
Abstract
Within the family of purinergic receptors, the P2X1 receptor is a ligand-gated ion channel that plays a role in urogenital, immune and cardiovascular function. Specifically, the P2X1 receptor has been implicated in controlling smooth muscle contractions of the vas deferens and therefore has emerged as an exciting drug target for male contraception. In addition, the P2X1 receptor contributes to smooth muscle contractions of the bladder and is a target to treat bladder dysfunction. Finally, platelets and neutrophils have populations of P2X1 receptors that could be targeted for thrombosis and inflammatory conditions. Drugs that specifically target the P2X1 receptor have been challenging to develop, and only recently have small molecule antagonists of the P2X1 receptor been available. However, these ligands need further biological validation for appropriate selectivity and drug-like properties before they will be suitable for use in preclinical models of disease. Although the atomic structure of the P2X1 receptor has yet to be determined, the recent discovery of several other P2X receptor structures and improvements in the field of structural biology suggests that this is now a distinct possibility. Such efforts may significantly improve drug discovery efforts at the P2X1 receptor.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
P2X1 receptors
P2 receptors
P2 receptors have been identified in all human organ systems; they mediate a wide array of physiological responses [1, 2]. In humans, there are seven subtypes of the P2X ligand-gated ion channel (P2X1-7) and eight subtypes of the P2Y G protein-coupled receptor (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, P2Y14). Studies utilising selective antagonists and genetic knockouts have identified P2 receptors as potential targets for multiple conditions [3, 4]. However, only a small proportion of P2X and P2Y receptors are current targets of clinically successful medicines (Table 1). There is, thus, a clear need for more selective drug-like compounds to validate and progress clinical opportunities. This is particularly true for the subject of this review, the P2X1 receptor. Although the P2X1 receptor has been discussed in several exemplar reviews [4,5,6,7,8,9], it has often been overlooked in favour of other P2X subtypes. Herein, we review what is known about the P2X1 receptor as a drug target and the currently available ligands that target the receptor. We also highlight the potential impact that advances in structural biology could have on P2X1 receptor drug development.
P2X receptors
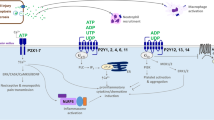
P2X receptors consist of three individual subunits that can be homotrimeric or heterotrimeric and together form a ligand-gated ion channel [10, 11]. Each P2X subunit contains two transmembrane spanning helices, a large extracellular domain and a small intracellular N- and C-terminus (Fig. 1a) except for the P2X7 receptor, which has a longer C-terminus. Endogenous ATP activates P2X receptors by binding to the three ATP binding sites that are located in the extracellular domain between subunits (Fig. 1b). Upon activation, P2X receptors form a non-selective pore that is permeable to cations; in the cellular context, these are typically calcium, potassium and sodium [12]. In addition, the P2X1, P2X2, P2X3, P2X4 and P2X7 receptors have been shown to transport large organic cations such as NMDG+ and spermidine across the membrane [8, 13,14,15]. The physiological importance of the transport of large cations has been difficult to study. However, a mutation in the P2X7 receptor which restricts large pore formation but not cation permeability was linked to chronic pain sensitivity [16]. The permeation of small cations, on the other hand, is critical in mediating specific cellular events. A few examples include smooth muscle contraction [17, 18], action potential propagation [19] and inflammation [20]. Following activation of P2X receptors, P2X1 and P2X3 receptors undergo rapid desensitisation while P2X2, P2X4, P2X5 and P2X7 receptors are minimally desensitised [12]. Trafficking studies show that the P2X1, P2X3, P2X4 and P2X7 receptors internalise upon activation and utilise the dynamin and clathrin-mediated pathways [21,22,23,24,25]. The P2X6 receptors are unique in that they have an uncharged N-terminal region that restricts the formation of functional homomeric channels and is retained within the endoplasmic reticulum [26]. P2X6 receptors can form functional heteromeric channels with P2X2, P2X4 and P2X7 receptors [11]. Glycosylation of P2X receptors is essential in the assembly and trafficking of functional receptors to the cell membrane [27,28,29,30], and enhanced P2X6 glycosylation has been shown to improve cell-surface expression and restore function [31]. In addition, the C-terminal of P2X receptors share a YXXXK motif that is important for membrane expression, while other regions, such as a tyrosine C-terminal motif at the P2X4 receptor and single residues within the C-terminus of the P2X7 receptor, control internalisation and trafficking [32,33,34,35,36]. Further information on the general properties of P2X receptors is available in a recent International Union of Basic and Clinical Pharmacology review [4].

a A monomeric subunit of the P2X receptor in the plasma membrane and b a functional trimeric P2X receptor shown as a cartoon. There are three ATP binding sites located in the extracellular domain between subunits. Upon receptor activation by ATP binding, there is an inward flux of calcium and sodium ions and an outward flux of potassium ions
P2X1 receptors
The P2X1 receptor has a unique pharmacological profile that distinguishes it from other P2X receptors. For example, in heterologous systems, ATP exhibits the highest potency at the P2X1 receptor, with values ranging from 56 to 300 nM (Table 2). Additionally, only the P2X1 and P2X3 receptors are rapidly desensitised on a sub-second timescale [37]. This fast desensitisation has been shown to reduce the apparent potency of ATP for the P2X1 receptor [38]. Fluorescent labelling and cell surface biotinylation studies show that, in contrast to other P2X receptors, the P2X1 receptor is quickly internalised and recycles back to the membrane [25, 39, 40]. The P2X1 receptor also has higher fractional calcium currents than all other P2X receptors and is ranked highly among other ligand-gated ion channels for having high calcium permeability [41].
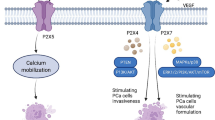
There are functional roles for the P2X1 receptor in the urogenital, immune and cardiovascular systems. Within the urogenital system, P2X1 receptors localise to smooth muscle cells of various organs and tubules, including the vas deferens and bladder [1, 42]. Knockout studies in mice showed reduced fertility in male mice due to decreased vas deferens contractility, demonstrating the importance of the P2X1 receptor on the vas deferens [43, 44]. In the cardiovascular system, P2X1 receptors are primarily located on arterial smooth muscle, and P2X1 receptor knockout mice showed a slight increase in blood pressure [1, 42, 44, 45]. P2X1 receptors are also localised to immune cells such as platelets, macrophages, neutrophils and mast cells [1, 42, 46, 47]. Further studies in knockout mice established a functional role as mice exhibited impaired thrombus formation [48].
Like most P2X receptors, the quaternary P2X1 receptor structure can be in a heterotrimeric or homotrimeric form. Biochemical methods, ligand sensitivity and desensitisation kinetics have shown differences between the homotrimeric and heterotrimeric forms of the P2X1 receptors [11]. The P2X1/2, P2X1/4 and P2X1/5 heterotrimeric receptors have been co-purified and functionally verified in heterologous systems [11]. In native systems, heterotrimeric P2X1 receptors were expressed in low quantities making it difficult to ascertain their functional implications [49,50,51]. The homotrimeric form of the P2X1 receptor is likely the prevalent form, particularly in systems like the vas deferens where only the P2X1 receptor is expressed [44].
P2X1 receptors as therapeutic drug targets
Male contraception
P2X1 receptors and α1A-adrenoceptors are co-localised on the smooth muscle of the vas deferens [43]. Following activation of these receptors, there are contractions of the vas deferens, which propel spermatozoa anterograde through the vas deferens to the ejaculatory duct, where they mix with glandular secretions and are expelled during ejaculation [52]. If these receptors are blocked, sperm cannot leave their storage site in the cauda epididymis rendering a male infertile. Validation for this concept comes from a combined genetic knockout of the P2X1 receptor and α1A-adrenoceptor that produced complete infertility in a dual knockout male mice population [43]. Furthermore, the contraction of the human and mouse vas deferens is controlled by the same adrenergic and purinergic receptors suggesting that the contraceptive efficacy seen in mice could translate to humans [43, 53]. Equally important was the observation that the dual knockout male mice were sexually, physiologically and behaviourally healthy. There was concern that the P2X1 receptor and α1A-adrenoceptor knockout mice could have cardiovascular complications as both receptors are located on the smooth muscle of blood vessels and mediate vasoconstriction. Fortunately, the dual knockout mice had only minor changes in their baroreflex response, resting arterial pressure and heart rate similar to the changes seen in α1A-adrenoceptor knockout mice. In addition, clinical trials using α1A-adrenoceptor antagonists such as silodosin and tamsulosin as oral male contraceptives have shown promise [54,55,56]. This is encouraging, as α1A-adrenoceptor antagonists have been used chronically by men to treat benign prostatic hyperplasia and have proven to be safe and effective [57, 58]. However, pharmacological in vivo studies are needed to further validate the P2X1 receptor as a male contraceptive target. Nevertheless, the P2X1 receptor is a promising target for the design of a non-hormonal oral contraceptive.
Thrombosis and inflammation
P2X1 receptors are also located on platelets and neutrophils, and within these cells, P2X1 receptors have important functional roles. P2X1 receptor activation can mediate platelet shape change, amplify platelet signalling and cause shear-induced aggregation of platelets [59, 60]. In mice models, pharmacologically blocking or genetically deleting the P2X1 receptor causes impairment in thrombus formation [48, 61]. Thrombosis is a cardiovascular condition in which a blood clot blocks blood flow. In theory, reducing platelet activation by antagonising the P2X1 receptor could alleviate thrombosis. P2X1 receptor activation also regulates activation and promotes chemotaxis of neutrophils [62, 63]. These two actions may be conflicting in inflammatory conditions, as seen in a model of acute colitis where P2X1 receptor knockout mice had high neutrophil levels, which contributed to thrombosis and intestinal bleeding [64]. In addition, studies into sepsis survival in pharmacological and genetic P2X1 receptor knockout mice have been conflicting, although most reported a reduction in survival rates signifying that the P2X1 receptor has a protective role [62, 65, 66]. Therefore, P2X1 receptors on neutrophils and platelets could be targeted for their regulatory role in severe inflammatory conditions such as sepsis. Further research is needed to describe if these conflicting functions of the P2X1 receptor will be detrimental for drug development. A functional role for the P2X1 receptor has also been identified for mast cells and macrophages, suggesting additional roles in inflammation and the immune system [46, 47].
Bladder dysfunction
ATP and acetylcholine co-released from parasympathetic nerves stimulate smooth muscle contractions of the bladder from P2X1 and muscarinic receptors, respectively, which causes the voiding of urine [67]. Post junctional muscarinic receptors are typically targeted to treat bladder dysfunction, but purinergic contractions in bladder conditions such as overactive bladder and interstitial cystitis can be enhanced [68, 69]. Prolonged exposure of isolated detrusor muscle to α,β-meATP completely abolished purinergic contractions; hence, a P2X1 receptor antagonist could theoretically be used to abolish the purinergic component of the bladder contractions [70]. However, the P2X receptor antagonists suramin and PPADS were unable to completely inhibit the non-cholinergic contractions suggesting a role for another purinergic receptor [70]. As such, specific P2X1 receptor antagonists are needed to further validate the role of the P2X1 receptor as a target for bladder dysfunction.
P2X1 drug discovery
Medicinal chemistry
Many compounds have been shown to modulate P2X1 receptors with varying levels of selectivity and potency (Table 2, see for references). Derivatives of ATP have been designed to increase selectivity and reduce metabolic breakdown by ectonucleotidases. α,β-meATP is one such compound with high selectivity for the P2X1 and P2X3 receptor and has slower enzymatic breakdown than ATP [71, 72]. Another ATP derivative, BzATP, is the most potent agonist at the P2X1 receptor. Diadenosine polyphosphates and similar compounds can be both antagonists or agonists of the P2X1 receptor depending upon the length of the phosphate chain and the substituents on the adenosine group [73, 74]. The most potent P2X1 receptor antagonist is NF449 with sub-nanomolar potency at human and rat P2X1 receptors and is selective over other purinergic receptors [75]. Due to high molecular weight and polarity, these compounds are poor starting points for drug development (Fig. 2) [71]. Nevertheless, these compounds have been useful for in vitro and in vivo studies to investigate the function of the P2X1 receptor [17, 48]. PPADS is a selective P2 receptor antagonist whose drug properties were improved to create a series of antagonists, including MRS2159 and compound 1, which are low molecular weight compounds that are selective for the P2X1 receptor [76, 77]. Several research groups have identified novel P2X1 receptor antagonists. These antagonists have inhibitory activity in the range of low micromolar to low nanomolar potency and will hopefully become building blocks for molecules to be used in preclinical and clinical studies. Currently, MRS2159, aurintricarboxylic acid (ATA), PSB-2001 and compound 1 are the most exciting antagonists of the P2X1 receptor due to their nanomolar potency and low molecular weight. Furthermore, ATA, PSB-2001 and compound 2 are reported as non-competitive antagonists due to their pharmacological profile and molecular modelling studies [78,79,80]. Allosteric ligands are of great interest as it is likely easier to design selective ligands by not targeting the conserved ATP binding sites. In addition, a few studies have also looked into designing positive allosteric modulators (PAMs) that target the P2X1 receptor. So far, only MRS2219 has been identified as a small molecule PAM, although phosphoinositides and gintonin have been shown to potentiate P2X1 activation [5, 81, 82].

Chemical structures of P2X1 receptor modulators. P2X1 receptor modulators are referred to by their most common abbreviated name except for compounds 1–5 which do not have a common name and have been assigned a number
P2X1 structural biology
Structural biology of P2X receptors
Structural biology delivers a unique view into understanding the function of macromolecules on a molecular level. In particular, structural biology techniques are highly complementary to drug discovery efforts and can greatly facilitate the discovery and improvement of new therapeutics [83,84,85]. Prior to 2009, there were no available P2X receptor structures and mutagenesis studies were the primary method to study the key components of P2X receptors. However, since then, 29 P2X receptor structures have been deposited in the Protein Data Bank (PDB; Table 3). These structures were solved using nuclear magnetic resonance (NMR) spectroscopy (1 structure), X-ray crystallography (26 structures) and cryogenic electron microscopy (cryo-EM; 2 structures) with a resolution range between 2.7 and 4.1 Å. Recent advancements in 3D protein prediction have been significant, highlighted by AlphaFold predicting structures for the entire human proteome with reasonable accuracy for many proteins [86, 87].
The primary structure of human P2X receptors (hP2X1-7) is between 388 and 595 amino acids and has 35–52% sequence similarity. The general structure of a P2X receptor was first described in the discovery of the zebrafish P2X4 receptor structure and the P2X4 subunit was zoomorphically described as the shape of a dolphin (Fig. 3a) [10]. Specifically, each P2X monomer can be described as having a head domain, left and right flipper, dorsal fin, upper body, lower body and fluke (Fig. 3a). All experimentally determined P2X receptor structures have exhibited the same structural architecture for the extracellular and transmembrane regions (Fig. 3a) [88, 89]. AlphaFold structure prediction of the P2X1, P2X2, P2X5 and P2X6 receptors also shows a similar architecture for the extracellular and transmembrane domains but not the intracellular region, this may reflect the fact that the N- and C-terminus are likely to be disordered, as all experimentally solved P2X receptor structures, except the cryo-EM P2X7 receptor structure, have truncated N- and C-termini (Fig. 3b) [86, 87].

a Experimentally determined P2X structures coloured by features of a dolphin (based on the zfP2X4 [10]) with the head domain in pink, the upper body in dark blue, the lower body in light blue, the left flipper in yellow, the right flipper in red, the dorsal fin in orange and the fluke in green. b AlphaFold generated P2X receptor structures with each residue coloured by the confidence of the prediction with dark blue a very high confidence prediction, light blue a high confidence prediction, yellow a low confidence prediction and orange a very low confidence prediction
P2X1 receptor structure
Currently, there are no experimentally determined P2X1 receptor structures; however, reasonable assumptions can be made from other solved P2X receptor structures, molecular modelling, protein predication and mutagenesis of the P2X1 receptor. The P2X1 receptor AlphaFold model has a well predicted extracellular domain, orthosteric binding site and transmembrane domain (Fig. 3b). A recent study generated P2X1 receptor homology models using the experimentally determined zfP2X4, hP2X3 and pdP2X7 receptor structures [78]. This homology model compares well with the AlphaFold model of a single subunit alignment that has a root-mean-square deviation of 2.38 Å. P2X1 receptor models such as these could serve as valuable tools for modelling and structure-based drug design projects.
Structures of P2X receptors have revealed three ATP binding sites where each monomer interfaces with the neighbouring monomer and is located approximately 40 Å above the transmembrane helices (Fig. 4a). The ATP molecules assume a U shape in the conserved orthosteric binding site (Fig. 4b). Mutagenesis of the ATP binding site in P2X1 receptors has revealed key residues. Alanine mutation of amino acid residues K68, K70, T186, N290, R292 and K309 in the P2X1 receptor caused large reductions in the potency of ATP [90, 91]. These residues are highly conserved, and ATP-bound P2X receptor structures reveal that these residues interact strongly with ATP (Fig. 4b) [88, 89, 92]. Several other P2X1 receptor residues have been linked to receptor activation, but most of these residues are unlikely to interact with ATP directly and instead may be important for either protein folding or the conformational rearrangement that occurs during activation (see review [93]). Another feature of the P2X1 orthosteric binding site may have been revealed by the hP2X3 receptor structures that contained a magnesium binding site adjacent to ATP [94]. This is supported by the ATP-Mg2+ complex being an agonist for the hP2X1 and hP2X3 receptors suggesting the magnesium binding site may also be located in the P2X1 receptor [95]. Although we have a good description of the ATP binding site for the P2X1 receptor, additional interactions could be uncovered by a high-resolution structure.

a ATP-bound hP2X3 receptor with one of the ATP binding locations emphasised (Protein Data Bank: 5SVK). b A close up view of the ATP molecule bound to the hP2X3 receptor with the interacting residues labelled, the interacting hP2X3 residues are reported in the table below with the sequence aligned P2X1 residues
Channel activation shares a common mechanism among P2X receptor structures in which the dorsal fin and head domains move toward each other, producing a cleft closure, while the left flipper domain moves away from the orthosteric binding site. A P2X1 receptor mutagenesis study revealed that, unsurprisingly, the head domain is important in receptor activation. The movements of the dorsal fin and head domains produce an outward flexing of the rigid lower body and the beta-sheet strands of the lower body translate the movement to the transmembrane domains (TM). TM2 rotates counterclockwise, and there is an outward flexing that opens the pore and allows the permeation of cations. Mutagenesis studies of the P2X1, P2X2, P2X3 and P2X4 receptors have identified that polar and acidic residues on the outer edge of the transmembrane regions contribute significantly to calcium permeability; however, further studies are needed to explain what residues contribute to differing cation permeability among P2X receptors [41, 96,97,98]. Another region of interest for the P2X1 receptor is the cytoplasmic region, of which the N-terminus has been shown to control receptor desensitisation [99]. A study using cysteine mutagenesis and cross-linking compounds at the P2X1 receptor suggested that the cytoplasmic domain remains in a cap-like structure in both the apo and desensitised states [100]. The open state structure of the hP2X3 receptor revealed a cytoplasmic cap containing β-strands from the N- and C-terminus, which contain residues responsible for modulating receptor desensitisation [89]. Full-length P2X receptor structures would be useful in defining the differences among P2X receptors for receptor desensitisation and ion permeability.
P2X3, P2X4 and P2X7 receptor structures have revealed novel allosteric binding sites located in the extracellular domain of the P2X receptor [101, 102]. The hP2X3 receptor bound to gefapixant (other names: AF-219 and MK-7264) revealed an allosteric binding site located between the lower body and left flipper [101]. A series of structurally distinct ligands were crystallised in the pdP2X7 structure and revealed another allosteric binding site found in the upper body of the pdP2X7 receptor [102]. Using the zfP2X4 receptor structure to model in ligands complimented with mutagenesis studies, BX430 and 5-BDBD were demonstrated to bind the P2X4 receptor at an allosteric site located at the upper and lower body of the P2X4 receptor [103, 104]. There are other P2X receptor ligands that have been described as non-competitive but need to be further validated using mutagenesis and molecular modelling studies to verify their binding location. Unfortunately, most allosteric ligands for other P2X receptors have low potency for the P2X1 receptor, suggesting that these allosteric binding sites may not be completely conserved in the P2X1 receptor [105,106,107]. Fortunately, non-competitive antagonists for the P2X1 receptor have been recently discovered and molecular modelling studies have revealed two putative allosteric binding sites [78,79,80]. One allosteric site is located in the upper body of the P2X1 receptor using PSB-2001 which was docked into a homology model of the P2X1 receptor [78]. The other allosteric site is located between the lower body and left flipper as binding data demonstrated that ATA did not bind to the orthosteric binding site and molecular modelling showed that ATA docks at the same location as gefapixant in the hP2X3 receptor structure. Identifying and targeting allosteric binding sites is highly desirable for the P2X receptors as these are likely to represent better locations for designing specific ligands compared to the highly conserved orthosteric binding site in P2X receptors. Overall, many significant questions remain surrounding P2X1 receptor structure, function and drug discovery that are ready to be answered by experimental structures of the P2X1 receptor.
Conclusion
The P2X1 receptor is still at the formative stage for the development of new therapeutics as promising (patho)physiology and clinical indications have been identified. The development of potent small-molecule ligands is progressing but has yet to produce the highly potent and well-validated compounds that are needed for in vivo studies. Improvement to the current suite of P2X1 receptor modulators is essential, which could be facilitated by structure-based discovery efforts. The recent determination of the P2X3, P2X4 and P2X7 receptor structures along with new models generated by AlphaFold provides a pathway toward the discovery of new P2X receptor structures [83, 84]. Ultimately, the P2X1 receptor needs better chemical tools to further validate its therapeutic potential in the treatment of thrombosis, inflammation, bladder dysfunction and as a male contraceptive.
Data availability
The data discussed can be found in the original publications referenced in the text.
References
Burnstock G, Knight GE (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. In: International review of cytology. Academic Press, pp 31–304
Burnstock G (2018) Purine and purinergic receptors. Brain Neurosci Adv 2:2398212818817494. https://doi.org/10.1177/2398212818817494
Burnstock G (2018) The therapeutic potential of purinergic signalling. Biochem Pharmacol 151:157–165. https://doi.org/10.1016/j.bcp.2017.07.016
Illes P, Müller CE, Jacobson KA et al (2021) Update of P2X receptor properties and their pharmacology: IUPHAR review 30. Br J Pharmacol 178:489–514. https://doi.org/10.1111/bph.15299
Stokes L, Bidula S, Bibič L, Allum E (2020) To inhibit or enhance? Is there a benefit to positive allosteric modulation of P2X receptors? Front Pharmacol 11:627. https://doi.org/10.3389/fphar.2020.00627
Oury C, Wéra O (2021) P2X1: a unique platelet receptor with a key role in thromboinflammation. Platelets 32:902–908. https://doi.org/10.1080/09537104.2021.1902972
Müller CE, Namasivayam V (2021) Recommended tool compounds and drugs for blocking P2X and P2Y receptors. Purinergic Signal 17:633–648. https://doi.org/10.1007/s11302-021-09813-7
Schmid R, Evans RJ (2019) ATP-gated P2X receptor channels: molecular insights into functional roles. Annu Rev Physiol 81:43–62. https://doi.org/10.1146/annurev-physiol-020518-114259
Sheng D, Hattori M Recent progress in the structural biology of P2X receptors. Proteins Struct Funct Bioinforma. https://doi.org/10.1002/prot.26302
Kawate T, Michel JC, Birdsong WT, Gouaux E (2009) Crystal structure of the ATP-gated P2X 4 ion channel in the closed state. Nature 460:592–598. https://doi.org/10.1038/nature08198
Saul A, Hausmann R, Kless A, Nicke A (2013) Heteromeric assembly of P2X subunits. Front Cell Neurosci. https://doi.org/10.3389/fncel.2013.00250
North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82:1013–1068
Peverini L, Beudez J, Dunning K et al (2018) New insights into permeation of large cations through ATP-gated P2X receptors. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2018.00265
Harkat M, Peverini L, Cerdan AH et al (2017) On the permeation of large organic cations through the pore of ATP-gated P2X receptors. Proc Natl Acad Sci U S A 114:E3786–E3795
Evans RJ, Lewis C, Virginio C et al (1996) Ionic permeability of, and divalent cation effects on, two ATP-gated cation channels (P2X receptors) expressed in mammalian cells. J Physiol 497:413–422. https://doi.org/10.1113/jphysiol.1996.sp021777
Sorge RE, Trang T, Dorfman R et al (2012) Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nat Med 18:595–599. https://doi.org/10.1038/nm.2710
Amobi NIB, Guillebaud J, Smith ICH (2012) Perspective on the role of P2X-purinoceptor activation in human vas deferens contractility. Exp Physiol 97:583–602. https://doi.org/10.1113/expphysiol.2011.063206
Burnstock G, Ralevic V (2014) Purinergic signaling and blood vessels in health and disease. Pharmacol Rev 66:102–192. https://doi.org/10.1124/pr.113.008029
Dunn PM, Zhong Y, Burnstock G (2001) P2X receptors in peripheral neurons. Prog Neurobiol 65:107–134. https://doi.org/10.1016/S0301-0082(01)00005-3
Burnstock G (2016) P2X ion channel receptors and inflammation. Purinergic Signal 12:59–67. https://doi.org/10.1007/s11302-015-9493-0
Bobanovic LK, Royle SJ, Murrell-Lagnado RD (2002) P2X receptor trafficking in neurons is subunit specific. J Neurosci 22:4814–4824. https://doi.org/10.1523/JNEUROSCI.22-12-04814.2002
Feng Y-H, Wang L, Wang Q et al (2005) ATP stimulates GRK-3 phosphorylation and β-arrestin-2-dependent internalization of P2X7 receptor. Am J Physiol Cell Physiol 288:C1342–C1356. https://doi.org/10.1152/ajpcell.00315.2004
Stokes L (2013) Rab5 regulates internalisation of P2X4 receptors and potentiation by ivermectin. Purinergic Signal 9:113–121. https://doi.org/10.1007/s11302-012-9336-1
Vacca F, Giustizieri M, Ciotti MT et al (2009) Rapid constitutive and ligand-activated endocytic trafficking of P2X3 receptor. J Neurochem 109:1031–1041. https://doi.org/10.1111/j.1471-4159.2009.06029.x
Lalo U, Allsopp RC, Mahaut-Smith MP, Evans RJ (2010) P2X1 receptor mobility and trafficking; regulation by receptor insertion and activation. J Neurochem 113:1177–1187. https://doi.org/10.1111/j.1471-4159.2010.06730.x
Ormond SJ, Barrera NP, Qureshi OS et al (2006) An uncharged region within the N terminus of the P2X6 receptor inhibits its assembly and exit from the endoplasmic reticulum. Mol Pharmacol 69:1692–1700. https://doi.org/10.1124/mol.105.020404
Torres GE, Egan TM, Voigt MM (1998) N-linked glycosylation is essential for the functional expression of the recombinant P2X2 receptor. Biochemistry 37:14845–14851. https://doi.org/10.1021/bi981209g
Rettinger J, Aschrafi A, Schmalzing G (2000) Roles of individual N-glycans for ATP potency and expression of the rat P2X1 receptor. J Biol Chem 275:33542–33547. https://doi.org/10.1074/jbc.M002918200
Vacca F, D’Ambrosi N, Nestola V et al (2011) N-glycans mutations rule oligomeric assembly and functional expression of P2X3 receptor for extracellular ATP. Glycobiology 21:634–643. https://doi.org/10.1093/glycob/cwq211
Lenertz LY, Wang Z, Guadarrama A et al (2010) Mutation of putative N-linked glycosylation sites on the human nucleotide receptor P2X7 reveals a key residue important for receptor function. Biochemistry 49:4611–4619. https://doi.org/10.1021/bi902083n
Jones CA, Vial C, Sellers LA et al (2004) Functional regulation of P2X6 receptors by N-linked glycosylation: identification of a novel αβ-methylene ATP-sensitive phenotype. Mol Pharmacol 65:979–985. https://doi.org/10.1124/mol.65.4.979
Royle SJ, Bobanović LK, Murrell-Lagnado RD (2002) Identification of a non-canonical tyrosine-based endocytic motif in an ionotropic receptor. J Biol Chem 277:35378–35385. https://doi.org/10.1074/jbc.M204844200
Chaumont S, Jiang L-H, Penna A et al (2004) Identification of a trafficking motif involved in the stabilization and polarization of P2X receptors. J Biol Chem 279:29628–29638. https://doi.org/10.1074/jbc.M403940200
Bradley HJ, Liu X, Collins V et al (2010) Identification of an intracellular microdomain of the P2X7 receptor that is crucial in basolateral membrane targeting in epithelial cells. FEBS Lett 584:4740–4744. https://doi.org/10.1016/j.febslet.2010.11.007
Wiley JS, Dao-Ung L-P, Li C et al (2003) An Ile-568 to Asn polymorphism prevents normal trafficking and function of the human P2X7 receptor. J Biol Chem 278:17108–17113. https://doi.org/10.1074/jbc.M212759200
Smart ML, Gu B, Panchal RG et al (2003) P2X7 receptor cell surface expression and cytolytic pore formation are regulated by a distal C-terminal region. J Biol Chem 278:8853–8860. https://doi.org/10.1074/jbc.M211094200
Bianchi BR, Lynch KJ, Touma E et al (1999) Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur J Pharmacol 376:127–138. https://doi.org/10.1016/S0014-2999(99)00350-7
Rettinger J, Schmalzing G (2004) Desensitization masks nanomolar potency of ATP for the P2X1 receptor. J Biol Chem 279:6426
Ennion SJ, Evans RJ (2001) Agonist-stimulated internalisation of the ligand-gated ion channel P2X1 in rat vas deferens. FEBS Lett 489:154–158. https://doi.org/10.1016/S0014-5793(01)02102-0
Dutton JL, Poronnik P, Li GH et al (2000) P2X1 receptor membrane redistribution and down-regulation visualized by using receptor-coupled green fluorescent protein chimeras. Neuropharmacology 39:2054–2066. https://doi.org/10.1016/S0028-3908(00)00058-7
Egan TM, Khakh BS (2004) Contribution of calcium ions to P2X channel responses. J Neurosci 24:3413–3420. https://doi.org/10.1523/JNEUROSCI.5429-03.2004
Mahaut Smith MP, Evans RJ, Vial C (2019) Development of a P2X1-eYFP receptor knock-in mouse to track receptors in real time. Purinergic Signal 15:397–402. https://doi.org/10.1007/s11302-019-09666-1
White CW, Choong Y-T, Short JL et al (2013) Male contraception via simultaneous knockout of α1A-adrenoceptors and P2X1-purinoceptors in mice. Proc Natl Acad Sci U S A 110:20825–20830. https://doi.org/10.1073/pnas.1318624110
Mulryan K, Gitterman DP, Lewis CJ et al (2000) Reduced vas deferens contraction and male infertility in mice lacking P2X1 receptors. Nature 403:86. https://doi.org/10.1038/47495
del Gonzalez-Montelongo M, C, Fountain SJ, (2021) Neuropeptide Y facilitates P2X1 receptor-dependent vasoconstriction via Y1 receptor activation in small mesenteric arteries during sympathetic neurogenic responses. Vascul Pharmacol 136:106810. https://doi.org/10.1016/j.vph.2020.106810
Wareham K, Vial C, Wykes RCE et al (2009) Functional evidence for the expression of P2X1, P2X4 and P2X7 receptors in human lung mast cells. Br J Pharmacol 157:1215–1224. https://doi.org/10.1111/j.1476-5381.2009.00287.x
Vargas-Martínez EM, Gómez-Coronado KS, Espinosa-Luna R et al (2020) Functional expression of P2X1, P2X4 and P2X7 purinergic receptors in human monocyte-derived macrophages. Eur J Pharmacol 888:173460. https://doi.org/10.1016/j.ejphar.2020.173460
Darbousset R, Delierneux C, Mezouar S et al (2014) P2X1 expressed on polymorphonuclear neutrophils and platelets is required for thrombosis in mice. Blood 124:2575–2585. https://doi.org/10.1182/blood-2014-04-571679
Harhun MI, Povstyan OV, Albert AP, Nichols CM (2014) ATP-evoked sustained vasoconstrictions mediated by heteromeric P2X1/4 receptors in cerebral arteries. Stroke 45:2444–2450. https://doi.org/10.1161/STROKEAHA.114.005544
Calvert JA, Evans RJ (2004) Heterogeneity of P2X receptors in sympathetic neurons: contribution of neuronal P2X1 receptors revealed using knockout mice. Mol Pharmacol 65:139–148. https://doi.org/10.1124/mol.65.1.139
Surprenant A, Schneider DA, Wilson HL et al (2000) Functional properties of heteromeric P2X1/5 receptors expressed in HEK cells and excitatory junction potentials in guinea-pig submucosal arterioles. J Auton Nerv Syst 81:249–263. https://doi.org/10.1016/S0165-1838(00)00123-5
Koslov DS, Andersson K-E (2013) Physiological and pharmacological aspects of the vas deferens—an update. Front Pharmacol 4:101. https://doi.org/10.3389/fphar.2013.00101
Banks FCL, Knight GE, Calvert RC et al (2006) The purinergic component of human vas deferens contraction. Fertil Steril 85:932–939. https://doi.org/10.1016/j.fertnstert.2005.09.024
Wang J, Zhao Y, Jiang S et al (2012) Assessment of tamsulosin as a potential male contraceptive in healthy volunteers. Urology 80:614–617. https://doi.org/10.1016/j.urology.2012.06.003
Hellstrom WJG, Sikka SC (2009) Effects of alfuzosin and tamsulosin on sperm parameters in healthy men: results of a short-term, randomized, double-blind, placebo-controlled, crossover study. J Androl 30:469–474. https://doi.org/10.2164/jandrol.108.006874
Bhat GS, Shastry A (2020) A prospective double-blind, randomized, placebo-controlled study to evaluate the efficacy of silodosin 8 mg as an on-demand, reversible, nonhormonal oral contraceptive for males: a pilot study. World J Urol 38:747–751. https://doi.org/10.1007/s00345-019-02806-7
Montorsi F, Moncada I (2006) Safety and tolerability of treatment for BPH. Eur Urol Suppl 5:1004–1012. https://doi.org/10.1016/j.eursup.2006.08.012
Wu YJ, Dong Q, Liu LR, Wei Q (2013) A meta-analysis of efficacy and safety of the new α1A-adrenoceptor-selective antagonist silodosin for treating lower urinary tract symptoms associated with BPH. Prostate Cancer Prostatic Dis 16:79–84. https://doi.org/10.1038/pcan.2012.36
Hechler B, Gachet C (2011) P2 receptors and platelet function. Purinergic Signal 7:293. https://doi.org/10.1007/s11302-011-9247-6
Mahaut-Smith MP, Jones S, Evans RJ (2011) The P2X1 receptor and platelet function. Purinergic Signal 7:341–356. https://doi.org/10.1007/s11302-011-9224-0
Oury C, Daenens K, Hu H et al (2006) ERK2 activation in arteriolar and venular murine thrombosis: platelet receptor GPIb vs. P2X1. J Thromb Haemost 4:443–452. https://doi.org/10.1111/j.1538-7836.2006.01745.x
Lecut C, Faccinetto C, Delierneux C et al (2012) ATP-gated P2X1 ion channels protect against endotoxemia by dampening neutrophil activation. J Thromb Haemost 10:453–465. https://doi.org/10.1111/j.1538-7836.2011.04606.x
Lecut C, Frederix K, Johnson DM et al (2009) P2X1 ion channels promote neutrophil chemotaxis through rho kinase activation. J Immunol 183:2801–2809. https://doi.org/10.4049/jimmunol.0804007
Wéra O, Lecut C, Servais L et al (2020) P2X1 ion channel deficiency causes massive bleeding in inflamed intestine and increases thrombosis. J Thromb Haemost 18:44–56. https://doi.org/10.1111/jth.14620
Maître B, Magnenat S, Heim V et al (2015) The P2X1 receptor is required for neutrophil extravasation during lipopolysaccharide-induced lethal endotoxemia in mice. J Immunol 194:739–749. https://doi.org/10.4049/jimmunol.1401786
Skals M, Greve A-S, Fagerberg SK et al (2019) P2X1 receptor blockers reduce the number of circulating thrombocytes and the overall survival of urosepsis with haemolysin-producing Escherichia coli. Purinergic Signal 15:265–276. https://doi.org/10.1007/s11302-019-09658-1
Vial C, Evans RJ (2000) P2X receptor expression in mouse urinary bladder and the requirement of P2X1 receptors for functional P2X receptor responses in the mouse urinary bladder smooth muscle. Br J Pharmacol 131:1489–1495. https://doi.org/10.1038/sj.bjp.0703720
Sjögren C, Andersson KE, Husted S et al (1982) Atropine resistance of transmurally stimulated isolated human bladder muscle. J Urol 128:1368–1371. https://doi.org/10.1016/s0022-5347(17)53509-0
Burnstock G (2014) Purinergic signalling in the urinary tract in health and disease. Purinergic Signal 10:103–155. https://doi.org/10.1007/s11302-013-9395-y
Kennedy C, Tasker PN, Gallacher G, Westfall TD (2007) Identification of atropine- and P2X1 receptor antagonist-resistant, neurogenic contractions of the urinary bladder. J Neurosci 27:845–851. https://doi.org/10.1523/JNEUROSCI.3115-06.2007
Catia L, Dal Diego B, Michela B et al (2015) Medicinal chemistry of P2X receptors: agonists and orthosteric antagonists. Curr Med Chem 22:915–928. https://doi.org/10.2174/0929867321666141215093513
Chen BC, Lin W-W (1997) Inhibition of ecto-ATPase by the P2purinoceptor agonists, ATPγS, α, β-methylene-ATP, and AMP-PNP, in endothelial cells. Biochem Biophys Res Commun 233:442–446. https://doi.org/10.1006/bbrc.1997.6478
Wildman SS, Brown SG, King BF, Burnstock G (1999) Selectivity of diadenosine polyphosphates for rat P2X receptor subunits. Eur J Pharmacol 367:119–123. https://doi.org/10.1016/S0014-2999(98)00976-5
King BF, Liu M, Pintor J et al (1999) Diinosine pentaphosphate (IP5I) is a potent antagonist at recombinant rat P2X1 receptors. Br J Pharmacol 128:981–988. https://doi.org/10.1038/sj.bjp.0702876
Rettinger J, Braun K, Hochmann H et al (2005) Profiling at recombinant homomeric and heteromeric rat P2X receptors identifies the suramin analogue NF449 as a highly potent P2X1 receptor antagonist. Neuropharmacology 48:461–468. https://doi.org/10.1016/j.neuropharm.2004.11.003
Jung K-Y, Cho J-H, Lee JS et al (2013) Synthesis and structure–activity relationships of carboxylic acid derivatives of pyridoxal as P2X receptor antagonists. Bioorg Med Chem 21:2643–2650. https://doi.org/10.1016/j.bmc.2013.01.073
Kim Y-C, Brown SG, Harden TK et al (2001) Structure−activity relationships of pyridoxal phosphate derivatives as potent and selective antagonists of P2X1 receptors. J Med Chem 44:340–349. https://doi.org/10.1021/jm9904203
Tian M, Abdelrahman A, Baqi Y et al (2020) Discovery and structure relationships of salicylanilide derivatives as potent, non-acidic P2X1 receptor antagonists. J Med Chem 63:6164–6178. https://doi.org/10.1021/acs.jmedchem.0c00435
Obrecht AS, Urban N, Schaefer M et al (2019) Identification of aurintricarboxylic acid as a potent allosteric antagonist of P2X1 and P2X3 receptors. Neuropharmacology 158:107749. https://doi.org/10.1016/j.neuropharm.2019.107749
Mathiew M, Dennis BM, Bennetts F et al (2020) Synthesis of 2-phenyl-5,6,7,8-tetrahydroquinoxaline derivatives and screening for P2X1-purinoceptor antagonist activity in isolated preparations of rat vas deferens, for translation into a male contraceptive. Biol Reprod 103:323–332. https://doi.org/10.1093/biolre/ioaa117
Jacobson KA, Kim Y-C, Wildman SS et al (1998) A pyridoxine cyclic phosphate and its 6-azoaryl derivative selectively potentiate and antagonize activation of P2X1 receptors. J Med Chem 41:2201–2206. https://doi.org/10.1021/jm980183o
Bernier L-P, Ase AR, Tong X et al (2008) Direct modulation of P2X1 receptor-channels by the lipid phosphatidylinositol 4,5-bisphosphate. Mol Pharmacol 74:785–792. https://doi.org/10.1124/mol.108.047019
Pinzi L, Rastelli G (2019) Molecular docking: shifting paradigms in drug discovery. Int J Mol Sci 20. https://doi.org/10.3390/ijms20184331
Batool M, Ahmad B, Choi S (2019) A structure-based drug discovery paradigm. Int J Mol Sci 20. https://doi.org/10.3390/ijms20112783
Westbrook JD, Burley SK (2019) How structural biologists and the Protein Data Bank contributed to recent FDA new drug approvals. Structure 27:211–217. https://doi.org/10.1016/j.str.2018.11.007
Jumper J, Evans R, Pritzel A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. https://doi.org/10.1038/s41586-021-03819-2
Varadi M, Anyango S, Deshpande M et al (2022) AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50:D439–D444. https://doi.org/10.1093/nar/gkab1061
McCarthy AE, Yoshioka C, Mansoor SE (2019) Full-length P2X7 structures reveal how palmitoylation prevents channel desensitization. Cell 179:659-670.e13. https://doi.org/10.1016/j.cell.2019.09.017
Mansoor SE, Lü W, Oosterheert W et al (2016) X-ray structures define human P2X3 receptor gating cycle and antagonist action. Nature 538:66. https://doi.org/10.1038/nature19367
Ennion S, Hagan S, Evans RJ (2000) The role of positively charged amino acids in ATP recognition by human P2X1 receptors. J Biol Chem 275:29361–29367. https://doi.org/10.1074/jbc.M003637200
Roberts JA, Evans RJ (2006) Contribution of conserved polar glutamine, asparagine and threonine residues and glycosylation to agonist action at human P2X1 receptors for ATP. J Neurochem 96:843–852. https://doi.org/10.1111/j.1471-4159.2005.03593.x
Motoyuki H, Eric G (2012) Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485:207. https://doi.org/10.1038/nature11010
Chataigneau T, Lemoine D, Grutter T (2013) Exploring the ATP-binding site of P2X receptors. Front Cell Neurosci 7:273. https://doi.org/10.3389/fncel.2013.00273
Li M, Wang Y, Banerjee R, et al (2019) Molecular mechanisms of human P2X3 receptor channel activation and modulation by divalent cation bound ATP. eLife 8. https://doi.org/10.7554/eLife.47060
Li M, Silberberg SD, Swartz KJ (2013) Subtype-specific control of P2X receptor channel signaling by ATP and Mg2+. Proc Natl Acad Sci 110:E3455–E3463. https://doi.org/10.1073/pnas.1308088110
Migita K, Haines WR, Voigt MM, Egan TM (2001) Polar residues of the second transmembrane domain influence cation permeability of the ATP-gated P2X(2) receptor. J Biol Chem 276:30934–30941. https://doi.org/10.1074/jbc.M103366200
Samways DSK, Egan TM (2007) Acidic amino acids impart enhanced Ca2+ permeability and flux in two members of the ATP-gated P2X receptor family. J Gen Physiol 129:245–256. https://doi.org/10.1085/jgp.200609677
Browne LE, Cao L, Broomhead HE et al (2011) P2X receptor channels show threefold symmetry in ionic charge selectivity and unitary conductance. Nat Neurosci 14:17–18. https://doi.org/10.1038/nn.2705
Allsopp RC, Evans RJ (2011) The intracellular amino terminus plays a dominant role in desensitization of ATP-gated P2X receptor ion channels. J Biol Chem 286:44691–44701. https://doi.org/10.1074/jbc.M111.303917
Fryatt AG, Dayl S, Stavrou A et al (2019) Organization of ATP-gated P2X1 receptor intracellular termini in apo and desensitized states. J Gen Physiol 151:146–155. https://doi.org/10.1085/jgp.201812108
Wang J, Wang Y, Cui W-W et al (2018) Druggable negative allosteric site of P2X3 receptors. Proc Natl Acad Sci 115:4939–4944. https://doi.org/10.1073/pnas.1800907115
Akira K, Toshimitsu K (2016) Structural basis for subtype-specific inhibition of the P2X7 receptor. eLife 5:https://doi.org/10.7554/eLife.22153
Ase AR, Therrien É, Séguéla P (2019) An allosteric inhibitory site conserved in the ectodomain of P2X receptor channels. Front Cell Neurosci. https://doi.org/10.3389/fncel.2019.00121
Bidula S, Nadzirin IB, Cominetti M et al (2022) Structural basis of the negative allosteric modulation of 5-BDBD at human P2X4 receptors. Mol Pharmacol 101:33–44. https://doi.org/10.1124/molpharm.121.000402
Ase AR, Honson NS, Zaghdane H et al (2015) Identification and characterization of a selective allosteric antagonist of human P2X4 receptor channels. Mol Pharmacol 87:606–616. https://doi.org/10.1124/mol.114.096222
Gever JR, Soto R, Henningsen RA et al (2010) AF-353, a novel, potent and orally bioavailable P2X3/P2X2/3 receptor antagonist. Br J Pharmacol 160:1387–1398. https://doi.org/10.1111/j.1476-5381.2010.00796.x
Donnelly-Roberts DL, Namovic MT, Surber B et al (2009) [3H]A-804598 ([3H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacology 56:223–229. https://doi.org/10.1016/j.neuropharm.2008.06.012
Wildman SS, Brown SG, Rahman M et al (2002) Sensitization by extracellular Ca2+ of rat P2X5 receptor and its pharmacological properties compared with rat P2X1. Mol Pharmacol 62:957–966. https://doi.org/10.1124/mol.62.4.957
Brown SG, Kim Y-C, Kim S-A et al (2001) Actions of a series of PPADS analogs at P2X1 and P2X3 receptors. Drug Dev Res 53:281–291. https://doi.org/10.1002/ddr.1197
Virginio C, Robertson G, Surprenant A, North RA (1998) Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol Pharmacol 53:969–973
Braun K, Rettinger J, Ganso M et al (2001) NF449: a subnanomolar potency antagonist at recombinant rat P2X1 receptors. Naunyn Schmiedebergs Arch Pharmacol 364:285–290. https://doi.org/10.1007/s002100100463
Hülsmann M, Nickel P, Kassack M et al (2003) NF449, a novel picomolar potency antagonist at human P2X1 receptors. Eur J Pharmacol 470:1–7. https://doi.org/10.1016/s0014-2999(03)01761-8
Lambrecht G, Rettinger J, Bäumert HG et al (2000) The novel pyridoxal-5′-phosphate derivative PPNDS potently antagonizes activation of P2X1 receptors. Eur J Pharmacol 387:R19–R21. https://doi.org/10.1016/S0014-2999(99)00834-1
Cho J-H, Jung K-Y, Jung Y et al (2013) Design and synthesis of potent and selective P2X3 receptor antagonists derived from PPADS as potential pain modulators. Eur J Med Chem 70:811–830. https://doi.org/10.1016/j.ejmech.2013.10.026
Jung Y-H, Kim YO, Lin H et al (2017) Discovery of potent antiallodynic agents for neuropathic pain targeting P2X3 receptors. ACS Chem Neurosci 8:1465. https://doi.org/10.1021/acschemneuro.6b00401
Hernandez-Olmos V, Abdelrahman A, El-Tayeb A et al (2012) N-substituted phenoxazine and acridone derivatives: structure–activity relationships of potent P2X4 receptor antagonists. J Med Chem 55:9576–9588. https://doi.org/10.1021/jm300845v
Beswick P, Wahab B, Honey MA et al (2019) A challenge finding P2X1 and P2X4 ligands. Neuropharmacology 157:107674. https://doi.org/10.1016/j.neuropharm.2019.107674
Jaime-Figueroa S, Greenhouse R, Padilla F et al (2005) Discovery and synthesis of a novel and selective drug-like P2X1 antagonist. Bioorg Med Chem Lett 15:3292–3295. https://doi.org/10.1016/j.bmcl.2005.04.049
Kasuya G, Fujiwara Y, Tsukamoto H et al (2017) Structural insights into the nucleotide base specificity of P2X receptors. Sci Rep 7:45208. https://doi.org/10.1038/srep45208
Igawa T, Abe Y, Tsuda M et al (2015) Solution structure of the rat P2X4 receptor head domain involved in inhibitory metal binding. FEBS Lett 589:680–686. https://doi.org/10.1016/j.febslet.2015.01.034
Kasuya G, Fujiwara Y, Takemoto M et al (2016) Structural insights into divalent cation modulations of ATP-gated P2X receptor channels. Cell Rep 14:932–944. https://doi.org/10.1016/j.celrep.2015.12.087
Kasuya G, Yamaura T, Ma X-B et al (2017) Structural insights into the competitive inhibition of the ATP-gated P2X receptor channel. Nat Commun 8:876. https://doi.org/10.1038/s41467-017-00887-9
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. The first draft of the manuscript was written by FMB and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflicts of interest
The authors declare no conflict of interest.
Ethical approval
Ethics approval not applicable.
Informed consent
All authors agree with the content of the manuscript and with the submission.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bennetts, F.M., Mobbs, J.I., Ventura, S. et al. The P2X1 receptor as a therapeutic target. Purinergic Signalling 18, 421–433 (2022). https://doi.org/10.1007/s11302-022-09880-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-022-09880-4