
Abstract
Low-pathogenic avian influenza viruses (LPAIVs) of the H5 subtype can mutate to highly pathogenic forms, potentially destabilizing the poultry industry. Wild migratory birds are considered a natural reservoir of LPAIVs capable of dispersing both high- and low-pathogenic forms of the virus. Therefore, surveillance and characterization of AIV in wild birds are essential. Here, we report on the isolation and genetic characterization of 10 AIVs of the H5N2 subtype obtained through surveillance in Hokkaido, Japan, during 2009 and 2011. Full-genome sequencing revealed that the H5 and N2 genes of these isolates are all closely related to each other, belonging to the Eurasian avian-like lineage, but they are unrelated to H5 highly pathogenic strains of clade 2.3.4.4. The internal genes of the isolates were found to be diverse, consistent with our hypothesis that these H5N2 strains have undergone multiple reassortment events. Even though all of the H5N2 isolates were characterized as LPAIV based on the amino acid sequences at the HA cleavage site, this analysis demonstrates a diverse pool of precursors that may seed future outbreaks in poultry and possible human transmissions, suggesting the need for high-quality surveillance.
Similar content being viewed by others


Introduction
Avian influenza virus (AIV) is a negative-sense, segmented, and single-stranded RNA virus that belongs to genus Influenza virus A of the family Orthomyxoviridae. Its genome is composed of eight segments encoding at least 11 proteins, including the surface glycoproteins hemagglutinin (HA) and neuraminidase (NA), transcriptase proteins (PB2, PB1, PB1-F2 and PA), matrix proteins (M1 and M2), nucleoprotein (NP), and non-structural proteins (NS1 and NS2) [1]. The segmented nature of the genome facilitates reassortment, which contributes to the rapid evolution of AIVs. Furthermore, this genomic reshuffling plays a major role in generating pandemic influenza strains [2]. In the past, reassortant viruses with avian lineage gene segments caused the influenza pandemics of 1957, 1968 and 2009 [3, 4]. Thus, for pandemic preparedness, it is important to monitor the evolution of AIVs in avian populations, including wild and domestic birds.
Wild birds, particularly waterfowl, are considered the main reservoir of AIVs [5–7], which usually cause either mild or subclinical disease in domestic poultry hosts. Generally, evolution of AIV involves mutations or reassortment caused by the exchange of gene segments [7, 8]. Surveillance studies and genetic characterization of AIVs in nature have been widely performed, as AIVs are prone to reassort in migratory birds, resulting in the appearance of different influenza viruses of either a new subtype or the same subtype [2, 9, 10]. Viruses that reassort in wild birds can result in rapid changes in viral genotype that are conducive to host switching, potentially causing serious disease in poultry and human hosts [11]. To date, only some viruses that belong to subtypes H5 and H7 have evolved into the highly pathogenic (HP) forms by acquiring genetic mutations in the HA cleavage site [12–14], but low-pathogenic (LP) forms remain a risk for epidemic spread. In 2014, Eurasian-origin H5 related to the Goose/Guangdong lineage spread rapidly to domestic flocks in East Asia, Western Europe and North America, consistent with movement by wild birds [15]. Thus, it is imperative to characterize newly isolated H5 AIVs, particularly in Asia, where biosecurity between wild and domestic birds is porous, in order to monitor viral distribution and any genetic changes that may alter the viral phenotype.
It has been hypothesized that LPAI H5N2 viruses are the precursor strains responsible for the outbreaks of H5N2 HPAIV in poultry in northern Europe [16], Mexico [17, 18], Italy [19], Texas [20] and Taiwan [21]. Also, the presence of avian-origin LPAI H5N2 viruses in domestic ducks and pigs in Korea [22, 23] and in food products in Singapore [24] has been reported. In addition, Japan experienced outbreaks in domestic poultry during 2005-2006, caused by LPAI H5N2 viruses of the North American lineage [25]. During these outbreaks, 41 farms were affected, in which 70 % of poultry were laying hens and 30 % were broilers [26]. Owing to past experiences with the LPAI H5N2 virus and its potential for mutating to HPAIV, the presence of H5 LPAIV as well as the H7 LPAIV in birds and poultry was incorporated in the list of Domestic Animal Infectious Diseases regulated by law in Japan and strictly monitored by the veterinary authorities (http://www.maff.go.jp/aqs/english).
Here, we report the genetic characterization of multiple reassortant H5N2 viruses isolated from wild birds in Hokkaido during 2009 and 2011 and propose a mechanism for their genesis. Molecular characteristics of these isolates were investigated in comparison with other H5N2 strains, including the previously reported Japanese strains from wild birds and chickens. In addition, phylogenetic analysis was conducted to estimate the evolutionary history of the isolates. Combined, this work helps us to understand the temporal-spatial scale for evolution of contemporary H5 viruses, which remain an agricultural and public-health concern.
Materials and methods
Viruses and virus identification
Ten viruses of the H5N2 subtype, isolated from wild birds in Hokkaido, the northern part of Japan, were included in this study. Isolation methods and surveillance details were reported previously [27]. Briefly, 10 % homogenous samples of feces were inoculated into 10-day-old embryonated chicken eggs by the allantoic route. The eggs were incubated at 37 °C for 4 days. The allantoic fluid was then collected and tested for hemagglutination activity. The presence of AIV was checked by real-time RT-PCR for detection of the M gene [28]. The AIV isolates were subtyped for HA and NA by RT-PCR and confirmed by serological tests according to the methods described previously [29–31]. Three of the 10 H5N2 viruses were isolated in Hakuryo Pond during 2009 from wild birds migrating through Hokkaido [27]. In 2011, the other seven strains of H5N2 were isolated from wild birds in Hakuryo Pond and the Obihiro River and identified as reported previously. Table 1 lists the information for all 10 isolates.
Nucleotide sequencing
Total RNA was extracted from allantoic fluids using ISOGEN-LS (NIPPON GENE CO., LTD., Tokyo, Japan). For nucleotide sequencing of AIV genes, the viral RNA was transcribed into cDNA using the universal 12-mer primer (5′-AGC RAA AGC AGG-3′) and Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) at 42 °C for 60 min, followed by 70 °C for 10 min. Using the cDNA as a template, PCR was conducted to amplify the entire length of all eight viral segments using segment-specific primers as described by Hoffmann et al. [32] and Obenauer et al. [33]. The resulting PCR products were separated by 1 % agarose gel electrophoresis and purified using either a QIAquick PCR Purification Kit (QIAGEN, Hilden, Germany) or a Gene Clean II Kit (Biogene, Inc., USA). The purified PCR products were used as templates for sequencing reactions using a BigDye Terminator ver. 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA). Sanger sequencing was performed using an Applied Biosystems 3500 Genetic Analyzer (Life Technologies, Carlsbad, CA). The primer sets described above as well as additional overlapping primers that were designed as needed (sequences available upon request) were used for nucleotide sequencing.
Molecular characterization and genotypic assignment
Viral nucleotide sequences were analyzed with the Genetyx Ver. 10 software (GENETYX Corp., Tokyo, Japan) and compared with other sequences available in GenBank identified by BLAST homology searches (http://www.ncbi.nlm.nih.gov/genomes/ FLU/FLU.html). Nucleotide and deduced amino acid sequences of all of the gene segments of the isolates were compared with those of other H5N2 strains, including previously reported Japanese strains, based on alignments made using Clustal W [34]. Genotyping of the H5N2 viruses was conducted using the influenza A virus genome segment genotyping tool available at http://www.flugenome.org/genotyping.php. The program was used to determine the individual genome segment lineage as described previously by Lu et al. [35]. Alleles for gene segments were determined using the BLAST method [36] with coverage >95 % and identity >90 %. Concatenating alleles for each gene segment created genotypes of each isolate.
Phylogenetic analysis
Phylogenetic analysis was performed by aligning nucleotide sequences of the H5N2 viruses from this study with those of other isolates of the Eurasian and American lineages and closely related isolates identified using BLAST. Multiple sequence alignment was performed using Clustal W [34]. The Tamura-Nei substitution model was identified as the best-fit model for the data and used for phylogenetic tree reconstructions of all eight gene segments. All positions containing gaps and missing data were eliminated. Phylogenetic trees were constructed using the maximum-likelihood (ML) method supported by 500 bootstrap replicates [37] using MEGA 6.06 software [38].
Bayesian phylogenetic analysis was conducted to determine the time to the most recent common ancestor (TMRCA) and date the divergence between LP and HP strains circulating in East Asia. Sequences of AIV from avian hosts collected globally were downloaded from the Influenza Research Database (IRD) on 15 July 2015 and aligned using MUSCLE [39], and any incomplete sequences were removed. Using PAUP 4.0 [40], neighbor-joining trees were generated using the HKY85 substitution model and taxa that formed outgroups were randomly down-sampled. The final number of sequences in each dataset was as follows: PB2, n = 450; PB1, n = 418; PA, n = 404; H5, n = 454; NP, n = 406; N2, n = 207; M, n = 386; NS, n = 894. Phylogenetic analysis was performed using a Bayesian Markov chain Monte Carlo (MCMC) method implemented in BEAST v1.8.0 [41]. The date of sample collection associated with each sequence was used to time calibrate the tree. The uncorrelated lognormal relaxed molecular clock and general time-reversible (I+G) substitution model were used with a Bayesian skyride coalescent tree prior. We performed four independent MCMC chains of 50 million generations that were diagnosed in Tracer 1.6 to ensure that the effective sample size of each run reached 200. Trees from each MCMC run were combined after appropriate burn-in (~10 %) to produce 10,000 trees. For TMRCA analysis, the most recent sequences were used to calibrate the root height of the tree and the nodes were used to estimate the divergence time between two sequences.
Results
Virus identification
Samples showing hemagglutination titers from 1:64 to 1:512 after harvest from inoculated eggs were confirmed for influenza A virus infection by real-time RT-PCR for detection of the M gene segment, which yielded Ct values ranging from 16.96 to 22.70 (Table 1). The 10 isolates were identified as H5N2 using RT-PCR and serological tests (data not shown) followed by sequencing of the viral genome. The entire genome sequences of the isolates obtained in this study were deposited in GenBank under the accession numbers shown in Table 1. The sequences including the HA, NA and M genes of three H5N2 viruses (9UO025, 9UO036 and 9UO139) isolated in 2009 were previously reported by Abao et al. [27], while the rest of the sequence data were obtained in this study.
Molecular characterization
Genomic features of the 10 H5N2 isolates are shown in Table 2. Analysis of the deduced amino acid sequences of the HA protein revealed that all of the H5N2 viruses had a PQRETR/GLF amino acid sequence motif at the HA cleavage site, however, a single virus (9UO139) had PQKETR/GLF. Both motifs are typical of LPAI H5 viruses [42]. All of the isolates had a Gln at position 222 and Gly at position 224 of the HA, suggesting that all of them retained affinity for alpha 2,3 avian-type glycan receptors [43, 44]. No NA stalk deletions, which are associated with adaptation to terrestrial poultry [45], were found in any of the Japanese H5N2 strains, including the 10 isolates described here, except in the H5N2 Ibaraki strain, which was isolated in 2005 [25]. Several amino acids that enhance pathogenicity in mice and/or humans were detected in genes of the new H5N2 isolates. An Ala residue at position 42 of the NS1 protein was found in the three isolates from 2009, as well as in the reference Japanese strains, including Ibaraki and Akita (Table 2). All of the H5N2 viruses reported in this study and most of the reference strains contained the PDZ binding sequence motif (-ESEV-), although the Ibaraki strain has -ESKV- in the NS1 gene sequence. The PB1-F2 gene of the 9UO036 and 9UO139 H5N2 isolates had an S66 mutation, while the other strains and reference strains retained N66. No other genetic markers associated with virulence in mammals such as mutations in the PB2 gene (E627K and D701N) [11, 46], were found in any of the isolates or selected Japanese strains. Moreover, no mutations conferring amantadine or oseltamivir resistance, such as H274Y in the NA gene, were found in any of the isolates or the selected reference strains shown in Table 2 [47–49], except for the Ibaraki strain, which had 26F and 31N in the M2 protein, indicating amantadine resistance.
Phylogenetic analysis
HA and NA genes
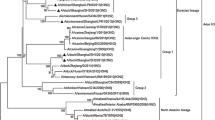
The ten HA gene sequences of the H5N2 isolates were analyzed to explore their evolutionary relationships relative to the Eurasian and American lineages and H5 viruses. The initial ML analysis revealed that the HA genes of the H5N2 isolates belong to the Eurasian lineage and clustered separately from the HPAI H5 viruses. The HA genes of our H5N2 isolates were closely related and clustered with viruses from wild birds in Japan, Korea and China sampled between 2005 and 2010 (Fig. 1). Bayesian analysis indicated that the H5N2 isolates diverged from the HPAIV clade circa 1990 and were distantly related to clade 2.3.4.4 (Fig. 4).

Maximum-likelihood phylogenetic trees of the H5 gene of avian influenza isolates in Japan together with reference strains of H5 subtypes available in the GenBank database. The H5N2 strains isolated in 2009 (●), 2011 at Hakuryo Pond (▲) and 2011 at Obihiro River (■) are indicated by markers. Numbers at each node indicate bootstrap values ≥60 %. Phylogenetic analysis was conducted using MEGA6.06. The scale bar indicates 0.05 nucleotide substitutions per site
All NA genes of the 10 H5N2 viruses fell within the Eurasian lineage. These sequences formed a monophyletic group structured according to year and location of isolation. All of the H5N2 isolates clustered with a lineage containing H4N2, H5N2, H9N2, H10N2, H13N2 and H6N2 viruses isolated in Japan, Korea and China. The N2 gene sequences of the 10 H5N2 isolates were most closely related to A/avian/Japan/8KI0148/2008(H4N2) (Fig. 2). The TMRCA date of the N2 viruses and clade 2.3.4.4. is approximately 1975-1976, demonstrating that the NA of these Japanese isolates do not contain an immediate precursor to circulating clade 2.3.4.4 virus.

Maximum-likelihood phylogenetic trees of the full N2 gene of avian influenza isolates in Japan together with reference strains of N2 subtypes available in the GenBank database. The H5N2 strains isolated in 2009 (●), 2011 at Hakuryo Pond (▲) and 2011 at Obihiro River (■) are indicated by markers. Numbers at each node indicate bootstrap values ≥60 %. Phylogenetic analysis was conducted using MEGA6.06. The scale bar indicates 0.05 nucleotide substitutions per site
Internal genes
ML analysis indicated that all of the internal genes of the H5N2 isolates belonged to the Eurasian-like avian lineage of AIV (Fig. 3A-F). The ML tree topology of the M gene was similar to those of the HA and NA trees, indicating possible co-carriage, as they clustered within a single branch with H3N8 viruses from Japan and Laos as well as H6N1 and H7N7 viruses from Korea (Fig. 3E). TMRCA analysis revealed that the H5N2 isolates diverged from the currently circulating clade 2.3.4.4 between 1977 and 1978 (Fig. 4).



Maximum-likelihood phylogenetic trees of the internal genes of H5N2 avian influenza isolates in Japan together with reference strains available in the GenBank database. Phylogenetic trees are shown for the nucleotide sequences for PB2, PB1, PA, NP, M and NS (A-F, respectively). The H5N2 strains isolated in 2009 (●), 2011 at Hakuryo Pond (▲) and 2011 at Obihiro River (■) are indicated by markers. Numbers at each node indicate bootstrap values ≥60 %. Phylogenetic analysis was conducted using MEGA6.06. The scale bar indicates nucleotide substitutions per site

Dated phylogenies of all six internal genes, the H5 gene, and the N2 gene belonging to the Eurasian lineage of influenza viruses using Bayesian inference (BEAST 1.8.0). The 10 isolates generated in this study are color coded according to purple (9UO), green (11UO) and blue (11OG) distinguishing the three separate sampling events in Japan. All eight gene trees indicate that the 10 isolates are only distantly related to the highly pathogenic H5 clade 2.3.4.4, with the most recent common ancestor dated at 1975-1996. The horizontal axis shows the calendar years
For PB2, the 10 H5N2 isolates clustered into two distinct subgroups. Six of the isolates obtained from Hakuryo Pond (UO samples) clustered with H9N2, H6N5, H6N8 and H3N8 viruses isolated from ducks in Japan and an H3N8 strain isolated from an environmental sample in Korea. The other four isolates sampled during 2011 grouped with isolates from chickens (H7N7) and domestic ducks (H4N8 and H6N1) from China, as well as the H10N4 virus of wild birds in Korea (Fig. 3A). Molecular dating indicated that these four H5N2 viruses diverged from the H5 clade 2.3.4.4 during 1996-1997 (Fig. 4).
The PB1 genes of the 10 H5N2 isolates were divided into four distinct clusters. Three isolates at Hakuryo Pond, including 11UO0008, 11UO0012 and 11UO0023, were close to each other and closely related to the isolates from Korea (H4N6), Vietnam (H11N9), Hong Kong (H11N9) and Japan (H6N5). The four isolates in the second cluster, 11OG1032, 11OG1038, 11OG1083 and 11OG1084, were close to the isolates from China (H6N1), Vietnam (H5N2), Korea (H4N6) and Japan (H4N2). The other isolates, including 9UO025, 9UO036 and 9UO139, formed a distinct cluster relative to the 2011 group (Figs. 3B and 4).
For the PA gene, the ML tree revealed that the 10 isolates were separated into four distinct clusters. Among those, 11UO samples were close to each other and formed a cluster distinct from the 11OG samples. The 11UO samples were closely related to Korean strains (H2N3 and H3N8). The 11OG samples were close to A/wild duck/Korea/SH5-26/2008(H4N6). The 9UO isolates were structured into two distant clusters in which 9UO036 and 9UO139 formed a cluster with Japanese H3N5 and H6N5 and Chinese H5N2 isolates. The 9UO025 isolate was parental to the 11UO isolates (Fig. 3C).
For the NP gene, the ML tree showed that the 10 isolates were separated into two distinct clusters. The three isolates of the 11UO group were slightly distant from other H5N2 isolates and clustered with the H3N8 isolated from a Vietnamese domestic duck and other Korean (H3N1, H3N6 and H3N8) and Chinese isolates (H6N2). Other H5N2 isolates, including the three isolates in the 9UO group and four isolates in 11OG group, clustered together, along with Japanese isolates of H3N5; H3N8, H5N1, H6N5 and H9N2 subtypes (Fig. 3D).
The NS gene tree was structured into two alleles: allele A, containing 11UO and 11OG, and allele B, containing 9UO isolates. The ML analysis showed that the 11UO and 11OG isolates were closely related to each other and closely related to the Korean H7N7, Chinese H11N9 and American H4N6 isolates. The TMRCA for the H5N2 and American isolates was relatively recent, circa ~2005 (Fig. 4). In contrast, the 9UO isolates were highly similar to each other and closely related to the H3N8 Japanese strains, Korean strains (H6N1, H3N8), Chinese H11N9 and Taiwanese strains (H4N6 and H3N8) (Fig. 3F).
Assignment of genotypes
Three unique genotypes were assigned to the 10 isolates of H5N2 AIVs (Table 3). These genotypes were different in the PB2 and NS gene segments, and each of the three genotypes was assigned to more than one isolate. These genotype assignments depended on either the year (2009 or 2011) or sampling site (Hakuryo Pond or Obihiro River) and resulted in at least five genomic patterns among the 10 H5N2 subtype isolates (Supplementary Table S1). All of the 11OG strains (four isolates) had the same pattern in which the genes were derived from at least six different origins. While the 11UO strains (three isolates) shared a common pattern in which the genes originated from seven AIVs with four subtypes. In contrast, 9UO strains showed three different genomic patterns in which the 9UO036 and 9UO139 strains were different only in the NS gene segment. However, 9UO025 seemed to be separately evolved from the 9UO036 and 9UO139 strains.
Discussion
Here, we report the results of full-genome sequencing and genetic analysis of 10 H5N2 viruses obtained during surveillance in wild birds in Japan during 2009 and 2011, revealing multiple genetic reassortments. Phylogenetic analysis of the eight genomic segments of the 10 H5N2 viruses indicated that the HA, NA and M genes were closely related to each other and other LPAIVs isolated in East Asia. In contrast, the other segments were derived from various subtypes of AIV in the Eurasian-like avian lineage, while the NS lineage was associated with America-to-Asia viral flow. Phylogenetic analysis indicated that the H5N2 viruses contain segments closely related to the corresponding genes of a range of subtypes, including those of viruses isolated from wild birds in Korea, Japan, China, Taiwan and Vietnam. The multiple reassortment H5N2 viruses sampled during the same year and location (9UO, 11UO and 11OG isolates) seems to imply a high frequency of reassortants among AIVs, as described previously [50, 51]. This observation suggests that these viruses could play a role as gene donors for H5N2 viruses that are found in Japan and provides further evidence of dynamic inter-subtype interaction in this region.
No mutations that increased virulence of the H5N2 viruses were found except in the NS and PB1-F2 genes. The NS1 protein of 9UO025, 9UO036 and 9UO139 displayed a mutation of S42A, which has been associated with an increase in virulence in chickens and mice [52]. In all of the H5N2 isolates in this study, the PDZ binding sequence at the C-terminus of the NS1 protein was ESEV and the residue at position 138 was Phe (F), suggesting increased virulence in mammals [33, 53]. In addition to the mutation in the NS1 gene, the mutation N66S, associated with increased virulence of influenza A viruses in mice [54], was found in the PB1-F2 gene of two isolates (9UO036 and 9UO139). Interestingly, the Ibaraki strain (a reference strain that caused an outbreak in Japan during 2005) did not contain either S66 or N66 in the PB1-F2 gene. Taken together, several critical residues that contribute to the pathogenicity of AIV were found in the NS1 and PB1-F2 genes of our H5N2 isolates.
In order to further characterize the 10 H5N2 strains, we analyzed the genome for molecular markers associated with resistance to existing antivirals. To date, amantadine and oseltmivir have been developed for prophylaxis and treatment of influenza A virus infection. However, a single substitution in the M2 or NA protein has been shown to confer resistance to amantadine and oseltamivir, respectively [47–49, 55]. In this study, the NA and M2 of the H5N2 viruses did not contain any of the amino acid substitutions associated with resistance to oseltamivir and amantadine (Table 2). On the other hand, the Ibaraki strain contained two amino acid substitutions at positions 26 (L26F) and 31 (S31N), suggesting a reduced susceptibility to amantadine (Table 2).
In this study, all H5N2 viruses were characterized as LPAIV derived from multiple reassortments. However, the presence of H5N2 viruses in migratory wild birds in Japan represents a continued risk for poultry, as H5N2 LPAIV may acquire high pathogenicity by consecutive passages in chickens [21, 56]. In this study, the NP genes of the H5N2 viruses isolated in 2011 (11UO samples) were from the same lineage as H3N8 viruses isolated from domestic ducks in Vietnam. This result implies exchange of virus between migratory birds and free-ranging domestic ducks in South Asia, followed by movement to Japan facilitated by wild migratory birds. The continued circulation and proximity of rapidly evolving viruses, as demonstrated with the group of H5N2 viruses analyzed here, calls for expanded and continued surveillance of LPAIVs in poultry to ensure early detection of infection and limit regional spread of the virus due to the migration of wild birds. The rapid evolution of H5 and H7 viral subtypes to HP strains [13, 14] is a mechanism that was involved in 10 past outbreaks in poultry. Among them, three outbreaks were associated with H5N2 viruses [12]. Despite their phylogenetic distance from the current clade 2.3.4.4 H5 virus, the circulation and persistent presence of both HPAIV and LPAIV pathotypes in wild birds and poultry highlights the need for full genetic characterization of H5 viruses to better understand the evolution and potential origin of high-risk strains. This includes the risk to human health, as there have been reports of H5N2 virus of poultry origin infecting humans [57, 58], and we currently have little ability to predict pathogenicity from the viral sequence.
References
Wright PF, Neumann G, Kawaoka Y (2007) Orthomyxoviruses. In: Knipe DM, Howley PM (eds) Fields virology, 5th edn. Lippincott Williams & Wilkins Co., Philadelphia), pp 1691–1740
Chen R, Holmes EC (2009) Frequent inter-species transmission and geographic subdivision in avian influenza viruses from wild birds. Virology 383:156–161
Kilbourne ED (2006) Influenza pandemics of the 20th century. Emerg Infect Dis 12:9–14
Neumann G, Noda T, Kawaoka Y (2009) Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939
Olsen B, Munster VJ, Wallensten A, Waldenstrom J, Osterhaus AD, Fouchier RA (2006) Global patterns of influenza a virus in wild birds. Science 312:384–388
Stallknecht DE, Shane SM (1988) Host range of avian influenza virus in free-living birds. Vet Res Commun 12:125–141
Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y (1992) Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179
Domingo E, Mas A, Yuste E, Pariente N, Sierra S, Gutierrez-Riva M, Menendez-Arias L (2001) Virus population dynamics, fitness variations and the control of viral disease: an update. Prog Drug Res 57:77–115
Ellis TM, Bousfield RB, Bissett LA, Dyrting KC, Luk GS, Tsim ST, Sturm-Ramirez K, Webster RG, Guan Y, Malik Peiris JS (2004) Investigation of outbreaks of highly pathogenic H5N1 avian influenza in waterfowl and wild birds in Hong Kong in late 2002. Avian Pathol 33:492–505
Hoye BJ, Munster VJ, Nishiura H, Klaassen M, Fouchier RA (2010) Surveillance of wild birds for avian influenza virus. Emerg Infect Dis 16:1827–1834
Hatta M, Gao P, Halfmann P, Kawaoka Y (2001) Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293:1840–1842
Abdelwhab el-SM, Veits J, Mettenleiter TC (2013) Genetic changes that accompanied shifts of low pathogenic avian influenza viruses toward higher pathogenicity in poultry. Virulence 4:441–452
De Marco MA, Foni E, Campitelli L, Delogu M, Raffini E, Chiapponi C, Barigazzi G, Cordioli P, Di Trani L, Donatelli I (2005) Influenza virus circulation in wild aquatic birds in Italy during H5N2 and H7N1 poultry epidemic periods (1998 to 2000). Avian Pathol 34:480–485
Donatelli I, Campitelli L, Di Trani L, Puzelli S, Selli L, Fioretti A, Alexander DJ, Tollis M, Krauss S, Webster RG (2001) Characterization of H5N2 influenza viruses from Italian poultry. J Gen Virol 82:623–630
Verhagen JH, van der Jeugd HP, Nolet BA, Slaterus R, Kharitonov SP, de Vries PP, Vuong O, Majoor F, Kuiken T, Fouchier RA (2015) Wild bird surveillance around outbreaks of highly pathogenic avian influenza A(H5N8) virus in the Netherlands, 2014, within the context of global flyways. Euro Surveill 20
Munster VJ, Wallensten A, Baas C, Rimmelzwaan GF, Schutten M, Olsen B, Osterhaus AD, Fouchier RA (2005) Mallards and highly pathogenic avian influenza ancestral viruses, northern Europe. Emerg Infect Dis 11:1545–1551
Horimoto T, Rivera E, Pearson J, Senne D, Krauss S, Kawaoka Y, Webster RG (1995) Origin and molecular changes associated with emergence of a highly pathogenic H5N2 influenza virus in Mexico. Virology 213:223–230
Lee CW, Senne DA, Suarez DL (2004) Effect of vaccine use in the evolution of Mexican lineage H5N2 avian influenza virus. J Virol 78:8372–8381
Capua I, Marangon S, dalla Pozza M, Terregino C, Cattoli G (2003) Avian influenza in Italy 1997–2001. Avian Dis 47:839–843
Lee CW, Swayne DE, Linares JA, Senne DA, Suarez DL (2005) H5N2 avian influenza outbreak in Texas in 2004: the first highly pathogenic strain in the United States in 20 years? J Virol 79:11412–11421
Soda K, Cheng MC, Yoshida H, Endo M, Lee SH, Okamatsu M, Sakoda Y, Wang CH, Kida H (2011) A low pathogenic H5N2 influenza virus isolated in Taiwan acquired high pathogenicity by consecutive passages in chickens. J Vet Med Sci 73:767–772
Kim HR, Park CK, Oem JK, Bae YC, Choi JG, Lee OS, Lee YJ (2010) Characterization of H5N2 influenza viruses isolated in South Korea and their influence on the emergence of a novel H9N2 influenza virus. J Gen Virol 91:1978–1983
Lee JH, Pascua PN, Song MS, Baek YH, Kim CJ, Choi HW, Sung MH, Webby RJ, Webster RG, Poo H, Choi YK (2009) Isolation and genetic characterization of H5N2 influenza viruses from pigs in Korea. J Virol 83:4205–4215
Yeo DS, Ng SH, Liaw CW, Ng LM, Wee EJ, Lim EA, Seah SL, Wong WK, Lim CW, Sugrue RJ, Tan BH (2009) Molecular characterization of low pathogenic avian influenza viruses, isolated from food products imported into Singapore. Vet Microbiol 138:304
Okamatsu M, Saito T, Yamamoto Y, Mase M, Tsuduku S, Nakamura K, Tsukamoto K, Yamaguchi S (2007) Low pathogenicity H5N2 avian influenza outbreak in Japan during the 2005–2006. Vet Microbiol 124:35–46
Nishiguchi A, Kobayashi S, Ouchi Y, Yamamoto T, Hayama Y, Tsutsui T (2009) Spatial analysis of low pathogenic H5N2 avian influenza outbreaks in Japan in 2005. J Vet Med Sci 71:979–982
Abao LN, Jamsransuren D, Bui VN, Ngo LH, Trinh DQ, Yamaguchi E, Vijaykrishna D, Runstadler J, Ogawa H, Imai K (2013) Surveillance and characterization of avian influenza viruses from migratory water birds in eastern Hokkaido, the northern part of Japan, 2009–2010. Virus Genes 46:323–329
Runstadler JA, Happ GM, Slemons RD, Sheng ZM, Gundlach N, Petrula M, Senne D, Nolting J, Evers DL, Modrell A, Huson H, Hills S, Rothe T, Marr T, Taubenberger JK (2007) Using RRT-PCR analysis and virus isolation to determine the prevalence of avian influenza virus infections in ducks at Minto Flats State Game Refuge, Alaska, during August 2005. Arch Virol 152:1901–1910
Lee MS, Chang PC, Shien JH, Cheng MC, Shieh HK (2001) Identification and subtyping of avian influenza viruses by reverse transcription-PCR. J Virol Methods 97:13–22
Qiu BF, Liu WJ, Peng DX, Hu SL, Tang YH, Liu XF (2009) A reverse transcription-PCR for subtyping of the neuraminidase of avian influenza viruses. J Virol Methods 155:193–198
WHO (2005) WHO Manual on Animal Influenza Diagnosis and Surveillance. http://www.who.int/vaccine_research/diseases/influenza/WHO_manual_on_animaldiagnsis_and_surveilance_2002_5.pdf. Accessed 20 Feb 2009
Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR (2001) Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 146:2275–2289
Obenauer JC, Denson J, Mehta PK, Su X, Mukatira S, Finkelstein DB, Xu X, Wang J, Ma J, Fan Y, Rakestraw KM, Webster RG, Hoffmann E, Krauss S, Zheng J, Zhang Z, Naeve CW (2006) Large-scale sequence analysis of avian influenza isolates. Science 311:1576–1580
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lu G, Rowley T, Garten R, Donis RO (2007) FluGenome: a web tool for genotyping influenza A virus. Nucleic Acids Res 35:W275–W279
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Swofford LD (2002) PAUP*. phylogenetic analysis using parsimony (*and other methods). Version 4. http://paup.csit.fsu.edu/Cmd_ref_v2.pdf. Accessed 25 Mar 2012
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973
Duan L, Campitelli L, Fan XH, Leung YH, Vijaykrishna D, Zhang JX, Donatelli I, Delogu M, Li KS, Foni E, Chiapponi C, Wu WL, Kai H, Webster RG, Shortridge KF, Peiris JS, Smith GJ, Chen H, Guan Y (2007) Characterization of low-pathogenic H5 subtype influenza viruses from Eurasia: implications for the origin of highly pathogenic H5N1 viruses. J Virol 81:7529–7539
Ha Y, Stevens DJ, Skehel JJ, Wiley DC (2001) X-ray structures of H5 avian and H9 swine influenza virus hemagglutinins bound to avian and human receptor analogs. Proc Natl Acad Sci USA 98:11181–11186
Keawcharoen J, Amonsin A, Oraveerakul K, Wattanodorn S, Papravasit T, Karnda S, Lekakul K, Pattanarangsan R, Noppornpanth S, Fouchier RA, Osterhaus AD, Payungporn S, Theamboonlers A, Poovorawan Y (2005) Characterization of the hemagglutinin and neuraminidase genes of recent influenza virus isolates from different avian species in Thailand. Acta Virol 49:277–280
Li J, Zu Dohna H, Cardona CJ, Miller J, Carpenter TE (2011) Emergence and genetic variation of neuraminidase stalk deletions in avian influenza viruses. PLoS ONE 6:e14722
Li Z, Chen H, Jiao P, Deng G, Tian G, Li Y, Hoffmann E, Webster RG, Matsuoka Y, Yu K (2005) Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J Virol 79:12058–12064
Hay AJ, Zambon MC, Wolstenholme AJ, Skehel JJ, Smith MH (1986) Molecular basis of resistance of influenza A viruses to amantadine. J Antimicrob Chemother 18 Suppl B:19–29
Li KS, Guan Y, Wang J, Smith GJ, Xu KM, Duan L, Rahardjo AP, Puthavathana P, Buranathai C, Nguyen TD, Estoepangestie AT, Chaisingh A, Auewarakul P, Long HT, Hanh NT, Webby RJ, Poon LL, Chen H, Shortridge KF, Yuen KY, Webster RG, Peiris JS (2004) Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 430:209–213
Pinto LH, Holsinger LJ, Lamb RA (1992) Influenza virus M2 protein has ion channel activity. Cell 69:517–528
Campitelli L, Di Martino A, Spagnolo D, Smith GJ, Di Trani L, Facchini M, De Marco MA, Foni E, Chiapponi C, Martin AM, Chen H, Guan Y, Delogu M, Donatelli I (2008) Molecular analysis of avian H7 influenza viruses circulating in Eurasia in 1999–2005: detection of multiple reassortant virus genotypes. J Gen Virol 89:48–59
Macken CA, Webby RJ, Bruno WJ (2006) Genotype turnover by reassortment of replication complex genes from avian influenza A virus. J Gen Virol 87:2803–2815
Jiao P, Tian G, Li Y, Deng G, Jiang Y, Liu C, Liu W, Bu Z, Kawaoka Y, Chen H (2008) A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J Virol 82:1146–1154
Fan S, Macken CA, Li C, Ozawa M, Goto H, Iswahyudi NF, Nidom CA, Chen H, Neumann G, Kawaoka Y (2013) Synergistic effect of the PDZ and p85beta-binding domains of the NS1 protein on virulence of an avian H5N1 influenza A virus. J Virol 87:4861–4871
Conenello GM, Tisoncik JR, Rosenzweig E, Varga ZT, Palese P, Katze MG (2011) A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J Virol 85:652–662
Le QM, Kiso M, Someya K, Sakai YT, Nguyen TH, Nguyen KH, Pham ND, Ngyen HH, Yamada S, Muramoto Y, Horimoto T, Takada A, Goto H, Suzuki T, Suzuki Y, Kawaoka Y (2005) Avian flu: isolation of drug-resistant H5N1 virus. Nature 437:1108
Ito T, Goto H, Yamamoto E, Tanaka H, Takeuchi M, Kuwayama M, Kawaoka Y, Otsuki K (2001) Generation of a highly pathogenic avian influenza A virus from an avirulent field isolate by passaging in chickens. J Virol 75:4439–4443
Ogata T, Yamazaki Y, Okabe N, Nakamura Y, Tashiro M, Nagata N, Itamura S, Yasui Y, Nakashima K, Doi M, Izumi Y, Fujieda T, Yamato S, Kawada Y (2008) Human H5N2 avian influenza infection in Japan and the factors associated with high H5N2-neutralizing antibody titer. J Epidemiol 18:160–166
Wu HS, Yang JR, Liu MT, Yang CH, Cheng MC, Chang FY (2014) Influenza A(H5N2) virus antibodies in humans after contact with infected poultry, Taiwan, 2012. Emerg Infect Dis 20:857–860
Acknowledgments
We thank Sachiko Matsuda, Obihiro University of Agriculture and Veterinary Medicine, for technical support. This work was partially supported by a Grant-in-Aid for the Bilateral Joint Projects of the Japan Society for the Promotion of Science, Japan, the Heiwa Nakajima Foundation, Japan. This work was also partially supported by the US National Institute of Allergy and Infectious Diseases (NIAID contracts HHSN266200700009C and HHSN266200700007C).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was partially supported by a Grant-in-Aid for the Bilateral Joint Projects of the Japan Society for the Promotion of Science, Japan, the Heiwa Nakajima Foundation, Japan. This work was also partially supported by the US National Institute of Allergy and Infectious Diseases (NIAID contracts HHSN266200700009C and HHSN266200700007C).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
S. Sultan and V. N. Bui equally contributed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sultan, S., Bui, V.N., Hill, N.J. et al. Genetic characterization of H5N2 influenza viruses isolated from wild birds in Japan suggests multiple reassortment. Arch Virol 161, 3309–3322 (2016). https://doi.org/10.1007/s00705-016-3023-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3023-4