-α-amino-L-butyric acid by Antoni et al. [76], Kaneko et al. later published the synthesis of [11C]MET from O-acetyl-L-homoserine and the [11C]methanethiol precursor obtained from [11C]CH3I [77]. As S-H compounds are very reactive, its C-11 isotopologue rapidly reacted with immobilized α-γ-cyano-α-aminobutyric acid synthase and the radioactivity eluted from the column was only [11C]MET. The tracer was obtained within 15 min after EOB with an enantiomeric purity of > 99 % and d.c. RCY of 70 % (based on [11C]CH3I) [77]. Other [11C]MET analogues, like [11C]ethionine and [11C]propionine, could also be prepared from the corresponding [11C]alkanethiols.
Finally, we should also mention that a series of coupled enzymatic reactions have been reported to radiosynthesize L-[3-11C]serine from [11C]methanol ([11C]MeOH), starting from [11C]CO2. The immobilized enzymes alcoholoxidase (EC 1.1.13.13) combined with catalase (EC 1.11.1.16) selectively oxidized [11C]MeOH to [11C]formaldehyde, which condensed with tetrahydrofolate and was subsequently treated with immobilized serine hydroxymethyltransferase. Accordingly, only the L-[3-11C]serine enantiomer was detected by LC-analysis and obtained in 1–2 % d.c. RCY (from [11C]CO2) after a synthesis time of 50–65 min with AM of 1.1–1.9 GBq μmol−1 [78].
In general it can be concluded that enzyme transformation reactions can indeed be highly stereoselective towards the naturally occurring L-enantiomer. On the other hand, obtaining the D-product still remains a special challenge. A disadvantage that may arise is the long reaction time and loss of the radioactivity, resulting in a low RCY when enzymes function as a resolving agent. Nevertheless, the specificity remains their main asset, even after several reuses of these specialized immobilized enzymes.
Synthesis Strategies for Carbon-11 Small Peptides
Next to the use of C-11 labeled AAs as PET tracers, there has been tremendous interest in using C-11-labeled peptidic radiopharmaceuticals, as peptides offer a maximum degree of freedom and flexibility [9]. Only a few peptides have been successfully labeled with C-11 and applied in tumor and other imaging studies. The earliest reports from the 1980s were conducted on several biologically active peptides by attaching an isotopologue methyl group using [11C]CH3I or [11C]formaldehyde [79,80,81].
Inspired by the prosperity of formaldehyde labeling, Straatman et al. labeled proteins with [11C]formaldehyde in a borate buffer at different pH and subsequently treated the reaction mixture with various concentrations of NaBH4 at room temperature. Using this methodology, the proteins human serum albumin (66.5 kDa), fibrinogen (340 kDa), and luteinizing hormone (LH, 42 kDa) have been labeled succesfully, all using similar reaction conditions. Albumin required slightly basic pH, and after the optimization it was concluded that higher amounts of protein in the reaction gave higher RCYs with a mean of 39 % (representive structure in Fig. 9a). On the other hand, [11C]fibrinogen was obtained in 33 % RCY and exhibited still similar biological behavior as the unlabeled fibrinogen [80]. Finally, methylated glycoprotein LH obtained via the reductive alkylation with on-line produced [11C]formaldehyde, retained its biological potency; however, the AM of 7.2 GBq μmol−1 hampered preclinical application [79]. Evidently, the successful synthesis route illustrated in Fig. 9a preserved the biological activity of protein despite its lack of specificity considering the location of the radioactive label and the amount nuclide per molecule. Consequently for C-11 LH, high AM is hardly achievable especially as high amounts of starting protein are needed to achieve a successful radiolabeling.

Early beginnings of simplified strategies for carbon-11-labeled peptides.
Carbon-11-labeled proteins provided an insight into their mechanism of action in the early development of PET in routine diagnostic use (early 2000s) and proved their value and biological importance. Confirmation of their safety and already acknowledged specificity of small peptides expanded the development of C-11 radiolabeling strategies to obtain peptidic radiotracers via fast and efficient routes.
Other early examples of C-11 peptide labeling is the carboxylation of α-lithioisocyanides to obtain dipeptides with a radiolabeled carboxylic acid functionality [25]. Initially, [11C]glycine was produced from methylisocyanide via C-11 carboxylation, as discussed in the “ Carbon-11 Amino Acid Labeling of [1- 11 C]Carboxyl-Function (Bucherer-Strecker Synthesis) ” section, which is subsequently reacted with a cyclic AA anhydride to obtain L-phenylalanyl[1-11C]glycine and L-leucyl[1-11C]glycine. Bolster et al. achieved the dipeptides in an isolated RCY of 3–6 % in a total synthesis time of 50 min, as illustrated in Fig. 9b [25]. In theory, many anhydrides, also more complex cyclic ones, could react with [11C]glycine providing longer peptidic structures where the chiral center is preserved, generating a single enantiomer.
Carbon-11 methylation was also described as a strategy for peptide radiolabeling, mainly for the alkylation of the thiol functionality of homocysteine in the peptide. The complete deprotection of benzyl-homocysteine followed by side-specific methylation performed in one-step in liquid ammonia. Initial studies applying this approach have been described by Långström et al., where a tripeptide with the sequence Z-Gly-L-Homocysteine(Bzl)-Gly-O-Bzl was methylated to obtain H-Gly-[11C]Met-Gly-OH with more than 90 % RCY in less than 25 min [82]. Since C-11 methylation of homocysteine residues proved to be successful, this methodology has been used to obtain radiolabeled substance P, enabling its thorough investigation in biological studies. Its undecapeptide and octapeptide were labeled using a homocysteine precursor in satisfactory 35 % RCY within 1 h production time, including purification. Even though deprotection problems of other protecting side groups and partial cleavage involving proline nitrogens arose, a high RCC could be obtained [83, 84]. Furthermore, neuropeptide MET-enkephalin, a 5-mer peptide, and its two metabolic fragments thereof, Gly-Phe-MET and Phe-MET, were labeled using the C-11 methylation strategy by Nagren et al. S-[11C]MET-enkephalin and corresponding C-11-labeled fragments were reported in a RCY of 50–80 %, after LC purification, in 35–50 min with AM ranging 0.4–7.2 GBq μmol−1, depending on the AM of [11C]CH3I [85]. Though this method of peptide radiolabeling proved to be highly reliable and high yielding, it should be noted that radiolabeling can only be conducted if a methionine residue is present in the peptidic structure. Though being an interesting approach, further optimization of this procedure, especially with the respect of deprotection and generation of the sulfide anion for the reaction, has not been published.
More recently, analogous to the methodology developed by Långström et al., advantage was obtained by making use of the more reactive and volatile [11C]MeOTf for the methylation of three peptides. Here, a thiol nucleophile from a cysteine residue was alkylated and thereby a high single-step regioselective C-11 methylation of cysteine residues was demonstrated [15]. [11C]MeOTf as an alkylating reagent for peptides has been further explored to label more complex peptides as well, such as C-11 labeled glutathione (GSH) by using excess of NaOH in dimethylsulfoxide, furnishing successful methylation in less than 1 min (Fig. 10a). Additionally, a decapeptide has been labeled and was obtained in a 59–65 % incorporation yield. The strategy was also possible with the challenging cyclic somatostatin analogue [11C]Cys(Me)-[Tyr3-octreotate], as described in the next section [15, 86].

Most recent novel carbon-11 radiolabeling strategies for small peptides.
Recently the use of C-11-labeled peptides has attracted considerable attention with three promising novel methodologies to label small peptides containing 2 to 10 amino acids. One approach utilizes direct incorporation of C-11 label via stereoselective alkylation, published as “native” peptide labeling. This methodology is comparable to the one described in the “Racemic and Asymmetric Synthesis with Schiff Base Precursor: N-(Diphenylmethylene) Glycine tert-Butyl Ester” section, where an N-terminal Schiff base peptide precursor was alkylated at the α-carbon, potentially in a stereoselective manner. Pekošak et al. described the first C-11-labeled peptides and afforded multiple tetrapeptides [87] employing chiral phase-transfer catalysts in a reliable RCC and high diastereoselectiviy, as depicted in Fig. 10b and discussed in more detail below in the “Carbon-11-Labeled Somatostatin or Octreotate Analogues” section. Follow-up studies of Filp et al. investigated the influence of the substrate peptidic backbone precursor and phase-transfer catalyst (see Fig. 4b) induction on stereoselectivity for various natural dipeptides labeled with either [11C]CH3I or [11C]CH2PhI [88]. Overall, excellent conversion rates of > 85 % and diasteremomeric excess up to 94 % were uniformly observed in only 5 or 7 min (Fig. 10b), thereby showing the procedure can be extended to various N-terminal peptides using other [11C]alkyl halides and used for rapid preclinical assessment of the peptidic-lead structure.
A second recent methodology to obtain C-11-labeled peptides depends on the use of palladium-mediated amino-carboxylation using [11C]CO. Figure 10c shows the production of N-[11C]acetylated peptides on terminal amine or lysine residues was reported by Andersen et al. The strength of this paper is that several N-acetylated peptides have been described, such as [11C]LULU-Phol (RCC 43 %; d.c. RCY 33 %), [11C]acetyl-cRGDfK (RCC 63 %; d.c. RCY 37 %), [11C]lacosamide (RCC 63 %; d.c. RCY 46 %) in excellent RCP of 99 % within 30 min, and also a larger peptide the [11C]SPF-5506-A4 (RCC 15–21 %; d.c. RCY 2 %). The C-11 acetylation provided the labeled peptides with high AM ([11C]LULU-Phol 281 GBq μmol−1) and proved to be N-terminal selective, however has a competing reaction on cysteine residues, namely the S-acetylation, might serve as an additional challenge for future applications [89].
A more recent paper by Zhao et al. reported a direct [11C]cyanide labeling of cysteine-containing unprotected peptides via palladium-mediated sequential cross-coupling reaction. This method enables “nucleophilic-nucleophilic” coupling of a peptide with [11C]HCN, as [11C]cyanide source via Pd-complex dihaloarene oxidative addition. In this manner, a cysteine residue is chemoselectively labeled in the presence of other potentially nucleophilic functional side groups. Outstandingly, low amounts of peptide precursor (20 nmol) are needed for this reaction which can be of added value. The potential of the methodology was shown on the RGD peptide ligands for the αVβ3 integrin receptor yielding 10 % n.d.c. RCY with AM 37 GBq μmol−1 within only 15 min (Fig. 10d) confirming the applicability of the method [90].
In recent years, the development in biotechnology and its applications has lead us towards combined and improved mechanistic possibilities of peptidic radiopharmaceutical as molecular imaging agents. An early research example of using a biotechnological method for the synthesis of radiolabeled peptides was exemplified by Harada et al. [91]. A cell-free system, so-called PURESYSTEM, was combined with [11C]MET to obtain interleukin-8 (IL-8). IL-8 is involved in neutrophils activation, and already showed potential in inflammation imaging with Tc-99m and I-131 [92, 93]. Briefly, PURESYSTEM is a novel reconstituted protein synthesis kit consisting of necessary enzymes for in vitro transcription, translation, and energy recycling. The in vitro biochemical reaction proceeded well in a RCY of 63 % with RCP of > 95 %. [11C]IL-8 was obtained in n.d.c. RCY of 13 % in 20 min, sufficient for a small animal imaging study. It is important to bear in mind that protein synthesis will stop when [11C]MET is exhausted; therefore, high molar activity can be reached.
Carbon-11-Labeled Somatostatin or Octreotate Analogues
In the emerging field of molecular imaging, radiolabeled somatostatin analogues have become indispensable tools for in vivo localization of tumors and real-time monitoring of therapeutic response. Many somatostain analogues have been developed and analyzed, whereas the radiolabeled analogue of the most well-known cyclic octapeptide—Octreotide—has become the gold standard for neuroendocrine tumors. Driven by the success of Octreotide and the significant potential of a large number of C-11 PET radiopharmaceuticals in clinical application, several C-11 somatostatin/octreotide analogues have been developed [1, 87, 94].
A chemoselective labeling strategy of carbohydrate analogue of Tyr3-octreotate based on the formation of oximes reacting with labeled prosthetic group has been proposed by Henriksen et al. [19]. Accordingly, 4-[11C]methoxy-benzaldehyde (Fig. 11a in green), derived from [11C]CH3I, reacted with aminooxy-functionalized peptide precursors of D-Phe1-Tyr3-Thr8-octreotide and a two-step synthesis yielded Cel-Dpr-[11C]MBOS-TOCA (Fig. 11a) in a d.c. RCY of 21 % within 60 min with a AM of 22–28 GBq μmol−1, starting from [11C]CO2 [19]. Another well-known example is a one-step regioselective [11C]methylation of cysteine residue with [11C]MeOTf towards [11C]Cys(Me)-[Tyr3-octreotate] (Fig. 11b), resulting in n.d.c. RCY of 11 % and AM of 96 MBq μmol−1, after purification within 30 min [15]. Very recently, the C-11-labeled “unmodified” peptide was described by asymmetric phase-transfer catalyzed alkylation with [11C]CH2PhI (Fig. 11c). As shown in Fig. 11c, the H-D-[11C]Phe-D-Trp-Lys-Thr-OH, the essential pharmacophore for somatostatin receptors, was labeled in a highly stereoselective manner using catalyst 8 (Fig. 4b) with a diastereomeric excess of 94 % with d.c. RCY of 9–10 % and AM of 15–35 GBq μmol−1 in less than 60 min [87].

Carbon-11-labeled analogues of octreotate.
Obviously, the advantage of the last strategy is to obtain a non-modified peptide as a highly enriched diastereomer, where further biological studies are not hampered comparing the radiolabeled compound to the natural peptide.
Reflections on the Preclinical and Clinical Applications of C-11-Labeled Amino Acids
Besides the radiosynthetic development, radiolabeled AAs and peptides have gained an increased interest for preclinical and clinical studies on the nature of various diseases. As diverse naturally occurring compounds accountable for many biological functions, [11C]AAs and peptides can be considered as attractive radiopharmaceutical tracers. These targeting vectors labeled with C-11 possess no structural changes whereas, with F-18 or radiometal-labeled tracers, structural differences arise. Notwithstanding, the radiosynthesis of F-18-labeled AAs and peptides is well developed and a comprehensive overview is given by Ermert et al. [95] and Richter et al. [95, 96]. Radiometals, such as gallium-68, require a corresponding bifunctional metal chelator, and due to the size of the chelation, these must be separated with the linker not to interfere the peptide binding region [11].
The advantages of C-11-labeled “native” compounds are (1) structurally preserved molecule, (2) equivalent biological properties, (3) favorable radionuclide characteristics, (4) low radiation burden for patient, and (5) feasible multiple tracer injections per day.
Regardless of the half-life and restricted use by PET centers with cyclotrons, the development towards C-11 “native” AAs and peptides has been strongly enhanced. Promising imaging studies are presented in the next paragraphs, starting with the top-most used PET AA radiotracer: [11C]MET. The in vivo evaluation of other [11C]AAs is limited and has been mostly performed with only small animals, where some studies even date back to the 1980s. Over the years, indeed some have progressed to small-scale human studies; however, no substantial trails have been conducted recently (Table 1).
[11C]Methionine
[11C]MET has been used extensively to visualize tumor metabolism. Nowadays, many different tumors are imaged using [11C]MET by increased fluxes into metabolic pathways and cellular proliferation, next to its involvement in the synthesis of phospholipids [97, 98]. The transport of [11C]MET into the brain is highly enhanced in tumor tissue (e.g., gliomas) across the reversible sodium-independent transport system L (LAT 1).
In 1985, the first report was published on the application of [11C]MET as a PET tracer in patients with supratentorial gliomas. It was proven that the uptake of [11C]MET is not only caused by passive diffusion and leakage in the blood-brain barrier (BBB), but should also be attributed to active AA-transport into the cell [99]. Since then, various follow-up studies compared the use of [11C]MET with [18F]FDG in glioma, meningioma, non-small lung cancer, and non-Hodgkin’s lymphoma patients to evaluate metabolism, as well as to detect and delineate tumors [103,104,105,106,107]. Both [11C]MET and [18F]FDG as PET tracers have been used complimentary; however, the advantage of [11C]MET was clear. Especially for brain tumor imaging, better tumor-to-background ratios can be achieved in addition to tumor staging and determination of tumor margins. Overall, the conclusion of the various studies is that [11C]MET is better suited for low and intermediate grade brain tumors compared to [18F]FDG [108]. Another breakthrough in the use of [11C]MET was a study in young children and adults with confirmed brain tumors, conducted by O’Tuama et al. The consumption of administered [11C]MET declined with patient age in the tumor, proposing that AA consumption deteriorates [109]. In a follow-up study, Harris et al. showed that the pancreas and liver are the organs with the highest uptake of [11C]Met also for pediatric patients [110]. All this suggests that the main metabolic pathway of [11C]MET is protein incorporation, as the high uptake might be related to the need of MET for protein synthesis, for example plasma proteins in the liver [98, 111].
In recent years, the clinical value of [11C]MET has been thoroughly investigated for brain tumors by Glaudemans et al. as well as in a meta-analysis by Zhao et al. Both studies showed an excellent diagnostic performance of [11C]MET in brain tumor differentiation where the threshold for determination of brain tumors, relying on tumor-to-background ratio, was determined to be acceptable between 1.5 and 1.9 [112, 113]. Furthermore, [11C]MET uptake in the normal brain in comparison to glioma patients offered significant advantages, also for white matter changes, which are normally missed in standard analysis methods [114].
With respect to imaging of other tumor types, [11C]MET has been extensively studied in urinary and gynecological cancers, as well as in lung cancer patients; however, in mentioned tumors, it is of critical importance to identify the exact metabolic mechanism and its use has limitations (e.g., influence of AA-transport and protein synthesis, alternative metabolic pathways) [98, 115, 116]. The development of [11C]MET has been summarized by Coope et al. They concluded that the future of quantitative analysis of [11C]MET uptake needs sophisticated computer programs and standardization of tumor-to-background cut-off values [114]. Moreover, contemporary PET/MRI imaging might significantly improve the diagnosis with more precise reference regions maps compared to CT [112].
[11C]Tyrosine
The application of a racemic [11C]tyrosine was assessed in preliminary studies in tumor-bearing rats by Vaalburg et al. in 1984 [29] and later Daemen et al. [104]. Both enantiomers of [11C]tyrosine showed tumor uptake, which was even slightly in favor of the D-enantiomer. With respect to the metabolism, [11C]CO2 appeared as the major breakdown product, meaning that [11C]tyrosine is firstly metabolized via decarboxylation. Advantageously, the metabolite [11C]CO2 can be rapidly cleared through the lungs, not disturbing PET measurements and potentially even increasing the signal-to-noise ratio [117, 118].
A small-scale study investigated the potential reduction on tumor growth after radiotherapy in rats and patients with prolactinomas, benign tumors in the brain overproducing the hormone prolactin and bromocriptine. L-[11C]Tyrosine proved superior to [18F]FDG for the visualization of this particular brain tumor after radiotherapy [104, 119]. Regardless of the successes achieved, the use of [11C]tyrosine has been limited.
[11C]Valine
[11C]Valine has been described as a potential pancreas-imaging agent in 1978 by Washburn et al. performing an extensive biodistribution study in rats and other small animals [31]. Tissue distribution of [11C]valine in all animals showed high uptake and fast clearance, although [11C]valine was also exposed to rapid decarboxylation. Nevertheless, studies performed in patients with pancreatic disease showed uptake in the diseased tissue of racemic [11C]valine and [11C]tryptophan [120]. Both AAs showed steady pancreatic uptake; however, they also found a high concentration in the kidneys, making the scan interpretation difficult. At that time, in 1979, injecting a racemic mixture of PET tracer has not been recognized as problematic as it was assumed that D-AAs are simply not metabolized and remain in the blood flow. By contrast, L-AAs are either taken up by tumor or metabolized earlier. Therefore, with the lack for the sufficient model for quantitative analysis, strong conclusions could not be made.
[11C]DOPA
Though most C-11-labeled AAs are applied for tumor imaging, advances have also been made for imaging of neurological diseases with L-[11C]DOPA. The development of this PET tracer proved challenging and the first successful synthesis was reported in 1973 by Fowler et al. [121]. Later, Hartvig et al. reported the initial biological evaluation and clinical use of L-[11C]DOPA [100, 122], consequently showing its importance for various neurological disorders such as Parkinson’s disease and schizophrenia, next to its well-known analogue [18F]fluoro-3,4-dihydroxyphenyl-l-alanine (FDOPA) [123]. The best position for incorporation of the C-11 label in [11C]DOPA molecule was evaluated by the metabolism study in an enzyme-activity study, by decarboxylation of [11C]DOPA to [11C]Dopamine in endocrine pancreatic tumors [124]. The biological evaluation revealed that the C-11 position is important, and the radiolabel is not metabolized in the β-position. L-[11C]DOPA is stored mainly in one type of secretory granula. However, in active tumors, it is constantly released, thereby diminishing the content of radioactivity. Its metabolism was reported to be slow with in vivo half-life of roughly 2 h. Furthermore, metabolites of DOPA play an insignificant role, since these are stored in vesicles in areas with high density of dopaminergic pathways in the brain (e.g., the striatal region).
[11C]Glutamine
In recent preclinical studies, it was demonstrated that glutamine metabolism is a vital process for tumor development and progression. With the better understanding of tumor metabolism and the fact that glutamine, a non-essential AA available in high concentrations in blood, is a very important nutrient for diseases in the so-called glutaminolysis pathway [101], the interest in glutamine as a PET tracer have grown. Further, in vivo PET imaging in tumor-bearing rats suggested a rapid tumor uptake and trapping. This led to the proposal to utilize [11C]glutamine in addition to negative [18F]FDG tumor studies to discriminate a tumor’s reliance on different nutrients for growth. Interestingly, this tracer utilizes a different mechanism of action in tumors and its uptake is complementing glucose metabolism. Cells are able to employ alternative energy sources, compared to all other available AAs tracer so far, which are mostly dependent on elevated levels of AA-transporters on the cell membrane [125]. All this suggest there is still a great interest in glutamine or its derivatives as PET tracer.
Taken together, these findings enhanced the understanding of AA metabolism, transport mechanism in cells, biological behavior of radiolabeled compounds, and applicability in diagnosis of tumors as well as for neurological disorders. The main weakness of [11C]tyrosine, [11C]valine and [11C]dopamine studies conducted more than 20 years ago is their obsolence and lack of more recent re-evaluation with state-of-the-art PET analysis techniques. Reasons for the reluctant use of these labeled compounds in general might be the underdeveloped radiosynthesis methods and obviously need of proximity to radiochemistry facilities with cyclotrons. Also, F-18-labeled tracers are in some cases preferred (e.g., L-6-[18F]fluoro-DOPA), especially as they can be distributed to other hospitals. On the other hand, as new C-11 synthetic radiolabeling strategies advance, the accessibility can be broadened and consequently open doors for new imaging agents. That can be especially interesting for biological evaluation of peptides where no additional toxicological studies have to be performed before (pre)clinical studies.
Application of Carbon-11-Labeled Peptides as Imaging Agents
Although peptides and AAs are alike in many ways, such as AAs scaffold and consequently position for radiolabeling or short in vivo half-life, it should be emphasized that the use of C-11-labeled peptides is, up to date, far more challenging. Their translation into PET imaging agents has been supported with specialized radiolabeling reactions in very recent years [12]. However, the application of C-11-labeled peptides remains largely unexplored. A challenge for the application might be associated with slow distribution for in vivo studies due to rapid metabolism.
The very first example of in vivo study using a C-11-labeled peptide/protein was human serum albumin microspheres. Turton et al. evaluated [11C]albumin in dogs and demonstrated ideal properties of labeled microspheres to study blood flow for investigation of pulmonary and BBB permeability [126]. In 2004, Henriksen et al. reported a convenient synthetic procedure to obtain a functionalized sugar-containing derivative of octreotate and assessed the product in vivo as well [19]. With a AM of 22–28 GBq μmol−1, biodistribution and PET imaging studies in pancreatic carcinoma-bearing mice showed good results, recommending its further use.
Conclusion and Future Perspective
In conclusion, advances in radiopharmaceutical chemistry have enabled the establishment of efficient and reliable syntheses of many C-11-labeled AAs and peptides. Starting with [11C]CO2, [11C]HCN, or [11C]CH3I, a multitude of AAs and their derivatives have been obtained as PET tracers. Next to that, based on these synthesis strategies, C-11-labeled peptides can be successfully achieved. The main challenge of all these radiolabeling methods remains their stereocontrol, which might diminish the yield. With the upcoming advances in asymmetric synthesis and the commercial availability of chiral phase-transfer catalysts, the radiosynthesis has progressed as far as having natural enantiopure AAs and even small peptides in hand, with reasonable synthesis steps and reaction times.
Ensuring appropriate systems at hospitals, C-11-labeled biologically active compounds have versatile functions in physiology and are thereof ideal for PET research. To improve diagnosis, C-11-labeled AAs could be used complimentary to other tracers such as [11C]MET and [18F]FDG. A combination of two PET scans in 1 day, firstly with a short-lived [11C]AAs followed by a [18F]FDG scan, could serve as an additional tool for primary and metastatic brain tumors; peripheral tumors such as lymphoma, lung, or breast tumors [127]; or inflammation, as confirmed by [11C]MET. This suggests that [11C]AAs tracers could be used complimentary since they are potentially more suitable than [18F]FDG for differentiation of tumors and inflammation. However, use of [11C]AAs tracers are limited to the PET centers with an in-house facility and cyclotron due to the short half-life of C-11. Flourine-18-labeled AAs might be more appropriate for distant shipment or longer scanning times. This can be seen in various developments in the field where [18F]AAs are developed and successfully applied as well. A concern in general when using F-18 is defluorination that has also been reported for the [18F]AAs [128, 129].
With respect to imaging with C-11-labeled peptides, further in-depth research is required. Nevertheless, in recent years, development in biotechnology and its applications has lead towards combined and improved mechanistic possibilities in peptide-based radiopharmaceutical as molecular imaging agents. Even more, the recent achievements furnished various methodologies to label peptides with C-11 which are now ready to be applied to biologically relevant peptides. This will only boost future application of peptides in imaging. It should be noted that a handful of radiosynthetic options have been developed, but the preclinical and clinical applications are running behind. Furthermore, despite the significant advances in the field of radiosynthesis, there is a continuous need for discovery of unprecedented molecular targets, such as peptide receptors overexpressed on common human cancers, and development of their appropriate imaging agents.
Notes
RCC: Radiochemical yield, determined by analytical HPLC of the crude reaction mixture, defined as radiochemical conversion. In some of the older papers, there was not a clear statement between isolated and non-isolated radiochemical yield.
References
Vallabhajosula S, Solnes L, Vallabhajosula B (2011) A broad overview of positron emission tomography radiopharmaceuticals and clinical applications: what is new? Semin Nucl Med 41:246–264
Jager PL, Vaalburg W, Pruim J et al (2001) Radiolabeled amino acids: basic aspects and clinical applications in oncology. J Nucl Med 42:432–445
Huang C, Mcconathy J (2013) Fluorine-18 labeled amino acids for oncologic imaging with positron emission tomography. Curr Top Med Chem 13:871–891
Christensen HN (1990) Role of amino acid transport and countertransport in nutrition and metabolism. Physiol Rev 70:43–77
Huang C, McConathy J (2013) Radiolabeled amino acids for oncologic imaging. J Nucl Med 54:1007–1010
Popkov A, Elsinga PH (2013) Asymmetric synthesis of carbon-11 labelled a-amino acids for PET. Curr Org Chem 17:2127–2137
Popkov A, De Spiegeleer B (2012) Chiral nickel(II) complexes in the preparation of 11C- and 18F-labelled enantiomerically pure α-amino acids. Dalton Trans 41:1430–1440
Lee S, Jin X, Xiaoyuan C (2010) Peptide-based probes for targeted molecular imaging. Biochemistry 49:1364–1376
Schottelius M, Wester HJ (2009) Molecular imaging targeting peptide receptors. Methods 48:161–177
Fani M, Maecke HR, Okarvi SM (2012) Radiolabeled peptides: valuable tools for the detection and treatment of cancer. Theranostics 2:481–501
Charron CL, Farnsworth AL, Roselt PD et al (2016) Recent developments in radiolabelled peptides for PET imaging of cancer. Tetrahedron Lett 57:4119–4127
Charron CL, Hickey JL, Nsiama TK et al (2016) Molecular imaging probes derived from natural peptides. Nat Prod Rep 33:761–800
Ambrosini V, Fani M, Fanti S et al (2011) Radiopeptide imaging and therapy in Europe. J Nucl Med 52:42S–55S
Graham MM, Menda Y (2011) Radiopeptide imaging and therapy in the United States. J Nucl Med 52:56S–63S
Chin J, Vesnaver M, Bernard-Gauthier V et al (2013) Direct one-step labeling of cysteine residues on peptides with [ 11C]methyl triflate for the synthesis of PET radiopharmaceuticals. Amino Acids 45:1097–1108
Cutler CS, Hennkens HM, Sisay N et al (2013) Radiometals for combined imaging and therapy. Chem Rev 113:858–883
Wester HJ, Schottelius M, Scheidhauer K et al (2003) PET imaging of somatostatin receptors: design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur J Nucl Med Mol Imaging 30:117–122
Fani M, Maecke HR (2012) Radiopharmaceutical development of radiolabelled peptides. Eur J Nucl Med Mol Imaging 39:S11–S30
Henriksen G, Schottelius M, Poethko T et al (2004) Proof of principle for the use of 11C-labelled peptides in tumour diagnosis with PET. Eur J Nucl Med Mol Imaging 31:1653–1657
National Research Council. 1982. Synthesis of Carbon-11, Fluorine-18, and Nitrogen-13 Labeled Radiotracers for Biomedical Applications. Washington, DC: The National Academies Press. https://doi.org/10.17226/19636.
Rotstein BH, Liang SH, Placzek MS et al (2016) 11C=O bonds made easily for positron emission tomography radiopharmaceuticals. Chem Soc Rev 45:4708–4726
Antoni G (2015) Development of carbon-11 labelled PET tracers-radiochemical and technological challenges in a historic perspective. J Label Compd Radiopharm 45:65–72
Miller PW, Long NJ, Vilar R, Gee AD (2008) Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew Chem Int Ed Engl 47:8998–9033
Liang SH, Vasdev N (2015) Total radiosynthesis: thinking outside “ the box”. Aust J Chem 68:1319–1328
Bolster JM, Vaalburg W, Elsinga PH et al (1986) Synthesis of carbon-11 labelled glycine and the dipeptides L-phenylalanylglycine and L-leucylglycine. Appl Radiat Isot 37:985–987
Vaalburg W, Beerling-Van Der Molen HD, Reiffers S et al (1976) Preparation of carbon 11 labelled phenylalanine and phenylglycine by a new amino acid synthesis. Int J Appl Radiat Isot 27:153–157
Bolster JM, Vaalburg W, Van Veen W et al (1983) Synthesis of no-carrier-added L- and D-[1-11C]-DOPA. Int J Appl Radiat Isot 34:1650–1652
Bolster JM, Vaalburg W, Paans AM et al (1986) Carbon-11 labelled tyrosine to study tumor metabolism by positron emission tomography (PET). Eur J Nucl Med 12:321–324
Hayes R, Washburn L, Wieland B et al (1976) Carboxyl-labeled acid, 11C-1-Aminocyclopentanecarboxyl acid, a potential agent for cancer detection. J Nucl Med 17:748–751
Vaalburg W, Bolster JM, Paans AMJ et al (1984) Carbon-11 labeled tyrosine for investigation of tumor metabolism by PET. J Label Compd Radiopharm 21:1177–1179
Washburn C, Wieland W, Butler A (1978) 11C-DL valine, a potential pancreas-imaging agent. J Nucl Med 19:77–84
Hayes RL, Washburn LC (1978) Synthesis and purification of 11C-carboxyl-labeled amino acids. Int J Appl Radiat Isot 29:186–187
Iwata REN, Ido T, Takahashi T (1987) Optimization of [11C]HCN production and acid. Synthesis 38:97–102
Studenov AR, Szalda DE, Ding Y-S (2003) Synthesis of no-carrier-added C-11 labeled D- and L-enantiomers of phenylalanine and tyrosine for comparative PET studies. Nucl Med Biol 30:39–44
Berge G, Mazière M, Prenant C, Comar D (1980) Synthesis of carbon-l 1-labelled acetone. Int J Appl Radiat Isot 31:577–578
Xing J, Brooks AF, Fink D et al (2016) High-yielding automated convergent synthesis of no-carrier-added [11C-carbonyl]-labeled amino acids using the Strecker Reaction. Synlett 27:371–375
Hienzsch A, Hesse R, Lankau H-J, et al (2017) Development of 11C-chemistry on a microfluidic platform. J Label Compd Radiopharm 60:S111–S640
Bolster JM, Vaalburg W, van Dijk TH et al (1985) Syntheses of carbon-11 labelled ornithine and lysine. Preliminary accumulation studies in rats with Walker 256 carcinosarcoma. Int J Appl Radiat Isot 36:263–267
Bolster JM, Ten Hoeve W, Vaalburg W et al (1985) The preparation of carbon-11 labelled proline for positron emission tomography. Preliminary distrubition studies in rats with walker 256 carcinosarcoma. Int J Appl Radiat Isot 36:339–343
Bolster JM, Vaalburg W, Paans AMJ et al (1986) Carbon-11 labelled tyrosine to study tumor metabolism by positron emission tomography (PET). Eur J Nucl Med 12:321–324
Bolster JM, Vaalburg W, Elsinga PH et al (1986) Synthesis of DL-[1-11C]methionine. Appl Radiat Isot 37:1069–1070
Bolster JM, Vaalburg W, Elsinga PH et al (1986) The preparation of 11C-carboxylic labelled L-methionine for measuring protein synthesis rates. J Label Compd Radiopharm 23:1081–1082
Jeanjean F, Fournet G, Le Bars D et al (1997) Asymmetic carboxylation of amino acids: synthesis of L-[1-11C]methionine. J Label Compd Radiopharm 40:722–724
Jeanjean F, Prol N, Gor J, Fournet G (1997) Asymmetric carboxylationin thesynthesisof L-methionine: a new tool for 11Cchemistry. Tetrahedron Lett 38:7547–7550
Jeanjean F, Fournet G, Le Bars D, Gore J (2000) Synthesis of highly enantio-enriched α -amino acids by carboxylation of N - ( α -Lithioalkyl ) oxazolidinones. European J Org Chem 2000:1297–1305
Antoni G, Långström B (1987) Synthesis of racemic [3-11C]-labelled alanine, 2-aminobutyric acid, norvaline, norleucine, leucine and phenylalanine and preparation of L-[3-11C]alanine and L-[3-11C]phenylalanine. J Label Compd Radiopharm 24:125–143
Kilbourn RM, Dischino DD, Welch JM (1984) Synthesis of DL-[3-11C]phenylalanine. Int J Appl Radiat Isot 35:603–605
Bjurling P, Antoni G, Watanabe Y, Langstrom B (1990) Enzymatic synthesis of carboxy-11C-labelled L-tyrosine, L-DOPA, L-tryptophan and 5-hydroxy-L-tryptophan. Acta Chem Scand 44:178–182
Antoni G., Kihlberg T., Långström B. (2011) 11C: Labeling Chemistry and Labeled Compounds. In: Vértes A., Nagy S., Klencsár Z., Lovas R.G., Rösch F. (eds) Handbook of Nuclear Chemistry. Springer, Boston, MA
Nagren K (1997) Asymmetric synthesis of L-[3-11C]alanine utilizing a Sultam-derived glycine equivalent. J Label Compd Radiopharm 40:758–759
Shirakawa S, Maruoka K (2013) Recent developments in asymmetric phase-transfer reactions. Angew Chemie Int Ed 52:4312–4348
Brak K, Jacobsen EN (2013) Asymmetric ion-pairing catalysis. Angew Chemie Int Ed 52:534–561
Filp U, Pekošak A, Poot AJ, Windhorst AD (2016) Enantioselective synthesis of carbon-11 labeled L-alanine using phase transfer catalysis of Schiff bases. Tetrahedron 72:6551–6557
Pekošak A, Filp U, Škrinjar J et al (2017) A rapid and highly enantioselective C- 11 C bond formation of L-[ 11 C]phenylalanine via chiral phase-transfer catalysis. Org Biomol Chem 15:570–575
Filp U, Pees AL, Taddei C et al (2017) Efficient synthesis of 11 C-acrylesters, 11 C-acrylamides and their application in Michael addition reactions for PET tracer development. European J Org Chem 2017:5154–5162
Langstrom B, Stridsberg B (1978) Syntheses of racemic [ l- 11C]-alanine partially resolved [ 3-11C]-alanine. Int J Appl Radiat Isot 30:151–153
Antoni G, Langstrom B (1986) Asymmetric synthesis of L-[3-11C]alanine. Acta Chem Scand 40:52–156
Antoni G, Langstrom B (1987) Asymmetric synthesis of L-2-amino[3-11C]butyric acid, L-[3-11C]norvaline and L-[3-11C]valine. Acta Chem Scand 41(B):511–517
Fasth K, Anonti G, Langstroem B (1990) Asymmetric synthesis of l-[beta-11C]alanine using a glycine derivative with two chiral handles, (1R,2S, 3R)-8-phneylmenthan-3-yl N-[(1R,2R,5R0-2-hydrozpinan-3-ylidene]glycinate)]. Acta Chem Scand 44:527–530
Fasth K, Antoni G, Langstrom B (1988) Asymmetric synthesis of L-[3-11C]alanine and L-[11C]phenylalanin by a phase-transfer alkylation reaction. J Chem Soc I:3081–3084
Oppolzer W, Robert M, Zhou C (1994) Asymmetric alkylations of a sultam-derived glycine equivalent: practical preparation of enantiomerically pure α-amino acids. Helv Chim Acta 77:2363–2380
Chaly T, Diksic M (1988) Synthesis of “no-carrier-added”alpha-[11C]methyl-L-tryptophan. J Nucl Med 29:370–374
Belokon YN, Bulychev AG, Vitt SV et al (1985) General method of diastereo- and enantioselective synthesis of P-hydroxy-a-amino acids by condensation of aldehydes and ketones with glycine. J Am Chem Soc 107:4252–4259
Popkov A, Hanusek J, Cermak J et al (2010) Development of metallocomplex amino acids synthons for the asymmetric preparation of alpha-amino acids by stereoselective introduction of a side chain. Evaluation of the model asymmetric preparation of alanine and beta-13C monolabelled alpha-aminoisobutyr. J Radioanal Nucl Chem 285:621–626
Fasth K, Långström B (1990) Asymmetric synthesis of L-[beta-11C]amino acids using a chiral nickel complex of the Schiff Base of (S)-o-[(N-benzylprolyl)amino]benzophenone and glycine. Acta Chem Scand 44:720–725
Popkov A, Nádvorník M, Kružberská P et al (2007) Towards stereoselective radiosynthesis of α-[11C]methyl-substituted aromatic α-amino acids—a challenge of creation of quaternary asymmetric centre in a very short time. J Label Compd Radiopharm 50:370–374
Popkov A, Itsenko O (2015) An asymmetric approach to the synthesis of a carbon-11 labelled gliotransmitter d-serine. J Radioanal Nucl Chem 304:455–458
Plenevaux A, Lemaire C, Delfiore G, Comar (1994) Enantioselective synthesis of n.c.a. (S)-L-([a-“C]methyl)-tryptophan*. Appl Radiat Isot 45:651–653
Comar D, Cartron J, Maziere M, Marazano C (1976) Labelling and metabolism of methionine-methyl-11 C. Eur J Nucl Med 1:11–14
Schmitz F, Plenevaux A, Del-Fiore G et al (1995) Fast routine production of l-[11C-methyl]methionine with Al2O3KF. Appl Radiat Isot 46:893–897
Ozaki D, Yoshinobu H, Hisashi D, Masaaki S (2013) Synthesis of S-[11C]leucine and its analogues by the combination of rapid C-[11C]methylation and rapid hydrogenation. J Label Compd Radiopharm 56:6
Gillings NM, Gee AD (2001) Synthesis of [4-11C]amino acids via ring-opening of aziridine-2-carboxylates. J Label Compd Radiopharm 44:909–920
Harada N, Nishiyama S, Sato K, Tsukada H (2000) Development of an automated synthesis apparatus for L-[3-11C] labeled aromatic amino acids. Appl Radiat Isot 52:845–850
Barrio J, Eqbert J, Henze E et al (1982) [4-11C]aspartic acid: enzymatic synthesis, myocardial uptake, and metabolism. J Med Chem 25:93–96
Cohen MB, Spolter L, Chang CC et al (1982) The varying tissue distribution of L-glutamic acid labelled at three different sites. Int J Appl Radiat Isot 33:613–617
Antoni G, Omura H, Bergström M et al (1997) Synthesis of L-2,4-diamino[4-11C]butyric acid and its use in some in vitro and in vivo tumour models. Nucl Med Biol 24:595–601
Kaneko S, Ishiwata K, Ishii SI et al (1999) Enzymatic synthesis of carbon-11 labeled methionine and its derivatives with immobilized gamma-cyano-alpha-aminobutyric acid synthase. Appl Radiat Isot 51:285–291
Svard H, Jigerius S, Langstrom B (1990) The enzymatic synthesis of L- [ 3-11C ] serine. Appl Radiat Isot 41:587–591
Marche P, Morgat J, Fromageot P (1975) 11C-labelleling of ovine luteinizing hormone by reductive methylation. Radiochem Radioanal Lett 21:53–59
Straatmann MG, Welch MJ (1975) A general method for labeling proteins with 11C. J Nucl Med Med 16:425–428
Hanyu M, Takada Y, Hashimoto H et al (2013) Carbon-11 radiolabeling of an oligopeptide containing tryptophan hydrochloride via a Pictet-Spengler reaction using carbon-11 formaldehyde. J Pept Sci 19:663–668
Langstrom B, Ragnarsson U (1981) A rapid and convenient method for specific 11C-labelling of synthetic polypeptides containing methionine. J Label C Radiopharm XVIII:479–487
Franzen HM, Ragnarsson U, Nagren K, Langstrom B (1987) 11C-labelling of substance P. Preparation of a homocysteine-containing precursor and its subsequent application in the synthesis of the labelled neuropeptide. J Chem Soc I:2271–2247
Franzen HM, Ragnarsson U (1988) Preperation and 11C-labelling of a substance P analogue containing D-tryptophan in positions 7 and 9. J Chem Soc 1:497–502
Någren K, Ragnarsson U, Långström B (1988) Synthesis of three 11C-labelled methionine-containing enkephal in analogues. J Label Compd Radiopharm 25:149–160
Nabulsi NB, Smith DE, Kilbourn MR (2005) [11C]Glycylsarcosine: synthesis and in vivo evaluation as a PET tracer of PepT2 transporter function in kidney of PepT2 null and wild-type mice. Bioorganic Med Chem 13:2993–3001
Pekošak A, Rotstein BH, Collier TL et al (2017) Stereoselective 11C-labeling of a “native” tetrapeptide using asymmetric phase-transfer catalyzed alkylation reactions. European J Org Chem 2017:1019–1024
Filp U, Pekošak A, Poot AJ, Windhorst AD (2017) Stereocontrolled 11C-alkylation of N-terminal glycine Schiff bases to obtain dipeptides. European J Org Chem 2017:5592–5596
Andersen TL, Nordeman P, Christoffersen HF et al (2017) Application of methyl bisphosphine-ligated palladium complexes for low pressure N - 11 C-acetylation of peptides. Angew Chemie Int Ed 56:4549–4553
Zhao W, Lee HG, Buchwald SL et al (2017) Direct CN-labeling of unprotected peptides via palladium- mediated sequential cross-coupling reactions direct 11CN-labeling of unprotected peptides via palladium-mediated sequential cross-coupling reactions. J Am Chem Soc 139:7152–7155
Harada R, Furumoto S, Yoshikawa T et al (2012) Synthesis of [ 11C]interleukin 8 using a cell-free translation system and l-[ 11C]methionine. Nucl Med Biol 39:155–160
Gross MD, Shapiro B, Fig LM et al (2001) Imaging of human infection with (131)I-labeled recombinant human interleukin-8. J Nucl Med 42:1656–1659
Bleeker-Rovers CP, Rennen HJJM, Boerman OC et al (2007) 99mTc-labeled interleukin 8 for the scintigraphic detection of infection and inflammation: first clinical evaluation. J Nucl Med 48:337–343
Wadsak W, Mitterhauser M (2010) Basics and principles of radiopharmaceuticals for PET/CT. Eur J Radiol 73:461–469
Ermert J, Coenen HH (2013) Methods for (11) C- and (18) F-labelling of amino acids and derivatives for positron emission tomography imaging. J Labelled Comp Radiopharm 56:225–236
Richter S, Wuest F (2014) 18F-labeled peptides: the future is bright. Molecules 19:20536–20556
Langstrom B, Antoni G, Gullberg P et al (1987) Synthesis of L- and D- [methyl-11C]methionine. J Nucl Med 28:1037–1041
Kuang Y, Wang F, Corn DJ et al (2014) Metabolism of radiolabeled methionine in hepatocellular carcinoma. Mol Imaging Biol 16:44–52
Lilja A, Bergstrom K, Hartvig P et al (1985) Dynamic study of supratentorial gliomas with L-methyl-11C-methionine and positron emission tomography. Am J Neuroradiol 6:505–514
Hartvig P, Agren H, Reibring L et al (1991) Brain kinetics of L-[beta-11C]dopa in humans studied by positron emission tomography. J Neural Transm Gen Sect 86:25–41
Qu W, Oya S, Lieberman BP et al (2012) Preparation and characterization of L-[5-11C]-glutamine for metabolic imaging of tumors. J Nucl Med 53:98–105
van Ginkel RJ, Kole AC, Nieweg OE et al (1999) L-[1-11C]-tyrosine PET to evaluate response to hyperthermic isolated limb perfusion for locally advanced soft-tissue sarcoma and skin cancer. J Nucl Med 40:262–267
Ogawa T, Inugami A, Hatazawa J et al (1996) Clinical positron emission tomography for brain tumors: comparison of fludeoxyglucose F 18 and L-methyl-11C-methionine. Am J Neuroradiol 17:345–353
Daemen BJG, Zwertbroek R (1991) PET studies with 11C-TYR and Methyl-MET and FDG in prolactinomas in relation to bromocryptine treatment. Eur J Nucl Med 18:453–460
Lindholm P, Leskinen-kalilo S, Minn H et al (1993) Comparison of fluorine-18-fluorodeoxyglucose and carbon-11-methionine in head and neck cancer. J Nucl Med 34:1711–1716
Nettelbladt OS, Sundin AE, Valind SO et al (1998) Combined fluorine-18-FDG and carbon-11-methionine PET for diagnosis of tumors in lung and mediastinum. J Nucl Med 39:640–647
Leskinen-kallio S, Ruotsalainen U, Nã K, Terã M (2017) Uptake of carbon-11 methionine and fluorodeoxyglucose lymphoma: a PET study. J Nucl Med 32:1211–1219
Juhasz C, Dwivedi S (2014) Comparison of amino acid positron emission tomographic radiotracers for molecular imaging of primary and metastatic brain tumors. Mol Imaging 13:1–16
O’Tuama LA, Phillips PC, Smith QR et al (1991) L-methionine uptake by human cerebral cortex: maturation from infancy to old age. J Nucl Med 32:16–22
Harris SM, Davis JC, Snyder SE et al (2013) Evaluation of the biodistribution of 11C-methionine in children and young adults. J Nucl Med 54:1902–1908
Ishiwata K, Hatazawa J, Kubota K et al (1989) Metabolic fate of L-[methyl-11C]methionine in human plasma. Eur J Nucl Med 15:665–669
Glaudemans AWJM, Enting RH, Heesters MAAM et al (2013) Value of 11C-methionine PET in imaging brain tumours and metastases. Eur J Nucl Med Mol Imaging 40:615–635
Zhao C, Zhang Y, Wang J (2014) A meta-analysis on the diagnostic performance of F18-FDG and C11-MET PET for differentiating brain tumors. Am J Neuroradiol 35:1058–65
Coope DJ, Cízek J, Eggers C et al (2007) Evaluation of primary brain tumors using 11C-methionine PET with reference to a normal methionine uptake map. J Nucl Med 48:1971–1980
Kubota K (1990) Diagnosis of lung cancer with positron emission tomography (PET). J Nucl Med 31:1927–1933
Cook GJR, Maisey MN, Fogelman I (1999) Normal variants, artefacts and interpretative pitfalls in PET imaging with 18-fluoro-2-deoxyglucose and carbon-11 methionine. Eur J Nucl Med 26:1363–1378
Ishiwata K, Vaalburg W, Elsinga PH et al (1988) Metabolic studies with L-[1-14C]tyrosine for the investigation of a kinetic model to measure protein synthesis rates with PET. J Nucl Med 29:524–529
Kole AC, Pruim J, Nieweg OE et al (1997) PET with L-[ 1-carbon-11]-tyro sine to visualize tumors and measure protein synthesis rates potential todiagnose cancer monitor. J Nucl Med 38:191–196
Daemen BJG, Elsinga PH, Paans AMJ et al (1992) Radiation-induced inhibition of tumor growth as monitored by PET using L-[1-11C]tyrosine and fluorine-18-fluorodeoxyglucose. J Nucl Med 33:373–379
Hubner KF, Andrews GA, Buonocore E et al (1979) Carbon-11-labeled a mino acids for the rectilinear and positron tomographic imaging of the human pancreas. J Nucl Med 20:507–514
Fowler JS, Ansari AN, Atkins H et al (1973) Synthesis and preliminary evaluation in animals of carrier-free 11C-1-dopamine hydrochloride: X. J Nucl Med 14:867–869
Hartvig P, Lindner KJ, Tedroff J et al (1992) Regional brain kinetics of 6-fluoro-(beta-11C)-L-dopa and (beta-11C)-L-dopa following COMT inhibition. A study in vivo using positron emission tomography. J Neural Transm Gen Sect 87:15–22
Kumakura Y, Cumming P (2009) PET studies of cerebral levodopa metabolism: a review of clinical findings and modeling approaches. Neurosci 15:635–650
Bergström M, Eriksson B, Oberg K et al (1996) In vivo demonstration of enzyme activity in endocrine pancreatic tumors: decarboxylation of carbon-11-DOPA to carbon-11-dopamine. J Nucl Med 37:32–37
Bhutia YD, Babu E, Ramachandran S, Ganapathy V (2015) Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res 75:1782–1788
Turton D, Brady F, Pike VW (1984) Preparation of human serum [methyl-11C]methylalbumin microspheres and human serum [methyl-11C]methylalbumin for clinical use. Int J Appl Radiat Isot 35:337–344
Stöber B, Tanase U, Herz M et al (2006) Differentiation of tumour and inflammation: characterisation of [methyl-3H]methionine (MET) and O-(2-[18F]fluoroethyl)-L- tyrosine (FET) uptake in human tumour and inflammatory cells. Eur J Nucl Med Mol Imaging 33:932–939
Wang L, Zha Z, Qu W et al (2012) Synthesis and evaluation of 18F labeled alanine derivatives as potential tumor imaging agents. Nucl Med Biol 39:933–943
Venneti S, Dunphy MP, Zhang H et al (2015) Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med 7:274ra17
Fasth K-J, Hornfeldt K, Langstrom B (1995) Asymmetric synthesis of 11C-labelled L and D Ammino Acids by alkylation of imidazolidinone derivates. Acta Chem Scand 49:301–304
Acknowledgements
The study was supported by the European Marie Curie Actions RADIOMI Initial Training Network (FP7-PEOPLE-2012-ITN) under project reference no. 316882.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pekošak, A., Filp, U., Poot, A.J. et al. From Carbon-11-Labeled Amino Acids to Peptides in Positron Emission Tomography: the Synthesis and Clinical Application. Mol Imaging Biol 20, 510–532 (2018). https://doi.org/10.1007/s11307-018-1163-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-018-1163-5