
Abstract
Five H3N2 avian influenza viruses (AIVs) were isolated from live poultry markets (LPMs) and poultry slaughterhouses in Shanghai, China in 2013. All viruses were characterized by whole-genome sequencing with subsequent genetic comparison and phylogenetic analysis. The hemagglutinin cleavage site of all viruses indicated that the five strains were low-pathogenic AIVs. Phylogenetic analysis of all eight viral genes showed that the five H3N2 viruses clustered in the Eurasian lineage of influenza viruses. The eight genes showed evidence of reassortment events between these H3 subtype viruses and other subtype viruses, especially H5 and H7 subtypes, probably in pigeons, domestic ducks, and wild birds. These findings emphasized the importance of AIV surveillance in LPMs and poultry slaughterhouses for understanding the genesis and emergence of novel reassortants with pandemic potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza A viruses are found in their natural hosts, wild aquatic birds, which are primordial reservoirs of the gene pool of all influenza A virus subtypes. Currently, 16 HAs and 9 NAs have been identified from aquatic birds [1, 2]. However, one of the most frequently isolated subtypes is the H3 influenza virus subtype [3, 4]. H3 has a wide host range from birds to various mammalian species including humans, pigs, dogs, and horses [5, 6]. H3 viruses have become established in domestic poultry, cause mild or severe disease, and pose the threat of zoonotic infection. In Italy, multiple H3N2 avian influenza virus (AIV) isolations have been reported from chickens with mild respiratory disease [7]. Recent studies reported that some novel H3 subtype viruses were reassortants of H5N1 and H9N2 in Korea and China [8–10]. Furthermore, H3 subtype viruses are transmitted between species to humans, pigs, and dogs. In 1968, the H3N2 Hong Kong/68 human pandemic influenza virus was derived directly from an H3 subtype AIV [11, 12]. Phylogenetic and epidemiologic analyses indicate that avian and human viruses are transmitted to pigs in nature [13, 14] and that they reassort in pigs [15] and can be transmitted to and cause disease in humans [16]. Recent studies in Korea and southern China have reported transmission of avian-origin canine H3N2 viruses [17, 18] and determined that the virus can spread among dogs by contact infection [19]. Moreover, reassortment events may have increased the pathogenicity and host ranges of H3N2 subtype viruses.
Since the early 1970s, live poultry markets (LPMs) have been suspected as prolific sources of AIV [20]. In the high-density conditions of LPMs, numerous host species of backyard poultry are housed together, creating an ideal environment for viral reassortment and interspecies transfer [21]. H3 subtype AIVs have been circulating and evolving in LPMs in China [9, 21, 22]. Slaughterhouses are also considered potential areas for the spread of avian influenza among commercial farms in the United Kingdom [23, 24]. In 2013, five H3N2 AIVs, A/environment/Shanghai/LPM1/2013 (LPM1), A/chicken/Shanghai/LPM2/2013 (LPM2), A/duck/Shanghai/SH1/2013 (SH1), A/duck/Shanghai/SH2/2013 (SH2), and A/duck/Shanghai/SH3/2013 (SH3), were isolated from domestic poultry and the environment. LPM1 was isolated from an environmental sample collected in a retail LPM that sold about 300 chickens a day, while LPM2 isolated from swabs of chickens sold in a wholesale LPM which sold about 3000 chickens a day. The other three strains (SH1, SH2, and SH3) were isolated from ducks in a slaughterhouse that slaughtered 800 ducks using semi-automation equipments a day. To better understand the genetic relationships between these H3 influenza viruses from China, all gene segments of the five H3N2 AIVs were sequenced and compared with sequences in GenBank. Phylogenic analysis was conducted on H3 and other subtype influenza viruses from different hosts, regions, and periods.
Materials and methods
Samples
A total of 145 samples were obtained from two sources from March 31 to August 30, 2013. From LPMs, 65 swab samples (a cloacal swab and tracheal swab from the same chicken, put into the same sample collection tube and counted as a single sample) and 20 environmental samples (water and soil with deposited fecal matter) were collected, and 60 swab samples were collected from ducks in a Shanghai slaughterhouse. Samples were placed in a transport medium of phosphate-buffered saline (PBS) containing penicillin (2000 U/ml) and streptomycin (2000 μg/ml) and maintained at 4 °C during transfer to the laboratory.
Virus isolation
Samples were suspended in antibiotic solution and centrifuged at 3000 rpm for 15 min. Supernatants were filtered with 0.22 um syringe filters and used to inoculate 10-day-old specific-pathogen-free embryonated eggs. After incubation at 37 °C for 48 h, allantoic fluid was harvested and tested by hemagglutination assay. Influenza A real-time reverse transcription PCR (rRT-PCR) kits (Qiagen, Shenzhen, China) were used to confirm that the observed HA was caused by AIV. Subtyping was performed as described [25, 26], and AIV positive allantoic fluid was stored at −80 °C.
RNA extraction and RT-PCR
RNA extraction from HA-positive allantoic fluid was conducted by Viral RNA minikits (Qiagen, Germany), according to the manufacturer’s instructions. Reverse transcription (RT) was under standard conditions using the Uni12 primer (AGCAAAAGCAGG), followed by PCR as described by Hoffmann and Stech [26] using primers specific for the eight RNA segments. PCR products were purified with Agarose Gel DNA Purification Kits (TaKaRa, Dalian, China) and cloned into the pMD18-T vector (TaKaRa, Dalian, China). Sequencing of three independent clones of each PCR product was performed to eliminate errors resulting from the RT-PCR or cloning steps.
Sequence analysis
Reference sequences were obtained from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). Comparisons of nucleotide and deduced amino acid sequences used the Lasergene sequence analysis software package 7.0 (DNASTAR, Madison, WI). Phylogenetic trees were generated with MEGA 4.0 software by the neighbor-joining method [27]. Bootstrap values were calculated based on 1000 replicates of the alignment. The eight gene segments of these isolates were sequenced, and homologies based on complete open reading frames were determined by comparison with sequences in GenBank.
Nucleotide sequence accession numbers
The nucleotide sequences of the five viruses determined in this study have been deposited in GenBank under accession numbers from KM222537 to KM222576.
Results
Molecular characterization
Five H3N2 viruses shared the same amino acid sequence, PEKQTR/GL(/denotes cleavage site), at the cleavage site between HA1 and HA2. This sequence was same as in other reported avian H3N2 viruses with the exception of A/duck/Shanghai/C84/2009. The cleavage site sequences of the viruses did not contain additional basic amino acids, suggesting low pathogenicity in poultry species [28].
Amino acids at receptor-binding sites, HA 98(Y), 153(W), 155(L), 183(H), 190(E), and 194(L), were highly conserved and did not show variation in any H3N2 AIVs. Amino acids of the five H3N2 viruses were RGQSG at position 224-228 and GGSGA at position 134-138. The receptor-binding sites of the five H3N2 viruses (Q226 and G228) were similar to all H3 AIVs in both Eurasian and North American lineages. This result suggested that these viruses preferentially bind to alpha 2-3-linked sialic acid receptors that are predominant in avian species [29]. In HA, six potential glycosylation sites at amino acids 8, 22, 38, 165, 285, and 483 were detected in four H3N2 viruses but not LPM1. The specific sequence for N-linked glycosylation is Asn-X-Ser/Thr, where X can be any amino acid except proline or aspartic acid [30].
Viral genetic characteristics associated with host adaptation, receptor binding, antiviral resistance, and pathogenesis were analyzed (Table 1). Lysine at position 627 of the polymerase PB2 protein is essential for efficient replication of avian influenza viruses in mammals. The influenza A virus PB1-F2 protein (encoded by the PB1 segment) is also associated with virulence [34]. Based on the NA protein sequences, all H3N2 viruses analyzed should be sensitive to neuraminidase inhibitors (Table 1). Other amino acids in the NS1 and matrix (M1; encoded by the M segment) proteins of the novel viruses were also associated with increased virulence (Table 1). All five H3N2 viruses had the S31 N substitution in the viral ion channel M2 (encoded by the M segment) (Table 1) that conferred resistance to ion channel inhibitors.
Genetic and phylogenetic analyses
All eight gene segments of the subtype H3N2 isolates were sequenced. The genomes of the five H3N2 viruses were identical and were compared to nucleotide sequences available in the GenBank database.
Phylogenetic analysis of the eight genes showed that the influenza viruses could be segregated into two distinct lineages, avian and human/swine influenza A viruses. All five H3N2 viruses were clustered in the Eurasian lineage with no close relationship between the newly isolated H3N2 viruses and the previously isolated H9N2, H7N9, and H5N1 from China.
Surface genes
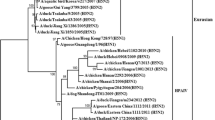
Phylogenetically, the HA genes of H3 subtype viruses were phylogenetically divided into North American and Eurasian lineages (Fig. 1a). The Eurasian lineage formed four groups: 1, 2, 3, and 4. Group 1 contained SH1 and SH2 strains, while Group 4 contained LPM2 and SH3. LPM1 and the other strain, namely A/environment/Hunan/S4304/2011 (H3N2), formed Group 3 to differentiate with other strains. Group 1 and 3 viruses were mainly Chinese viruses, while Group 4 viruses were a mixture from poultry and wild birds and were from a wider geographic region including Japan, Korea, Vietnam, and China. This diversity might have resulted from the poultry trade or wild bird migration. The long-distance flights of migratory birds might be important for transmission of Group 4, with local poultry trades accounting viral spread in Groups 1 and 3.


Phylogenetic trees for the HA (a) and NA (b) genes of the H3N2 AIVs. The nucleotide coding regions tree was generated by the neighbor-joining method and bootstrapped with 1000 replicates using the MEGA software version 4.0. Viruses in the present study are highlighted by a triangle. The scale bar represents the distance unit between sequence pairs
Like HA, the NA phylogenic tree was divided into North American and Eurasian lineages (Fig. 1b). The Eurasian lineage was further divided into three groups: 1, 2, and 3. LPM1 and LPM2 were in group 1 along with viruses from China and the neighboring regions of Mongolia, Korea, Japan, and Vietnam. SH1, SH2, and SH3 were in Group 3. Viruses in Groups 2 and 3 were mostly from China. These findings supported the hypothesis that migratory birds and the poultry trade might be important for transmission patterns of avian H3 influenza viruses in nature. Based on the HA and NA phylogenic trees, the H3N2 viruses circulating in Shanghai in 2013 were reassortants of viruses circulating in aquatic birds in Eastern and Southeastern Asia and viruses previously circulating in ducks and pigeons in China.
Internal genes
Phylogenic analysis of internal genes showed that all five isolates belonged to the Eurasian lineage (Fig. S1c-h) and had internal gene similarity of 87.1–100 %. In general, all internal gene trees were divided into three groups except for the PA and M genes. PA gene phylogeny revealed multiple genetic lineages from both the Eurasian lineage I and Eurasian lineage II cocirculating in the H3 subtype gene pool (Fig. S1c). The M genes of the five H3N2 viruses belonged to two different groups in the Eurasian lineage (Fig. S1d). LPM1 belonged to Group 1 which represented a wider geographic region and species range that included canines, ducks, and wild birds. The other four H3N2 M genes from LPM2, SH1, SH2, and SH3 were in Group 2 along with viruses from China and Vietnam.
Similar phylogenies were observed for the PB2 and NP genes. The PB2 genes of the five isolates were diverse, falling into two distinct groups in the phylogenetic tree (Fig. S1e). The PB2 genes of four H3N2 viruses, LPM1, SH1, SH2, and SH3, fell into a distinct group far from Group 3. One of the four H3N2 viruses, LPM1, shared 99.4 % identity with its closest relative in GenBank, A/duck/Hunan/S11090/2012(H4N6). The PB2 genes of two isolates, SH1 and SH2, had the highest identities with recently isolated H7N3 A/duck/Zhejiang/10/2011(H7N3). The PB2 gene of SH3 possessed the highest similarity with the H7N7 viruses recently isolated in Hong Kong. The single PB2 gene in Group 3, LPM2, shared 98.3 % identity with its closest relative in GenBank, A/mallard/Sweden/101900/2009(H4N3).
The NP genes of the five H3N2 viruses clustered into two groups, with nucleotide similarities of 91.6 % (Fig. S1f). As with the PB2 gene, the NP genes of four H3N2 viruses, LPM1, SH1, SH2, and SH3, formed a distinct group far from Group 3. One of the four H3N2 viruses, LPM1, shared 98.3 % identity with its closest relative in GenBank, A/mallard/Sweden/93225/2009(H4N6). The NP genes of three isolates, SH1, SH2, and SH3, had the highest identities with recently isolated H1N3 A/duck/Zhejiang/0611-8/2011(H1N3). The single NP gene in Group 3, LPM2, shared 98.5 % identity with its closest relative in GenBank, A/duck/Eastern China/1/2008(H6N1).
Phylogenetic analyses of the PB1 genes revealed three groups in the tree (Fig. S1g). Group 1 included LPM1, LPM2, and SH3, and was from a mix of canine, domestic, and wild birds. This group included H5N2 isolated from migratory birds in Northeast China. The PB1 gene of SH3 had the highest similarity with H5N2 viruses recently isolated in Northeast China. The two PB1 genes in Group 2, from SH1 and SH2, shared 99.9 % identity but had 99.0 and 99.0 % identity, respectively, with the closest PB1 gene in GenBank from A/duck/Jiangsu/10-d4/2011(H11N3).
In the NS gene tree, all five isolates formed an independent branch within the phylogenic tree (Fig. S1h). LPM1 and LPM2 shared 99.3 % identity but had 99.3 and 99.1 % identity, respectively, with the closest NS gene in GenBank from A/wild bird/Korea/A330/2009(H7N7). SH1 and SH2 shared 99.5 % identity but had 96.7 and 97.2 % identity, respectively, with the closest NS gene in GenBank from A/duck/Primorie/2633/2001(H5N3). SH3 shared 98.6 % identity with its closest relative in GenBank, A/canvasback/Mongolia/2-69/2007(H10N6).
Extensive reassortment in the internal genes among the H3N2 isolates and other subtype influenza viruses was observed. The nucleotide homologies of the internal genes were closely related to other influenza viruses subtypes isolated from Eurasian ducks or wild birds.
Discussion
H3N2 virus in chickens from LPMs was first detected in Central China in 2001 [21]. Since 2009, H3N2 AIVs have been regularly reported in China [10, 22, 42–44]. However, little was known about the molecular characteristics of H3N2 AIVs in China. In this study, five H3N2 AIVs were isolated in LPMs and slaughterhouses in Shanghai, Eastern China, in 2013. Whole-genome sequences of the five H3N2 AIVs were determined. Genetic analysis showed that the five H3N2 AIVs displayed low-pathogenic avian influenza characteristics. Phylogenetic analysis of all eight viral genes showed that the viruses clustered in the Eurasian lineage of influenza viruses. All phylogenetic trees showed that the two lineages corresponded to avian and human and swine influenza A viruses. The H3 AIVs were not close to the H3N2 viruses with human or swine origin.
Many mallards and waterfowl move easily into China from neighboring countries by migration or the live poultry trade. We found that migratory birds and the poultry trade might be important in the transmission patterns of avian H3 influenza viruses in nature. The Eurasian lineage included a mixture of viruses from wild birds and poultry, indicating exchange among viruses. These findings demonstrated that migratory birds and poultry trade might have spread AIV to Eastern China from neighboring countries and contributed to the complexity of the AIV ecology in this region. Viruses isolated previously in the neighboring countries of Vietnam, Mongolia, Korea, and Japan showed a high similarity in surface and internal genes to the five H3N2 AIVs. These results indicated frequent exchange of influenza viruses among Southeastern Asian countries, possibly from wild bird migration or the international poultry trade.
Reassortment among influenza viruses is considered the main mechanism for the emergence of novel viruses that could lead to an influenza pandemic. H3N2 subtype viruses infect avian and mammalian hosts [45, 46]. The H3N2 virus that causes mild respiratory disease in chicken flocks in Italy was a reassortant [7]. All these findings confirm that genetic reassortment among AIVs facilitates viral variation and the emergence of novel strains. We found that AIV subtypes other than H3N2 served as gene donors for H3N2 viruses in Eastern China, increasing the genetic diversity of H3N2 viruses [10, 42, 44]. Results on eight genes showed reassortment events, probably between viruses from pigeon, domestic ducks, and wild birds and H3 subtype viruses and other subtype viruses, especially H5 and H7. Reassortment between H3N2 and other influenza viruses increases genetic diversity and allows the emergence of new pathogenic influenza strains that might infect human beings [47].
LPMs are a major source of influenza virus dissemination and potential influenza virus reassortment [21]. Moreover, Dent et al. [23, 24] considered slaughterhouses as potential agents of AI spread among commercial farms in the United Kingdom. The ecological background of LPMs and slaughterhouses provides a crucial, suitable condition for viral reassortment. In this study, although we found no evidence of reassortment among the human, canine, and avian hosts in LPMs and slaughterhouses, we propose that extensive reassortment of the H3 subtype is a threat to avian and human health. Closure of LPMs is not a complete solution to reducing the emergence of novel reassortant influenza viruses with potential threats to humans. It is likely to have negative consequences for the healthy development of the poultry industry. Therefore, it is important to strengthen AIV surveillance work in avian, swine, canine, and human hosts.
References
R.A. Fouchier, V. Munster, A. Wallensten, T.M. Bestebroer, S. Herfst, D. Smith, G.F. Rimmelzwaan, B. Olsen, A.D. Osterhaus, J. Virol. 79(5), 2814–2822 (2005)
B. Olsen, V.J. Munster, A. Wallensten, J. Waldenström, A.D. Osterhaus, R.A. Fouchier, Science 312(5772), 384–388 (2006)
E.F. Kaleta, G. Hergarten, A. Yilmaz, Dtsch. Tierarztl. Wochenschr. 112(12), 448–456 (2005)
J. Pasick, H. Weingartl, A. Clavijo, J. Riva, H. Kehler, K. Handel, E. Watkins, K. Hills, Avian Dis. 47, 1208–1213 (2003)
W.J. Bean, M. Schell, J. Katz, Y. Kawaoka, C. Naeve, O. Gorman, R.G. Webster, J. Virol. 66(2), 1129–1138 (1992)
R.G. Webster, W.J. Bean, O.T. Gorman, T.M. Chambers, Y. Kawaoka, Microbiol. Rev. 56(1), 152–179 (1992)
L. Campitelli, C. Fabiani, S. Puzelli, A. Fioretti, E. Foni, A. De Marco, S. Krauss, R.G. Webster, I. Donatelli, J. Gen. Virol. 83, 413–420 (2002)
M.S. Song, T.K. Oh, H.J. Moon, D.W. Yoo, E.H. Lee, J.S. Lee, C.J. Kim, G.J. Yoo, H. Kim, Y.K. Choi, J. Gen. Virol. 89, 949–957 (2008)
J. Pu, Q.F. Liu, Y.J. Xia, Y.L. Fan, E.G. Brown, F.L. Tian, J.H. Liu, Virus Genes 38(1), 136–142 (2009)
H. Zhou, A. Zhang, H. Chen, M. Jin, Arch. Virol. 156(6), 1045–1048 (2011)
Y. Kawaoka, S. Krauss, R.G. Webster, J. Virol. 63(11), 4603–4608 (1989)
E.L. Stephen, J.C. Nancy, K. Alexander, Int. Congr. 1263, 184–190 (2004)
O.T. Gorman, W.J. Bean, Y. Kawaoka, I. Donatelli, Y. Guo, R.G. Webster, J. Virol. 65(7), 3704–3714 (1991)
U. Schultz, W.M. Fitch, S. Ludwig, J. Mandler, C. Scholtissek, Virology 183(1), 61–73 (1991)
M.R. Castrucci, I. Donatelli, L. Sidoli, G. Barigazzi, Y. Kawaoka, R.G. Webster, Virology 193(1), 503–506 (1993)
E.C. Claas, Y. Kawaoka, J.C. de Jong, N. Masurel, R.G. Webster, Virology 204(1): 453-457
S. Li, Z. Shi, P. Jiao, G. Zhang, Z. Zhong, W. Tian, L.P. Long, Z. Cai, X. Zhu, M. Liao, X.F. Wan, Infect Genet Evol 10(8), 1286–1288 (2010)
D. Song, B. Kang, C. Lee, K. Jung, G. Ha, D. Kang, S. Park, B. Park, J. Oh, Emerg. Infect. Dis. 14(5), 741–746 (2008)
D. Song, C. Lee, B. Kang, K. Jung, T. Oh, H. Kim, B. Park, J. Oh, Emerg. Infect. Dis. 15(1), 56–58 (2009)
K.F. Shortridge, Semin. Respir. Infect. 7(1), 11–25 (1992)
M. Liu, S. He, D. Walker, N. Zhou, D.R. Perez, B. Mo, F. Li, X. Huang, R.G. Webster, R.J. Webby, Virology 305(2), 267–275 (2003)
B.F. Qiu, W.J. Liu, D.X. Peng, S.L. Hu, Y.H. Tang, X.F. Liu, J. Virol. Methods 155(2), 193–198 (2009)
J.E. Dent, R.R. Kao, I.Z. Kiss, K. Hyder, M. Arnold, BMC Vet Res 4, 27 (2008)
J.E. Dent, I.Z. Kiss, R.R. Kao, M. Arnold, BMC Vet Res 7, 59 (2011)
L.P. Phipps, S.C. Essen, I.H. Brown, J. Virol. Methods 122(1), 119–122 (2004)
E. Hoffmann, J. Stech, Y. Guan, R.G. Webster, D.R. Perez, Arch. Virol. 146(12), 2275–2289 (2001)
K. Tamura, J. Dudley, M. Nei, S. Kumar, Mol. Biol. Evol. 24(8), 1596–1599 (2007)
D.A. Steinhauer, Virology 258(1), 1–20 (1999)
J. Liu, D.J. Stevens, L.F. Haire, P.A. Walker, P.J. Coombs, R.J. Russell, S.J. Gamblin, J.J. Skehel, Proc. Natl. Acad. Sci. 106(40), 17175–17180 (2009)
A. Helenius, M. Aebi, Annu. Rev. Biochem. 73, 1019–1049 (2004)
E.K. Subbarao, W. London, B.R. Murphy, J. Virol. 67(4), 1761–1764 (1993)
Z. Li, H. Chen, P. Jiao, G. Deng, G. Tian, Y. Li, E. Hoffmann, R.G. Webster, Y. Matsuoka, K. Yu, J. Virol. 79(18), 12058–12064 (2005)
R. Zell, A. Krumbholz, P. Wutzler, C. Campese, C. Pelaz, Emerg. Infect. Dis. 12(10), 1607–1608 (2006)
G.M. Conenello, D. Zamarin, L.A. Perrone, T. Tumpey, P. Palese, PLoS Pathog. 3(10), 1414–1421 (2007)
M. Okomo-Adhiambo, G.J. Demmler-Harrison, V.M. Deyde, T.G. Sheu, X. Xu, A.I. Klimov, L.V. Gubareva, Antimicrob. Agents Chemother. 54(5), 1834–1841 (2010)
J.L. McKimm-Breschkin, A. Sahasrabudhe, T.J. Blick, M. McDonald, P.M. Colman, G.J. Hart, R.C. Bethell, J.N. Varghese, J. Virol. 72(3), 2456–2462 (1998)
S. Fan, G. Deng, J. Song, G. Tian, Y. Suo, Y. Jiang, Y. Guan, Z. Bu, Y. Kawaoka, H. Chen, Virology 384(1), 28–32 (2009)
L.H. Pinto, L.J. Holsinger, R.A. Lamb, Cell 69(3), 517–528 (1992)
P. Jiao, G. Tian, Y. Li, G. Deng, Y. Jiang, C. Liu, W. Liu, Z. Bu, Y. Kawaoka, H. Chen, J. Virol. 82(3), 1146–1154 (2008)
S.H. Seo, E. Hoffmann, R.G. Webster, Virus Res. 103(122), 107–113 (2004)
J.C. Obenauer, J. Denson, P.K. Mehta, X. Su, S. Mukatira, D.B. Finkelstein, X. Xu, J. Wang, J. Ma, Y. Fan, K.M. Rakestraw, R.G. Webster, E. Hoffmann, S. Krauss, J. Zheng, Z. Zhang, C.W. Naeve, Science 311(5767), 1576–1580 (2006)
Q. Teng, T. Hu, X. Li, G. Li, Z. Li, J. Virol. 86(21), 11944 (2012)
G. Deng, D. Tan, J. Shi, P. Cui, Y. Jiang, L. Liu, G. Tian, Y. Kawaoka, C. Li, H. Chen, J. Virol. 87(17), 9452–9462 (2013)
Q. Li, Q. Zhao, M. Gu, J. Zhu, X. Gu, G. Zhao, Q. Liu, X. Wang, X. Liu, X. Liu, Genome Announc 1(2), e0022112 (2013)
H.M. Yassine, C.W. Lee, D.L. Suarez, Y.M. Saif, Vaccine 26(7), 966–977 (2008)
J.G. Choi, H.M. Kang, M.C. Kim, M.R. Paek, H.R. Kim, B.S. Kim, J.H. Kwon, J.H. Kim, Y.J. Lee, Vet. Micro 155, 147–157 (2012)
A. Kreibich, O. Stech, J. Hundt, M. Ziller, T.C. Mettenleiter, J. Stech, PLoS ONE 8(11), e79165 (2013)
Acknowledgments
This study was financially supported by Grant No. 2013 QLG001 from Shanghai Municipal Health & Family Planning Commission.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by William Dundon.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, D., Liu, J., Ju, H. et al. Genetic analysis of H3N2 avian influenza viruses isolated from live poultry markets and poultry slaughterhouses in Shanghai, China in 2013. Virus Genes 51, 25–32 (2015). https://doi.org/10.1007/s11262-015-1198-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-015-1198-5