
Abstract
Three Chinese chestnut bacterial artificial chromosome (BAC) libraries were developed and used for physical map construction. Specifically, high information content fingerprinting was used to assemble 126,445 BAC clones into 1,377 contigs and 12,919 singletons. Integration of the dense Chinese chestnut genetic map with the physical map was achieved via high-throughput hybridization using overgo probes derived from sequence-based genetic markers. A total of 1,026 probes were anchored to the physical map including 831 probes corresponding to 878 expressed sequence tag-based markers. Within the physical map, three BAC contigs were anchored to the three major fungal blight-resistant quantitative trait loci on chestnut linkage groups B, F, and G. A subset of probes corresponding to orthologous genes in poplar showed only a limited amount of conserved gene order between the poplar and chestnut genomes. The integrated genetic and physical map of Chinese chestnut is available at www.fagaceae.org/physical_maps.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chestnuts (Castanea spp.) and their relatives, including oaks (Quercus spp.) and beeches (Fagus spp.), comprise the Fagaceae, a plant family that dominates much of the climax hardwood forests of the Northern Hemisphere (Manos et al. 2008). On the North American continent, the American chestnut (Castanea dentata) was once an abundant and important element of the eastern hardwood forests, serving significant roles, both ecologic and economic (Hill 1994). However, from 1905 to the 1950s, a devastating invasion of the chestnut blight fungus (Cryphonectria parasitica) eliminated the American chestnut as a dominant forest tree species (Anagnostakis 1987).
Several strategies to restore the American chestnut forests, including biological control and hybrid breeding programs (MacDonald and Fulbright 1991; Hebard 2006), have been explored. A biological control approach that involved the introduction of exotic hypovirulent strains of Cryphonectria parasitica into local fungal communities successfully reduced the virulence of Cryphonectria strains in Europe (Heiniger and Rigling 1994). In North America, this approach was unsuccessful primarily due to the complexity of the vegetative incompatibility genetic system that prohibited fungal anastomosis (mycelial fusion) required for efficient transmission of the hypovirulent virus (Anagnostakis et al. 1986). For this reason, groups in the USA have pursued an interspecies backcross breeding strategy to introgress blight-resistant genes from Chinese chestnut (Castanea mollissima) into the American chestnut (Hebard 2006). Multiple American chestnut germplasm populations have been sampled to preserve as much of the genetic diversity of the species as possible (Hebard 2006). The long generation time and space requirements for chestnut breeding are significant barriers to the rapid development of resistant trees; however, the introduction of molecular markers and marker-assisted breeding and genetic engineering could accelerate the resistance improvement process (Merkle et al. 2007; Wheeler and Sederoff 2009).
An integrated structural genomic resource comprised of substantial expressed sequence tags (ESTs) and integrated (i.e., cross-referenced) genetic map and physical maps is ideal for high-resolution mapping of important traits and map-based cloning of candidate genes. In rice, Arabidopsis, and poplar, the model systems for monocots, dicots, and woody plants, respectively, such integrated resources serve as a platform for comparative genomics studies and provide scaffolds for gene mining from related but less studied species (Flavell 2009; Koornneef and Meinke 2010).
The Fagaceae Genome Project (discussed in Wheeler and Sederoff 2009) was launched in 2006 to develop genomic research tools to identify genes for chestnut blight resistance, establish chestnut (Castanea spp.) as a model for the Fagaceae, and to compare it to northern red oak (Quercus rubra), white oak (Quercus alba), and American beech (Fagus grandifolia). Specifically, the four main objectives of the project were to (1) develop DNA sequence-based resources in the form of ESTs and SSR markers and EST-based SNP and SSR markers; (2) develop a high-density, high-resolution resolution genetic map of the Chinese chestnut genome, positioning quantitative trait loci (QTLs) for chestnut blight resistance; (3) develop a whole genome physical map anchored to the genetic map; and (4) discover and analyze candidate genes for blight resistance located with the QTL intervals.
Prior to this project, our understanding of the chestnut genome, like most forest trees, was limited. In comparison to annual field crops and other model plants such as Arabidopsis, availability of highly informative genetic maps in forest trees having defined intervals for important phenotypic traits is limited. Most forest trees take years to flower, and progeny trials require a significant commitment of time, space, and labor for genetic analysis. For chestnut, a small number of genetic maps are currently available. These include (1) a genetic map based on a three-generation pedigree of an interspecific cross of American chestnut and Chinese chestnut comprising 196 RAPD and RFLP markers covering 530.1 cM (Kubisiak et al. 1997); (2) an updated version of the interspecific map, adding 275 AFLP markers, 24 SSR markers, and the 5S rDNA locus (Sisco et al. 2004); (3) a new genetic map of Chinese chestnut created from 1,401 new SSR and SNP markers (Kubisiak et al., companion manuscript) that served as a reference map for anchoring the physical map described here; and (4) a genetic map of European chestnut (Castanea sativa Mill.) using a two-way pseudo-testcross on a population of 96 F1 full-sib individuals (Casasoli et al. 2001).
In parallel to these genetic mapping efforts, a whole genome physical map (i.e., bacterial artificial chromosome (BAC) contig assembly) of the Chinese chestnut was constructed and anchored to a genetic map (Kubisiak et al., companion manuscript) via marker hybridization to BAC clones arrayed on filters. Besides integrating the genetic and physical maps, the marker hybridizations verified and improved the physical map through manual editing. Here we report on the physical mapping efforts including BAC library construction and analysis, high information content fingerprinting (HICF) and BAC contig assembly, integration of the physical map with the genetic map through marker hybridization, and comparative analyses with the poplar genome.
Materials and methods
BAC library construction
Three Chinese chestnut BAC libraries were prepared. Two BAC libraries, CMCMBb and CMCMBd, were created at the Clemson University Genomics Institute. The BAC library CM_MBc was constructed by LGG and differences in protocol for that library are noted in Additional File 1. Also, CUGI generated the BAC library CDC_Ba from the American chestnut (Castanea dentata) to be used for Castanea mollissima versus Castanea dentata comparative analysis. The BAC library construction protocol at CUGI is described below. Briefly, nuclei were isolated from young leaves and embedded in agarose plugs by the method of Peterson et al. (2000). A twofold increase in antioxidants was amended to the extraction buffer due to the high phenolic content of chestnut leaf tissue. The agarose plugs were treated with proteinase K supplemented with 0.2 % beta-mercaptoethanol to reduce oxidation of phenolic compounds. To fractionate genomic DNA, plugs were macerated with a razor blade followed by partial digestion with HindIII (libraries CMCMBb and CDC_Ba) or EcoRI (library CMCMBd) and size-selected in a clamped homogeneous electrical field apparatus (CHEF-DR III, Bio-Rad, Berkeley, CA, USA) (Peterson et al. 2000). After two rounds of size selection, DNA was recovered from the agarose by electroelution and cloned into the vector pIndigoBAC536. The ligation reactions were heated at 65 °C for 20 min to inactivate enzymes and desalted on a Millipore membrane (type VSWP, 0.025 μm) on 5 % PEG (polyethylene glycol) 8,000 at room temperature for 2 h before transformation into DH10B competent cells (Invitrogen, Life Technologies, Carlsbad, CA, USA) by electroporation. Transformed cells were plated on LB agar supplemented with chloramphenicol (12.5 μg/ml), 0.5 mM IPTG, and 50 μg/ml X-gal at 37 °C for 24 h. Library clones were picked (Genetix QBot, Molecular Devices, New Milton, UK), grown in LB amended with 7 % glycerol and chloramphenicol (12.5 mg/l) in 384-well plates, and stored at −80 °C. To evaluate the average insert size, 96 clones were randomly selected from each library and grown in Terrific Broth (TB) supplemented with 12.5 μg/ml chloramphenicol before DNA extraction. BAC DNA was then digested with NotI and resolved by electrophoresis in a 1.0 % agarose gel in 0.5× TBE on the CHEF system (Peterson et al. 2000) using a 5–15-s ramping pulse and 6 V/cm running voltage for 15 h at 14 °C. The gels were stained with ethidium bromide. Both Lambda ladder and MidRange I PFG Markers (New England BioLabs, Ipswich, MA, USA) were used as size references. Whole library copies, individual clones, or macrofilters for the three libraries may be obtained through the Clemson University Genomics Institute (http://www.genome.clemson.edu/online_orders).
BAC fingerprinting
All fingerprinting was performed using the four-color HICF SNaPshot method of Luo et al. (2003). BAC DNA was isolated from 1.2 ml cultures in TB supplemented with 12.5 μg/ml chloramphenicol by alkaline lysis in a 96-well format. DNA was digested with five enzymes. Four 6-bp cutters, BamHI, HindIII, XbaI, and XhoI, were chosen to digest DNA over a wide range of GC content, allowing for an unbiased fragmentation of the BAC DNA. One 4-bp cutter, HaeIII, was chosen to reduce the DNA fragment size for HICF analysis. Restriction digestions were incubated at 37 °C for 2 h, and the DNA was labeled using a ABI PRISM SNaPshot multiplex kit and was subsequently loaded on an ABI 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA). Two rice BAC clones with a known fragment pattern were included as internal controls.
The fingerprints were processed using GeneMapper v4.0 (Applied Biosystems), FPPipeliner 4.0, and FPMiner 2.0 (Bioinforsoft, Beaverton, OR, USA) to call fragment sizes and remove spurious peaks, vector bands, and the internal control clones. Clones yielding less than 10 bands would be statistically unlikely to be accurately placed on the physical map and were excluded from further analysis due to lack of information. Clones with more than 250 bands were excluded because they are likely to have multiple clone contamination.
Physical map construction
The fingerprinting data were analyzed using FPC v9.3 (“FingerPrinted Contigs”; Soderlund et al. 2000; Nelson et al. 2007) for contig assembly. The initial contig assembly was created with high stringency parameters (tolerance 3 and Sulston score of 1e−50). All contigs with more than 10 % questionable (Q) clones were split by the “DQer” function of FPC to reduce misassembly. An incremental re-build strategy was used to sequentially lower the stringency of the build parameters. In each round of calculation, the Sulston score cutoff was reduced by 1e−5 and an “Ends to Ends” merge with parameters “match” 2 and “from end” 34 was performed. This was followed by “Singles to Ends” merge at the same stringency until the final assembly Sulston score was at 1e−30. The average restriction band size was calculated by the average BAC insert size divided by the average number of bands per fingerprint.
Marker hybridization
Sanger and 454 DNA sequencing were used to generate 450 million bases of EST sequence from Chinese chestnut, American chestnut, northern red oak, white oak, and American beech (http://www.fagaceae.org/sequences), including 847,952 reads from Chinese chestnut and 688,198 reads from American chestnut. Computational analysis of the transcriptomes generated from chestnut blight cankers and healthy tissues revealed ESTs preferentially expressed during canker development in both Chinese chestnut and American chestnut and provided a list of potential candidate genes for blight resistance (Barakat et al. 2009, 2012). The collection of ESTs was analyzed for both SSR and SNP markers to develop a highly informative genetic map. In addition, this rich unigene resource was mined for gene sequences to use for overgo probe design.
The protocol for the pooled overgo hybridizations discussed in the “Results” section is detailed below. A separate set of 23 overgo markers was hybridized against the CM_Mbc library by LGG using a slightly different protocol. The process of selection and the methods used for these markers are detailed in Additional File 1. Low complexity sequences and known repetitive elements were masked from the EST sequences by RepeatMasker (Smit et al. 1996–2010) and Tandem Repeat Finder (Benson 1999). Repeat libraries from RepBase (Jurka et al. 2005) were used by RepeatMasker, with the species set to “viridiplantae” to utilize all repeats from green plants in the database. Overgo probe (Ross et al. 1999) design was performed on the resulting masked sequences using OligoSpawn (Zheng et al. 2006). All oligomers were synthesized by IDT (Integrated DNA Technologies, Coralville, IA, USA).
High-density BAC filters were created on Hybond N(+) membranes (GE Healthcare Life Sciences, Pittsburgh, PA, USA) by an automated QBot (Genetix, New Milton, UK). Each filter contained a total of 18,432 clones double-spotted in a 4 × 4 pattern in a six-field grid. The experimental design consisted of a previously developed 3D pooling strategy that enables resolution of 125 probes through 15 hybridizations (Fang et al. 2010). A pool of probes was used for each of the 15 hybridizations, named X1, X2, X3, X4, and X5 in the X dimension; Y1, Y2, Y3, Y4, and Y5 in the Y dimension; and Z1, Z2, Z3, Z4, and Z5 in the Z dimension. Each probe was assigned to one X pool, one Y pool, and one Z pool, with each probe having a unique address such as (X1, Y1, Z1) or (X3, Y4, Z2), etc., indicative of the pools to which it was included. An automated setup script was used to create the pool addresses for each probe and is available online at http://www.genome.clemson.edu/software/hybdecon/exp_setup. Overgo probes were individually labeled with 32-P at 37 °C for 1 h (McPherson et al. 2001), and the labeled probes were mixed according to the pooling formula, denatured at 95 °C for 10 min and hybridized at 60 °C overnight. Filters were washed with 0.1 % SDS, 1× SSC at 60 °C and exposed to phosphor screens for 24 to 48 h.
The images were recorded by a Typhoon 9400 Imager (GE Healthcare Life Sciences, Uppsala, Sweden). All images were examined using the Hybdecon software package to score the addresses of positively hybridized clones, and the deconvolution function from the software was used to resolve the hits for each probe. Hybdecon is an in-house expanded version of the software HybSweeper (Lazo et al. 2005) and can be downloaded from http://www.genome.clemson.edu/software/hybdecon. A BAC with a successful hybridization in one X pool, one Y pool, and one Z pool was scored as a successful hybridization of the probe assigned to those same three original pools. A BAC with more than one hybridization result in a single dimension (X, Y, or Z) or no hybridization result in a dimension was discarded. This is a very stringent method of hybridization because the probe and BAC must have a successful hybridization three times, one for each dimension of the experiment. Loss of data occurs if the hybridization fails in one of the dimensions or if a BAC is hybridized by more than one probe.
Manual editing of physical map
Two types of evidence were used to merge physical map contigs. BAC clones on the ends of each contig that strongly matched BAC clones at the end of another BAC contig (“overlapping” BACs) were identified using the FPC software. Contigs with clones that hybridized to the same overgo probe were also considered candidates for merging because the majority of overgo sequences are expected to be a single copy. The initial round of editing used parameters of tolerance 3 and Sulston score cutoff at 1e−20. Contigs were merged if they fulfilled the following criteria: (a) overlapped by three or more BACs from both contigs or (b) overlapped by two BACs from both contigs with at least one pair matching at a cutoff value equal to or more significant than 1e−30. For a contig pair with only one shared BAC from one contig matching to multiple BACs in the other, a common overgo marker hybridized to both contigs must further support the merge. The second round of manual contig editing was done at a lower stringency of tolerance 3 and cutoff 1e−15 using the requirements of (a) multiple overlapped clones from both contigs, with the first contig sharing three or more clones with two or more clones from the second contig, and (b) consensus band (CB) map computation recalculated at tolerance 3, cutoff at 1e−20 or lower if the number of overlapping clones did not fulfill the minimal requirement of the first rule.
Comparative genome analyses
To identify poplar orthologs for the comparative genomics study, all chestnut unigenes (i.e., EST contigs, CCall unigene version 2) were compared using BLAST against the poplar (Populus trichocarpa) gene sequences (http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.home.html). Putative orthologs were defined as reciprocal best matches between a chestnut unigene and a poplar gene sequence. The putatively orthologous chestnut unigene sequences were used to design overgos and hybridize as described above. The poplar paralog, expected from the recent whole genome duplication, was found using the VISTA-Point pairwise alignment browser with Poplar v2.0 aligned to Poplar v2.0(dupl.) (Frazer et al. 2004). If a physically mapped contig shared hybridizations to both an orthologous poplar marker and to one or more genetically mapped markers, the location of the poplar ortholog was inferred to be at the same location on the genetic map.
Genetic mapping of chestnut blight resistance identified three QTLs (referred to as Cbr1, Cbr2, and Cbr3 on linkage groups B, F, and G, respectively) (Kubisiak et al., companion manuscript). In order to identify chestnut BAC contigs originating from the QTL regions of the genome, three genetic map markers with high LOD scores (4.2, 4.3, and 3.5, for Cbr1, Cbr2, and Cbr3, respectively) were used for overgo design. The overgos were hybridized against the BAC libraries to identify BAC clones and physical BAC contigs corresponding to the QTLs.
Results
BAC library construction
The production of the Chinese chestnut physical map began with the creation of three large insert BAC libraries with a total of 36× genome coverage. A HindIII library, named CMCMBb, was created from Chinese chestnut (Castanea mollissima ‘Vanuxem’) and contains 73,728 clones arrayed in 192 plates. The average insert size was estimated to be 123 kb. Considering the estimated Chinese chestnut genome size of 794 Mb (Kremer et al. 2007), CMCMBb clones comprise 11× coverage of the genome. A second BAC library, CMCMBd, was created from the same genotype with EcoRI and includes 92,160 clones in 240 plates. With an estimated average insert size of 115 kb, the library contains approximately 13 genome equivalents. A third Chinese chestnut BAC library, CM_MBc, was also constructed using the HindIII enzyme and has 110,592 clones of average size 90 kb, yielding an estimated 12× coverage. A fourth (nonfingerprinted) BAC library from American chestnut (Castanea dentata) was constructed for the purposes of global genomic comparison and comparative studies of the candidate genes for blight resistance in the susceptible chestnut species. Designated as CDC_Ba, this library was also made by partial digestion of genomic DNA with HindIII (73,728 clones with an average insert size of 140 kb) and consisted of 13× genome equivalents (Table 1).
BAC fingerprinting
The HICF method was chosen for fingerprinting because it provides superior sensitivity over the agarose method, makes use of automated sequencing technology to increase throughput, and is able to create a more contiguous physical map (Nelson et al. 2005). A total of 166,161 BACs were fingerprinted: 73,728 from CMCMBb, 92,160 from CMCMBd, and 273 from CM_MBc. Quality control filtering yielded a final set of 126,176 high quality fingerprints, representing an estimated 18× genome coverage.
Physical map construction
The initial assembly of the HICF fingerprints generated 8,708 contigs and 43,570 singletons. The contigs were iteratively rebuilt at four consecutive levels of reduced stringency parameters, yielding 4,279 contigs and 11,820 singletons. The contig assembly was manually edited utilizing the marker hybridization data to merge contigs (hybridizations further discussed below). The final build consisted of 1,377 contigs and 12,919 singletons. The average restriction fragment band size was calculated at 1.74 kb, yielding an estimate of the average length of the contigs at 951 kb (Table 2). This build contained a total of 753,432 consensus bands across all contigs, covering an estimated physical span of 1,311 Mb. This is equal to 1.6× of the chestnut genome estimated size. The physical map may be found at http://www.fagaceae.org/physical_maps and will be updated as newer versions are developed.
Genetic marker hybridization
A total of 1,125 probes were designed and tested against the Chinese chestnut BAC libraries CMCMBb (HindIII) and CMCMBd (EcoRI) libraries. In order to assess chestnut–poplar macrosynteny, 260 overgo probes were designed from Chinese chestnut unigene sequences orthologous to poplar genes. The rest of the probes were designed from Chinese chestnut unigene sequences that contain SSR and SNP markers that were being screened for placement on the genetic map (Kubisiak et al., companion manuscript). This included a set of 43 RFLP marker sequences kindly provided by Dr. Paul Sisco and a set of 23 sequences previously screened against the CM_MBc library (LLG unpublished data, see Additional File 1). The overgo probes were hybridized against the BAC libraries in nine pools of 125 probes each. For each pool, an average of 90 % of the overgo probes successfully hybridized to one or more BAC clones, providing an efficient transfer of genetic information to the physical map. The 10 % failure rate was primarily due to the loss of information inherent in the 3D pooling strategy. Additional factors include the following: the overgo sequence spanning an intron splice site, no or very low BAC coverage in a particular region of the genome, and uneven growth of BAC clones on the filters.
Overall, a total of 1,026 overgo probes were successfully placed on the physical map. On average, each probe hybridized to 17.5 BAC clones, close to the estimated 18× genome coverage of the clones. Of the 1,026 overgos hybridized, 831 corresponded to EST contigs that provided the sequences for 878 markers (Table 3) that were used in genetic mapping (Kubisiak et al., companion manuscript). Of these 878 markers, 691 were successfully genetically mapped (i.e., placed on linkage groups), and they correspond to 376 BAC contigs (Additional Files 2a, 2b). These 376 physical contigs covered a total length of 621 Mb (~0.78× of the genome) across the 12 linkage groups, with physical sizes ranging from 38.9 Mb across linkage group K to 84.4 Mb across linkage group A (Table 4). Overall, 49.4 % of the consensus genetic map markers were placed on the physical map and 47 % of the length of the physical map was placed on the genetic map. With respect to the consensus genetic map (Kubisiak et al., companion manuscript), 572 genetic markers placed 350 BAC contigs on the physical map (Fig. 1). Results from the hybridizations were integrated into the Chestnut mapping project database and updated at the Fagaceae genomics public website at http://www.fagaceae.org/physical_maps. The relationship between the genetic and physical maps can be explored online with CMap software (http://www.fagaceae.org/cgi-bin/cmap/viewer).

The consensus Chinese chestnut genetic map, spanning 742.4 cM across 12 linkage groups (A–L), has been integrated with the BAC-based physical map from the same species. Overgo probes representing 572 genetic map markers anchored 350 BAC contigs to genomic locations. Only successfully anchored genetic map markers are shown in the figure
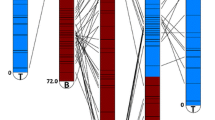
In addition to mapping physical contigs to genetic loci, results from overgo probe hybridization also provided evidence for the fidelity of contig assembly. Positively hybridizing clones for each probe show overlap within the contig, suggesting the consensus bands successfully placed the BACs in relation to one another (Fig. 2). Thus, results from marker hybridizations, based on sequence similarity, confirm the contig assembly that is based on fragment pattern DNA fingerprints. Among the 878 genetic markers (831 probes), 46 % mapped to a single contig and another 26 % mapped to only two contigs (Table 3). Many of the markers mapping to two or more physical map contigs were preferentially anchored to a single contig with only a single, possibly spurious, BAC hybridization to other locations, suggesting that 83 % of markers likely anchor a single genome location. Only 103 (12.6 %) markers were mapped to four or more contigs, suggesting a low level of gene duplication. The majority of genes are contained in one or two physical map locations. Attempts to further merge the contigs hybridized by the same marker using a lower Sulston score of 1e−20 were not successful in most cases. Some of the probes may have legitimately hybridized to multiple genomic loci due to (a) shared short repeats with the probe sequence, (b) segmental genome duplication of loci, or (c) the heterozygosity of the genome having complex diversified alleles resulting in heterogeneous unmerged fingerprints at these genomic locations.

Hybridizations with three strongly associated blight-resistant QTL markers anchored to three physical map contigs. The top track shows the overgos that hybridized to the contig with the QTL-associated overgo highlighted in blue. Each BAC clone that hybridized to the QTL-associated overgo is highlighted in green. The images were generated by the FPC software package. a Physical map contig ctg2730 was anchored by an overgo from EST CCall_contig40076_v2. This EST also contains CmSNP00927, mapped to 38.6 cM on linkage group B. Contig ctg2730 hybridized to nine other overgo probes, three of which correspond to genetic map markers mapped within 1 cM of CmSNP00927 (overgos CCall_contig44994_v2, CCall_contig25072_v2, CmSI0495). b Physical map contig ctg11964 hybridized to overgo probe CCall_contig6157_v2, which corresponds to CmSNP1333 on linkage group F at 53.1 cM. This physical map contig has 14 other overgo probe locations including CCall_contig8104, which maps to linkage group F, location 54.7. The QTL-associated overgo, CCall_contig6157_v2, appears to map to BACs that do not overlap. This is probably a result of a slight misalignment in the BACs (i.e., they should actually be overlapping and do indeed contain the same segment of DNA), but it could also indicate a tandem duplication in this region. c Physical map contig ctg2356 hybridized to overgo probe CCall_contig8443_v2, which corresponds to CmSNP1477 on linkage group G. Contig 2356 with six other overgo probes including CCall_contig15944_v2 was also placed on linkage group G
Comparative genome organization at low resolution
Macrosynteny between poplar and chestnut was assessed utilizing a set of 260 overgos chosen from chestnut unigenes with reciprocal best hits to poplar genes. For each chestnut overgo, two poplar loci were examined to account for the recent whole genome duplication discovered in the poplar genome (Tuskan et al. 2006). The chestnut unigenes selected for this analysis corresponded to putative poplar orthologs spanning the 19 poplar genome linkage groups and 11 unmapped poplar scaffolds. Contigs that could be localized to a chestnut linkage group via a genetically mapped marker and also to two poplar locations via an ortholog probe were used to assess how the chestnut and poplar genomes compare in organization. Results from 229 successful probes provided comparative data for a total of 260 physical map contigs (Additional File 3). Among these was a set of 131 contigs that could be anchored to chestnut linkage groups by genetically mapped markers.
High levels of macrosynteny between chestnut and poplar were not detected; the order of genes in poplar was, in general, not conserved in chestnut. Twenty-two chestnut physical map contigs hybridized to two or more poplar ortholog overgos. In eight of these contigs, there was some evidence that the markers may also be close together (less than 10 Mb) in at least one area of the poplar genome (Table 5). The distance between these poplar genes ranged from 13 kb to 5 Mb, suggesting regions of the chestnut genome in this size range may still share collinearity with the poplar genome. In two of these physical map contigs, the order of markers is conserved from chestnut to both of the paralogous areas of the poplar genome created through the recent whole genome duplication (Fig. 3).

Low resolution mapping of orthologous sequences from chestnut unigenes to the poplar genome shows small areas of conservation of gene order. In two cases, a set of two markers demonstrate conserved proximity across the recent poplar duplication as well as between chestnut and poplar. a Two markers on chestnut physical map contig 2223 map to approximately 3 Mb apart on linkage groups 1 and 9 in poplar; b two markers on chestnut physical map contig 12076 map to less than 1 Mb apart on linkage groups 8 and 10 in poplar. These syntenic blocks highlight the potential for comparative genomics between these two tree genomes
Confirmation of these syntenic blocks and a more in-depth understanding of the evolutionary rearrangements between chestnut and poplar will require finer mapping and more sequence data. This analysis is unable to establish the relative size of conserved syntenic blocks between the two genomes. The size of the physical map contigs limits the effective block size that could be detected, and the small sample size of 260 overgos prevents the discovery of small conserved blocks because of the large distance between each gene. Further research utilizing the entire set of overgo markers and additional sequence resources is ongoing and may yield more comparative information.
Physical mapping of blight-resistant QTLs
The main focus of the genomic characterization of chestnut is to identify candidate genes for chestnut blight resistance and to introgress the resistance into American chestnut. Kubisiak et al. (companion manuscript) mapped three major blight-resistant QTLs (Cbr1, Cbr2, and Cbr3) with EST-based genetic markers. Probes designed for these markers identified BAC clones and their contigs resulting in an integrated genetic physical map for each QTL. For example, the probe for marker CmSNP00927 (Ccall_contig40076_v2) on linkage group B (Cbr1) hybridized to 15 BAC clones, all members of physical map ctg2730. Similarly, probes designed for markers CmSNP01333 (linkage group F, Cbr2) and CmSNP01477 (linkage group G, Cbr3) hybridized to eight BAC clones in ctg11964 and six BAC clones in ctg2356 (Fig. 2). Based on the estimation of 1.74 kb for 1 CB unit, the lengths of these three BAC contigs are 4.4 Mb for ctg2730, 4.4 Mb for ctg11964, and 1.2 Mb for ctg2356. Further comparative mapping of the QTL contigs using genetic map marker sequences against the poplar and peach genomes (http://www.phytozome.net/) identified microsynteny at all three of these regions. These microsyntenies are further described in the companion genetic map manuscript.
Discussion
The new BAC-based genetically anchored physical map for Chinese chestnut is expected to facilitate map-based cloning and identification of genes involved in blight resistance. These genes can then be introduced through introgressive interspecies breeding strategies (Hebard 2006) or transgene technologies into American chestnuts (Merkle et al. 2007) with the eventual goal of restoring chestnut forests in North America. The physical map will also guide the whole genome sequence assembly and facilitate comparative genomics studies in other tree species. Because of the relatively conserved genome structure in the Fagaceae, detailed information from the chestnut genome is expected to serve as a model for the study of genome organization and the evolution of the Fagaceae as well as provide avenues for study of the genes and gene activity underlying the conservation and evolution of important traits such as disease resistance, phenological adaptation, and wood formation.
The collective span of the physical map contigs is 1,311 Mb, 1.6× of the estimated 794-Mb genome (Kremer et al. 2007). The inflated length from the physical map might be due to the obligate outcrossing nature of the species that resulted in a highly heterozygous genome, thus increasing the complexity of the fingerprints derived from alleles of any region. Consequently, BACs from variable regions of separate parental haplotypes, while often still overlapping enough to assemble into the same contig, would generate extra restriction fragments that appear to not be nonoverlapping and therefore contribute to the expanded size estimate from the physical contig assembly. Similar expansion in physical size estimated from physical maps was also reported from other heterozygous plant species such as grapevine (Moroldo et al. 2008) and poplar (Kelleher et al. 2007). Results from this physical map appear to indicate a relatively complex heterozygous genome. However, heterozygosity does not appear to interfere with marker hybridization in which more than 80 % of the genetic markers mapped preferentially to single contigs. The hybridization pattern confirms that heterozygous but overlapping BACs are usually correctly assembled into the same physical map contig. However, as the markers were derived from ESTs that in general are more conserved than nongenic regions in plant genomes, the effects of heterozygosity might be minimized in the genic regions where hybridizations were localized. Further investigation through the comparison with the whole genome sequence, which is under assembly (http://www.foresthealthinitiative.org/genomics.html), should provide more insights into the genome heterozygosity and provide references to improve the physical map.
The comparative mapping of the chestnut and poplar genomes provides a framework for further study of genome evolution across these lineages. It has been reported that many angiosperms have been through at least two rounds of whole genome duplication (WGD); an ancient WGD occurred early in the evolution of angiosperms followed by a second wave of large-scale genome duplication during further divergence into different lineages (Bowers et al. 2003; Paterson et al. 2004; De Bodt et al. 2005; Cui et al. 2006). In addition to the WGD, segmental duplications are commonly observed in plant genomes (Van de Peer et al. 2009). Collectively, the WGD and segmental duplications followed by translocation, substitution, and deletion play a critical role in the diversity of plant genomes and provide morphological and functional advantages in environmental adaptation (Wendel 2000; Adams and Wendel 2005). Results from the overgo hybridizations do not support a recent large-scale genome duplication in the chestnut lineage. Of the 831 overgo probes (representing 878 genetic markers), 83 % strongly preferentially hybridized to single physical map contigs, suggesting that these markers hybridized to unique loci. Results from more comparative genomics will provide informative clues to better understand the genome variation in this plant lineage.
A better understanding of the specific mechanism and genetic location of blight resistance is expected in the near future due to the robust resources now available for chestnut. Blight resistance is controlled by at least three major QTLs on linkage groups B, F, and G (Cbr1, Cbr2, and Cbr3, respectively), in blight-resistant Chinese chestnut (Kubisiak et al. 1997; Wheeler and Sederoff 2009). Markers flanking the QTL regions were used to design overgo probes to identify the physical map contigs containing potential genes for resistance. While the QTL regions and the corresponding physical map contigs are large, prior research into the biological response of the Chinese chestnut to canker blight will provide guidance for candidate gene selection. The physiologically complex disease resistance reactions have been studied in canker tissue and are known to involve signal sensing for programmed cell death. Expression of sets of “effector genes” carry out the synchronized cell death and activate other defense reactions such as lignin deposition from surrounding cells to restrain further enlargement of canker tissue and block growth of the fungal mycelial fans (Hebard et al. 1984; Anagnostakis 1987; Barakat et al. 2009). Genes found in the BAC sequences will be functionally profiled and those with activities in these particular cellular processes will be prioritized for further research.
Minimum tiling paths of the BAC contigs that span the three blight-resistant QTLs (Cbr1, Cbr2, and Cbr3) are being sequenced by the FHI initiative (J.E.C., M.E.S. unpublished data, http://www.foresthealthinitiative.org/). Strategies for utilizing the sequenced region to identify the actual source of resistance include (a) comparing the sequences to the corresponding homologous regions in peach, poplar, and grape genomes with particular attention to previously identified fungal disease resistance genes; (b) focusing on genes that are preferentially expressed in canker tissue; and (c) selecting, sequencing, and comparing the homologous regions of the American chestnut genome for allelic variation in these two chestnut species. This latter strategy will be enabled by the availability of the American chestnut BAC library. Further association mapping analysis with multiple Chinese chestnut genotypes exhibiting different levels of resistance may reveal the strength of each QTL for conferring resistance as well as reveal any regions controlling resistance that have been overlooked. Confirmation of the candidates for blight resistance may include expression profiling of the genes in different tissues, expression interference or gene disruption in Chinese chestnut followed by evaluation for disease responses, and transformation of American chestnut germplasm for improved disease resistance.
The physical map will continue to be a critical asset for clone-based mapping and sequencing of ecologically important phenotypes. Resistance to Cryphonectria parasitica is not the only trait required for restoration of the American chestnut. Phytophthora cinnamomi, an exotic fungal root pathogen also introduced from China, is a major threat to trees living in lower elevation regions (Jeffers et al. 2009). The chestnut gall wasp (Dryocosmus kuriphilus Yasumatsu) is another devastating pest (Stehli 2006). Natural sources for resistance to either the pest or pathogen can be found in existing germplasm and introgressed using the approach and resources pioneered for the canker blight. Given the discovery that Fagaceae species appear to be conserved in genome structure (Kremer et al. 2007), information from this genome resource can be transferred to other less studied Fagaceae species to improve the quality of other major forest trees as well.
References
Adams KL, Wendel JF (2005) Polyploidy and genome evolution in plants. Curr Opin Plant Biol 8:135–141
Anagnostakis SL (1987) Chestnut blight: the classical problem of an introduced pathogen. Mycologia 79:23–37
Anagnostakis SL, Hau B, Kranz J (1986) Diversity of vegetative compatibility groups of Cryphonectria parasitica in Connecticut and Europe. Plant Dis 70:536–538
Barakat A, DiLoreto DS, Zhang Y, Smith C, Baier K, Powell WA, Wheeler N, Sederoff R, Carlson JE (2009) Comparison of the transcriptomes of American Chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biol 9:51
Barakat A, Staton M, Cheng C-H, Park J, Buang N, Yassin M, Ficklin S, Yeh C-C, Hebard F, Baier K, Powell W, Schuster S, Wheeler N, Abbott A, Carlson J, Sederoff R (2012) Chestnut resistance to the blight disease: insights from transcriptome analysis. BMC Plant Biol 12:38
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2):573–580
Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unraveling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422:433–438
Casasoli M, Mattioni C, Cherubini M, Villani F (2001) A genetic linkage map of European chestnut (Castanea sativa Mill.) based on RAPD, ISSR and isozyme markers. Theor Appl Genet 102:1190–1199
Cui L, Wall PK, Leebens-Mack JH, Lindsay BG, Soltis DE, Doyle JJ, Soltis PS, Carlson JE, Arumuganathan K, Barakat A, Albert VC, Ma H, dePamphilis CW (2006) Widespread genome duplications throughout the history of flowering plants. Genome Res 16:738–749
De Bodt S, Maere S, Van de Peer Y (2005) Genome duplication and the origin of angiosperms. Trends Ecol Evol 20:591–597
Fang GC, Blackmon BP, Henry DC, Staton ME, Saski CA et al (2010) Genomic tools development for Aquilegia: construction of a BAC-based physical map. BMC Genomics 11:621
Flavell R (2009) Role of model plant species. Methods Mol Biol 513:1–18
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32(Web Server issue):W273–W279
Hebard FV (2006) The backcross breeding program of the American chestnut foundation. In: Steiner KC, Carlson JE (eds) Restoration of American chestnut to forest lands—proceedings of a conference and workshop. The North Carolina Arboretum Natural Res Rep NPS/NCR/CUE/NRR-2006/001, National Park Service
Hebard FV, Griffin GJ, Elkins JR (1984) Developmental histopathology of cankers incited by hypovirulent and virulent isolates of Endothia parasitica on susceptible and resistant chestnut trees. Phytopathology 74:140–149
Heiniger U, Rigling D (1994) Biological control of chestnut blight in Europe. Annu Rev Phytopathol 32:581–599
Hill JM (1994) Wildlife value of Castanea dentata past and present, the historical decline of the chestnut and its future use in restoration of natural areas. In: Double ML, MacDonald WL (eds) Proceeding of the International Chestnut Conference. West Virginia University Press, West Virginia, pp 186–193
Jeffers SN, James JJ, Sisco PH (2009) Screening for resistance to Phytophthora cinnamomi in hybrid seedlings of American chestnut. In: Goheen EM, Frankel SJ (eds) Phytophthoras in forests and natural ecosystems: Proceedings of the Fourth Meeting of the International Union of Forest Research Organizations (IUFRO) Working Party S07.02.09. Monterey, CA, 334 p
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogentic Genome Res 110:462–467
Kelleher CT, Chiu R, Chin H, Bosdet IE, Krzywinski MI et al (2007) A physical map of the highly heterozygous Populus genome: integration with the genome sequence and genetic map and analysis of haplotype variation. Plant J 50:1063–1078
Koornneef M, Meinke D (2010) The development of Arabidopsis as a model plant. PLANT J 61:909–921
Kremer A, Casasoli M, Barreneche T, Bodenes C, Sisco P, Kubisiak T, Salfi M, Leonardi S, Bakker E, Buiteveld J, Romero-Severson J et al (2007) Ch 5: Fagaceae trees. In: Kole C (ed) Genome mapping and molecular breeding in plants, vol 7. Springer, Heidelberg, pp 161–187
Kubisiak TL, Hebard FV, Nelson CD, Zhang J, Bernatzky R, Huang H, Anagnostakis SL, Doudrick RL (1997) Molecular mapping of resistance to blight in an interspecific cross in the genus Castanea. Phytopathology 87:751–759
Lazo GR, Lui N, Gu YQ, Kong X, Coleman-Derr D, Anderson OD (2005) Hybsweeper: a resource for detecting high-density plate gridding coordinates. Biotechniques 39(3):320–324
Luo MC, Thomas C, You FM, Hsiao J, Quyang S, Buell CR, Malandro M, McGuire PE, Anderson OD, Dvorak J (2003) High throughput fingerprinting of bacterial artificial chromosomes using the snapshot labeling kit and sizing of restriction fragments by capillary electrophoresis. Genomics 82:378–389
MacDonald WL, Fulbright DW (1991) Biological control of chestnut blight: use and limitations of transmissible hypovirulence. Plant Dis 75:656–661
Manos PS, Cannon CH, Sang-Hun Oh S-H (2008) Phylogenetic relationships and taxonomic status of the paleoendemic Fagaceae of western North America: recognition of a new genus, Notholithocarpus. Madrono 55(3):181–190
McPherson JD, Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, Sekhon M, Wylie K, Mardis ER, Wilson RK et al (2001) A physical map of the human genome. Nature 409:934–941
Merkle SA, Andrade GM, Hair CJ, Powell WA, Maynard CA (2007) Restoration of threatened species: a noble cause for transgenic trees. Tree Genet Genomes 3:111–118
Moroldo M, Paillard S, Marconi R, Fabrice L, Canaguier A et al (2008) A physical map of the heterozygous grapevine ‘Cabernet Sauvignon’ allows mapping candidate genes for disease resistance. BMC Plant Biol 8:66
Nelson WM, Bharti AK, Butler E, Wei F, Fuks G et al (2005) Whole-genome validation of high-information-content fingerprinting. Plant Physiol 139:27–38
Nelson WM, Dvorak J, Luo MC, Messing J, Wing RA, Soderlund C (2007) Efficacy of clone fingerprinting methodologies. Genomics 89:160–165
Paterson AH, Bowers JE, Chapman BA (2004) Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc Natl Acad Sci U S A 101:9903–9908
Peterson DG, Tomkins JP, Frisch DA, Wing RA, Paterson AH (2000) Construction of plant bacterial chromosome (BAC) libraries: an illustrated guide. J Agric Gen 5:www.ncgr.org/research/jag (http://wheat.pw.usda.gov/jag/)
Ross MT, LaBrie S, McPherson J, Stanton VP (1999) Screening large-insert libraries by hybridization. In: Dracopoli NC et al (eds) Current protocols in human genetics. Wiley, New York, pp 5.6.1–5.6.32
Sisco PH, Kubisiak TL, Cassasoli M, Clark C, Barreneche T, Hebard FV, Sederoff RR, Kremer A, Villani F (2004) An improved genetic map for Castanea dentata/Castanea mollissima and its relationship to the genetic map of Castanea sativa. Acta Horticult 693:491–496
Smit, AFA, Hubley, R, Green, P. RepeatMasker Open-3.0. 1996–2010 <http://www.repeatmasker.org>
Soderlund C, Humphrey S, Dunham A, French L (2000) Contigs built with fingerprints, markers, and FPC V4.7. Genome Res 10:1172–1187
Stehli R (2006) Oriental chestnut gall wasp in Ohio—an update. The Nutshell 60:27
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Van de Peer Y, Fawcett JA, Proost S, Sterck L, Vandepoele K (2009) The flowering world: a tale of duplications. Trends Plant Sci 14:680–688
Wendel JF (2000) Genome evolution in polyploids. Plant Mol Biol 42:225–249
Wheeler N, Sederoff R (2009) Role of genomics in the potential restoration of the American chestnut. Tree Genet Genomes 5:181–187
Zheng J, Svensson JT, Madishetty K, Close TJ, Jiang T, Lonardi S (2006) Oligospawn: a software tool for the design of overgo probes from large unigene datasets. BMC Bioinforma 7:7
Acknowledgments
This study was supported by the Plant Genome Research Program (PGRP) from the US National Science Foundation (NSF-PGRP 0605135) awarded to RRS. The Forest Health Initiative (FHI) awarded to AGA supported part of the map integration and comparative analyses. Plants used in this research were obtained from Greg Miller at the Empire Chestnut Company. The authors thank Dr. Paul Sisco for early support through The American Chestnut Foundation (TACF) and for markers in the earliest stages of this project and Dr. Bill Powell and Kathleen Baier for full-length cDNA sequence and in-depth discussion.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. Grattapaglia
Guang-Chen Fang and Barbara P. Blackmon contributed equally to this study.
Thomas L. Kubisiak—retired.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional file 1
Description of BAC library CM_MBc and additional overgo probes. The 273 BAC clones listed here were integrated into the physical map using 23 overgo probes developed from an earlier chestnut genetic map. This file gives the methods and details of these resources. (DOCX 145 kb)
Additional file 2
Summary of integrated maps for Chinese chestnut. The file lists the physically anchored genetic markers and the genetically mapped BAC contigs. The complete genetic map (including 1156 mapped and 409 unmapped markers) can be found in Kubisiak et al. (companion manuscript). (xlsx 59 kb)
Additional file 3
Comparative hybridization with 260 poplar orthologs. The file lists the chestnut overgo probes used for the poplar/chestnut synteny analysis. These chestnut unigenes were compared to the poplar gene set to identify orthologs across all 19 poplar linkage groups. The poplar paralog, probably resulting from the recent whole genome duplication, is also included. Anchored physical map contigs and their genetic map locations are listed. (xlsx 31.6 kb)
11295_2012_576_MOESM5_ESM.png
{kind=link}
The consensus Chinese chestnut genetic map, spanning 742.4 cM across 12 linkage groups (A–L), has been integrated with the BAC-based physical map from the same species. Overgo probes representing 572 genetic map markers anchored 350 BAC contigs to genomic locations. Only successfully anchored genetic map markers are shown in the figure (PNG 208 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fang, GC., Blackmon, B.P., Staton, M.E. et al. A physical map of the Chinese chestnut (Castanea mollissima) genome and its integration with the genetic map. Tree Genetics & Genomes 9, 525–537 (2013). https://doi.org/10.1007/s11295-012-0576-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-012-0576-6