
Abstract
Two solid complexes, fac–[Cr(gly)3] and [Cr(gly)2(OH)]2, (where gly is glycinato ligand) were prepared and their acid-catalysed aquation products were identified. The structure of [Cr(gly)3] was solved by X-ray diffraction, revealing a cationic 3D sublattice with perchlorate anions inside its cavities. Acid-catalysed aquation of [Cr(gly)3] and [Cr(gly)2(OH)]2 leads to the same inert product, [Cr(gly)2(H2O)2]+, in a two-stages process. At the first stage, intermediate complexes, [Cr(gly)2(O–glyH)(H2O)]+ and [Cr(gly)2(H2O)–OH–Cr(gly)2(H2O)]+, are formed respectively. Kinetics of the first aquation stage of [Cr(gly)3] were studied in HClO4 solutions. The dependencies of the pseudo first-order rate constants on [H+] are as follows: k obs1H = k 0 + k 1 K p1[H+], where k 0 and k 1 are rate constants for the chelate-ring opening via spontaneous and acid-catalysed reaction paths, respectively, and K p1 is the protonation constant. The proposed mechanism assumes formation of the reactive intermediate as a result of proton addition to the coordinated carboxylate group of the didentate ligand. Some kinetic studies on the second reaction stage, the one-end bonded glycine liberation, were also done. The obtained results were analogous to those for stage I. In this case, the proposed reactive species are intermediates, protonated at the carboxylate group of the monodentate glycine. Base hydrolysis of two complexes, [Cr(gly)2(O–gly)(OH)]− and [Cr(gly)2(OH)2]−, was studied in 0.2–1.0 M NaOH. The pseudo first-order rate constants, k obsOH, were [OH−] independent in the case of [Cr(gly)2(O–gly)(OH)]−, whereas those for [Cr(gly)2(OH)2]− linearly depended on [OH−]. The reaction mechanisms were proposed, where the OH− -catalysed reaction path was rationalized in terms of formation of the reactive conjugate base, [Cr(gly)2(OH)(O)]2−, as a result of OH− ligand deprotonation. Activation parameters were determined and discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since 1955, chromium(III) has been considered an essential element for mammals [1–5]. However, to date, the exact composition and structure of its biologically active forms, GTF (glucose tolerance factor) and chromodulin are not known. In his recent review on biologic role of chromium, Vincent [5] concludes that numerous studies resulted with “controversial and conflicting results”. Nevertheless, chromium nutritional supplements are still only second to calcium among mineral supplements. The most popular artificial source of chromium is tris-picolinatochromium(III), even so it is hardly soluble and exhibits a very low bioavailability (ca. 1–3%). For this reason, our group have undertaken studies on preparation and characterization in aqueous solution of new chromium(III) complexes with biologically active ligands, which could be more efficient biochromium sources. Previously, series of complexes with pyridinemonocarboxylato and pyridinedicarboxylato ligands were obtained and kinetic studies on their acid- and base-catalysed aquation were performed [e.g. 6–10]. Additional studies on their effect toward 3T3 fibroblasts revealed that they are not toxic and may be good candidates as biochromium sources [6, 10]. On the other hand, it is likely that a higher bioavailability characterizes chromium(III) complexes with amino acids, as naturally existing biomolecules. However, though many chromium–amino acids complexes are known [11–16], to date no studies on their kinetics in aqueous solutions or their cytotoxicity were performed. In this work, studies on chromium-glycinato complexes are described. Two compounds, [Cr(gly)3] and [Cr(gly)2(OH)]2, were obtained earlier [11–13] and their structures were determined by X-ray diffraction. The tris-complex was found to be the fac-isomer [12]. In the dimer [15], the chromium atoms, connected by two hydroxo bridges, have two gly coordinated in cis positions such that introduction of the third gly yields the fac-isomer. The complexes were practically insoluble in aqueous solutions at pH 7, and therefore only spectroscopic characteristics for Nujol mulls were done [12, 16]. Some preliminary studies on their acid-catalysed aquation [16] demonstrated that they form the same, exceptionally inert (in acidic media) product of [Cr(gly)2(H2O)2]+ formulae. Based on the yeast fermentation bioassay results [16], it was suggested that this complex exhibits biological activity. However, no further studies were undertaken.
In this paper, the structure of fac–[Cr(gly)3], obtained by a new method, is presented. Products of acid-catalysed aquation of [Cr(gly)3] and [Cr(gly)2(OH)2] are identified. Results of kinetic studies on the acid-catalysed aquation of [Cr(gly)3] are discussed, and the mechanism of the first gly substitution is proposed. For two of its aqua-derivatives, [Cr(gly)2(O–glyH)(H2O)]+ and [Cr(gly)2(H2O)2]+, studies on base hydrolysis, at pH > 13, are described. The impact of pH on the rate of gly liberation is discussed.
Experimental
Glycine 99% (Aldrich), 70% HClO4 (Aldrich), NaClO4·H2O (Merck), NaOH (POCh, Gliwice), and other chemicals were used without further purification. Sephadex DEAE A-25 (ClO4 −) and Sephadex C-25 (H+) were used for chromatographic separations.
Chromium was determined spectrophotometrically as CrO 2−4 at 372 nm (ε372 = 4,830 M−1 cm−1) after decomposition of the complexes with 0.1 M NaOH followed by oxidation of the chromium(III) species by H2O2. Glycine was determined spectrophotometrically at 570 nm (ε570 = 7,500 M−1cm−1) applying the ninhydrin method described by Bailey [17]. The molar absorption coefficient of glycine was determined from the dependence of absorbance on glycine concentration as follows: a mixture containing 0.5 cm3 of 0.5–2.0 × 10−3 M glycine aqueous solution, 1.5 cm3 of 0.5 M acetate buffer and 2.0 cm3 of ninhydrin solution (2 g of ninhydrin in 50 cm3 of 50% ethanol) was placed in a 10 cm3 flask and boiled for 30 min. Then, after cooling, the flasks were filled with 50% ethanol and absorbance at 570 nm was registered. The collected data fulfilled Beer’s law very well. The same procedure was applied in order to determine glycine in the obtained complexes. Electronic spectra were recorded using an HP 8453 Spectrophotometer.
Single-crystal structure determination
The X-ray diffraction data of fac–[Cr(gly)3]·0.5NaClO4 were collected at room temperature for the single crystal (0.21 × 0.19 × 0.12 mm) using Oxford Diffraction KM4 CCD diffractometers, Mo Kα radiation λ = 0.71073 Å. The reflections were measured with ω-2θ method, and the numerical absorption correction was applied for the crystal studied (RED171 package of programs, Oxford Diffraction, 2,000, [18]). The structure of [Cr(gly)3]·0.5NaClO4 was solved by direct methods and refined with full-matrix least-squares procedure on F 2 (SHELX-97 [19]). Heavy atoms were refined with anisotropic thermal displacement parameters. Positions of hydrogen atoms were found from difference Fourier synthesis, and they were refined with isotropic thermal displacement parameters fixed to a value of 20% higher than those of the corresponding atoms to which they are bound. All figures were prepared in ORTEP-3 [20]. The results of the data collections and refinement have been summarized in Table 1.
CCDC 788768 contains the supplementary crystallographic data for fac–[Cr(gly)3]·0.5NaClO4. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieveing.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U; Fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
Preparation of [Cr(gly)3] and [Cr(gly)2(OH)] 02
A mixture of Cr(ClO4)3 (4 mmol) in 0.05 M HClO4 (30 cm3) and 3 g of glycine (40 mmol) was heated at 70 °C for 30 min. Meanwhile, 2 M NaOH was gradually added up to pH 6–7. As the reaction proceeded, the color changed from dark blue to red and some red precipitate appeared. After cooling, the deposit divided into two layers: the upper, pink-violet and the lower, red. Then the red solution was removed, and the upper layer was taken off by flotation with ethanol. The pale red residue was dried in the air, and its composition was established by comparison of the visible spectrum of its Nujol mull (Table 2) with that described by Bryan [12] and by determination of the Cr:gly molar ratio. The obtained results indicated that the analyzed precipitate was composed of [Cr(gly)3] and free glycine (Cr:gly = 1:6). Therefore, in order to obtain a pure product, some modification of the synthesis was applied; the reaction mixture was maintained at pH 5–6 and the solution was kept for 24 h, which resulted in formation of small red crystals of the same visible spectrum like the former product and the proper Cr:gly molar ratio (1:3). As in any case the obtained product exhibited the same reactivity in HClO4 solutions, the kinetic measurements were performed for the one obtained immediately during the synthesis.
The dimer, [Cr(gly)2(OH)]2, was prepared as described in [11]. The complex precipitated as a pink-violet powder, the composition of which was confirmed by the determined 1:2 Cr:gly molar ratio and thermal gravimetric analysis (TGA) using an SDT2960 Simultaneous DSC-TGA Instrument; the complex is stable up to 400 °C and then decomposes with the weight loss of 63.2%. The weight of remaining residue was in accord with Cr2O3 (35.0%), assuming the [Cr(gly)2(OH)]2 formulae (MW = 434).
Aquation products of [Cr(gly)3] and [Cr(gly)2(OH)]2
A suspension of 1 mmol of the appropriate complex in 20 cm3 of 0.1 M HClO4 (50 cm3) was heated at 40 °C for ca. 5–10 min. Then, the process was quenched by addition of equivalent amount of 3 M KOH (up to pH 3–4) and, after freezing for 30 min., the mixture of deposited KClO4 and unreacted substrate was filtered off and the filtrate was passed through a column of Sephadex Sp C-25 (H+). Two distinct bands were observed on the resin: the first, red-pink was eluted with 0.1 M NaClO4 + 0.05 M HClO4 and the second, pink-reddish was eluted with 0.1 M NaClO4 + 0.1 M HClO4. The chromium content depended on reaction conditions; the longer aquation time and higher [H+], the smaller the first band and larger the second one. Usually content of chromium(III) in the first band was ca. 70% (from [Cr(gly)3]) and 30% (from [Cr(gly)2(OH)]2). Identification of the eluted complexes was based on their spectroscopic characteristics (Table 2) and the determined Cr:gly molar ratio. The obtained results indicate that both the complexes undergo two stages aquation to give the same product, [Cr(gly)2(H2O)2]+ (fraction 2) via different intermediates (fraction 1): [Cr(gly)2(O–glyH)(H2O)]+ (from [Cr(gly)3]) and [Cr(gly)2(H2O)–OH–Cr(gly)2(H2O)]+ (from [Cr(gly)2(OH)]2).
Products of base hydrolysis of [Cr(gly)2(O–gly)(OH)]− and [Cr(gly)2(OH)2]−
Products of base hydrolysis of [Cr(gly)2(O–gly)(OH)]− were separated from the reaction mixture, obtained after ca. t 1/2 (600 s) at 30 °C. The reaction was performed in 0.4 M KOH (10 cm3) at the complex concentration ca. 5 mM. Then the reaction was quenched by HClO4 addition to pH ca. 3. After cooling, the KClO4 deposit was filtered off and the resulting solution was separated chromatographically using the cationic exchanger. Only two bands were observed on the column: the first of lower affinity to the resin was eluted with 0.1 M NaClO4 + 0.05 M HClO4 and contained unreacted substrate (ca. 50%), and the second, greenish, of much higher affinity to the resin which contained chromates(III), e.g. polynuclear μ-hydroxo-aquachromium(III) complexes [21].
A similar procedure was applied to determine the products of base hydrolysis for [Cr(gly)2(OH)2]−. The reaction was conducted for 200 s at 15 °C. Two chromium(III) complexes were separated: the unreacted substrate (ca. 40%) and polynuclear μ-hydroxo-aquachromium(III) complexes.
Electric charge of the aqua-complexes studied depends on pH of the solution; at pH < 5, they are monocations, whereas at pH > 13 they exist as monoanions due to deprotonation of the coordinated H2O ligands and the monodentate gly.
Kinetic measurements
Kinetics of acid-catalysed aquation of [Cr(gly)3] and [Cr(gly)2(O–glyH)(H2O)]+ and base hydrolysis of [Cr(gly)2(O–gly)(OH)]− and [Cr(gly)2(OH)2]− were studied spectrophotometrically (Spectrophotometer HP 8453 equipped with HP89090 Peltier temperature controller or with an external Julabo F25 thermostat) within the visible spectrum range (Figs. 1–4), under pseudo first-order conditions. The concentration of the chromium was 4–5 × 10−3 M, the HClO4 concentration was varied in the 0.1–0.9 M range, the NaOH concentration was varied in the 0.2–0.9 M range, and the ionic strength was 1.0 M (NaClO4). The registered scans were analyzed with SPECFIT software for a simple first order A → B reaction model.

Spectroscopic changes during the first aquation stage of [Cr(gly)3] in 0.8 M HClO4; [Cr] = 5 × 10−3 M, I = 1.0 M, T = 293 K, scans every 40 s

Spectroscopic changes during the second aquation stage of [Cr(gly)3] in 0.8 M HClO4; [Cr] = 4 × 10−3 M, I = 1.0 M, T = 313 K, scans every 45 s

Spectroscopic changes during base hydrolysis of [Cr(gly)2(O–gly)(OH)]− in 0.9 M NaOH; [Cr] = 4.5 × 10−3, I = 1 M, T = 303 K, scans every 200 s

Spectroscopic changes during base hydrolysis of [Cr(gly)2(OH)2]− in 0.9 M NaOH; [Cr] = 5 × 10−3 M, I = 1 M, T = 288 K, scans every 45 s
Measurements were performed in a 1 cm tandem-cell of equal volumes (0.85 cm3) of the complex and appropriate medium solutions. The process was initiated by mixing the thermostated contents of the cell. The tris-complex solution was obtained immediately before measurements as a result of [Cr(gly)3] dissolution in cold water, followed by filtration of the remaining precipitate, and the [Cr(gly)2(O–glyH)(H2O)]+ species were freshly separated using the cationic exchanger.
Each kinetic run was repeated twice. The relative standard errors of the calculated pseudo-first-order rate constants were usually ca. 1–2%.
Results and discussion
Tris-gycinatochromium(III) was obtained via anation of [Cr(H2O)6](ClO4)3 by glycine. The crystal structure was resolved by X-ray diffraction method. The fac–[Cr(gly)3]·0.5NaClO4 complex crystallized in the cubic Pa-3 space group. Chromium, sodium, chlorine, and O8 oxygen atoms are positioned in special positions on threefold axes. Thus, the asymmetric unit contains only one glycine ligand bound bidentately (via nitrogen and oxygen atoms) to chromium atom and monodentately (via oxygen atom) to sodium cation, and a 3D cationic network with perchlorate anions inside its cavities is formed. The coordination spheres of chromium and sodium atoms adopt slightly distorted octahedron geometry. The sodium is surrounded by six O4 oxygen atoms coming from six different complex molecules, whereas the chromium is surrounded by three oxygen and three nitrogen atoms in fac- conformation (Fig. 5) with Cr–O distances significantly shorter (by 0.1 Å) than Cr–N bond lengths (Table 3). The geometry of the chromium coordination sphere is very similar in both related complexes, [Cr(gly)3]·0.5NaClO4 and [Cr(gly)3]·H2O [12]. Cr–N distances are identical within experimental error, and Cr–O bond lengths differ only by 0.01 Å being at the limit of significance. C–O bond lengths in the carboxylic group depend on the kind of interaction formed by oxygen atoms. Coordination to chromium in both cases leads to an elongation by approximately 0.06 Å of the C–O bond, irrespective of whether the second oxygen atom is involved in extensive hydrogen network formation or even in coordination to the Na ion.

Molecule of fac–[Cr(gly)3]·0.5NaClO4 with the numbering scheme; the thermal ellipsoids are at 20% probability
The structure is completed by chlorate anions characterized by two Cl–O distances, the longer corresponding to O8 atom positioned along the threefold axis and shorter for O10 oxygen found in general position. The chlorine is positioned at an inversion center placed on threefold axis and surrounded by oxygen atoms found in 8 positions adopting distorted cube geometry. Thus, the perchlorate anion reveals rotational disorder with two set of positions related by threefold inversion axis.
Solubility of the complex obtained by us is much higher than that of the compound prepared from Cr(NO3)3 [12]. This property facilitates kinetic studies on [Cr(gly)3] aquation, which require spectrophotometric measurements within the visible region, at [Cr] > 10−3 M.
The other solid product formed in reaction between [Cr(H2O)6](ClO4)3 and glycine was a dimer of [Cr(gly)2(OH)]2 formula, the solubility of which was too low to obtain a solution suitable for kinetic measurements.
It is already known that acid-catalysed aquation of fac–[Cr(gly)3] and [Cr(gly)2(OH)]2 yields the same, extremely inert (in acidic media) product, [Cr(gly)2(H2O)2]+ [16]. However, no kinetic studies on these reactions were done. Our studies revealed that in each case, intermediate complexes (unknown before) exist in the reaction mixture. They were separated chromatographically and identified as [Cr(gly)2(O–glyH)(H2O)]+ (for [Cr(gly)3]) and [Cr(gly)2(H2O)–(OH)–Cr(gly)2(H2O)]+ (for [Cr(gly)2(OH)]2), based on the determined Cr:gly molar ratio, their affinity to the cationic exchanger and spectroscopic characteristics (Table 2).
Spectra of the starting complexes were recorded in Nujol mulls and in the case of [Cr(gly)3] also in aqueous solution. As seen (Table 2), the position of the lower energy d–d transition band for the [Cr(gly)3] depends on the medium and is shifted toward longer wavelengths after complex dissolution. This red shift can be attributed to destruction of the crystal lattice present in a solid form of the complex. Moreover, no effect of pH change (from 2 to 13) on the [Cr(gly)3] spectrum implicates lack of H2O in the chromium(III) coordination sphere and supports the same composition of the inner coordination sphere of the dissolved and solid complex. The tris-glycinatochromuim(III) is not stable in aqueous solution and undergoes a slow gly substitution accompanied by spectroscopic changes (509 → 526 and 526 → 535 nm), which were attributed to the successive Cr–N and Cr–O bonds breaking and formation of the [Cr(gly)2(O–glyH)(H2O)]+ intermediate and the final product, [Cr(gly)2(H2O)2]+, respectively. The proposed sequence of the metal-donor atom bonds breaking is in accord with the position of N- and O-donor ligands in the spectrochemical series and with the observed red shift of the lower energy d–d transition band (Figs. 1, 2). The complex with the one-end bonded gly, via carboxylate oxygen atom, was identified by Kallen as an intermediate during acid-catalysed aquation of [Cr(ox)2(gly)]2− [22].
Insolubility of the dimer affected the yield of its first-stage aquation product, because while sufficient amount of the dimer was transformed into the intermediate, a larger part of the latter underwent the next reaction stage to give [Cr(gly)2(H2O)2]+. For this reason (too low concentration), kinetics of neither the dimer nor its intermediate could be studied spectrophotometrically in the visible region.
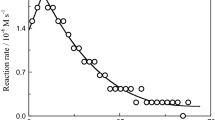
Kinetics of the first stage of acid-catalysed aquation of [Cr(gly)3] were studied within 0.1–0.9 M HClO4 range. The spectral changes, attributed to the Cr–N bond breaking and formation of the intermediate, are shown in Fig. 1. One isosbestic point at 540 nm is observed, which is consistent with lack of a substantial concentration of the final product in the system studied. The calculated pseudo first-order rate constants, k obs1,H, are collected in Table 4 and their dependences on [H+] are shown in Figs. 6 and 7.

Dependence of k obs1,H on [H+] for the first aquation stage of [Cr(gly)3] in HClO4

Dependence of k obs2,H on [H+] for the second aquation stage of [Cr(gly)3] in HClO4
As seen, these data fit a linear equation very well:
The rate expression (Eq. 1) and the identified reaction product (the intermediate complex) are consistent with the following mechanism (Scheme 1, stage I).

The reaction sequence during the first glycine dissociation from [Cr(gly)3] in acidic medium
The mechanism assumes formation of the reactive form of the substrate (SH) as a result of protonation of the didentate glycine at the carboxylate group. Both forms of the substrate (S and SH) are in equilibrium (characterized by K p1) shifted to the left, which is consistent with the found linear dependence of k obs1,H on [H+]. The rate law derived from the reaction sequence shown in Scheme 1 is of the form:
which simplifies to a linear expression if K p1[H+] ≪ 1:
Comparison of the experimental (1) and derived (3) rate laws gives the meaning of the parameters a 1 and b 1:
where k 0 and k 1 are the first-order rate constants for the spontaneous aquation of the conjugate base (S) and acid (SH), respectively. Linear dependence of k obs1,H on [H+] makes impossible separation of k 1 and K p1 from the product k 1 K p1, and therefore only apparent activation parameters could be calculated (Table 5). Values of the intercepts (k 0) were determined with large errors and cannot be used for calculation of activation parameters.
Comparing the rate constants for spontaneous and acid-catalysed reaction paths demonstrates that catalytic process predominates at [H+] > 0.1 M and at [H+] = 1 M over 95% of the reaction proceeds via the [H+] catalysed path.
For the second aquation stage, spectroscopic changes were much smaller than those for stage I and for this reason only some kinetic runs, at one temperature, were performed (Table 4, Fig. 8). The obtained kinetic parameters (Table 4) should be treated as rough estimations.

Electronic spectra of [Cr(gly)2(O–glyH)(H2O)]+ (I) and [Cr(gly)2(O–gly)(OH)]− (II); [Cr] = 7 × 10−3 M
The dependence of k obs2,H on [H+] is analogous to that for the first stage:
The proposed mechanism (Scheme 1, stage II) assumes protonation of the one-end bonded glycine (with protonated terminal amine, NH3 +) at the carboxylate group. So, in this case, the meaning of the determined a 2 and b 2 parameters is as follows:
where k ′0 and k 2 are first-order spontaneous and acid-catalysed rate constants, respectively for the Cr–O bond breaking of the one-end bonded glycine, and K p2 is the protonation constant of the coordinated carboxylate group of the monodentate bonded gly. Comparison of the rate constants for stages I and II (Table 5) demonstrates a similar contribution of the spontaneous and the acid-catalysed reaction paths.
In alkaline media, no protolytic reaction is observed spectrophotometrically for [Cr(gly)3] because of low acidity constant for the coordinated NH2 group. Moreover, the complex is exceptionally inert in NaOH solutions, which additionally confirms practical absence of its conjugate base. On the other hand, distinct spectroscopic changes are found upon NaOH addition to acidic solutions of [Cr(gly)2(O–glyH)(H2O)]+ and [Cr(gly)2(H2O)2]+ (Figs. 8, 9). The observed red shift of the lower energy d–d transition band is consistent with deprotonation of the coordinated H2O ligands and formation of appropriate monohydroxo- and dihydroxo-complexes. Both the hydroxo-complexes are labile and undergo base hydrolysis to give chromates(III) and free glycinate anions (Figs. 3, 4). Lack of intermediate products indicates that each consecutive reaction step is much faster than the preceding one.

Electronic spectra of [Cr(gly)2(H2O)2]+ (I) and [Cr(gly)2(OH)2]− (II); [Cr] = 12 × 10−3 M
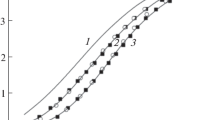
Kinetics of base hydrolysis were studied spectrophotometrically within 0.2–0.9 M NaOH range, because at lower [OH−], precipitation of chromium hydroxide takes place. Scans registered within the 480–580 nm range for [Cr(gly)2(O–gly)(OH)]− (Fig. 3) and 380–700 nm range for [Cr(gly)2(OH)2]− (Fig. 4) were analyzed with help of SPECFIT software. Calculations confirmed a simple first-order A → B reaction model for both reactants. The obtained pseudo first-order rate constants (k obs1,OH and k obs2,OH) are collected in Table 6 and their dependences on [OH−] are shown in Figs. 10 and 11.

Dependence of k obs1,OH on [OH−] for base hydrolysis of [Cr(gly)2(O–gly)(OH)]−

Dependence of k obs2,OH on [OH−] for base hydrolysis of [Cr(gly)2(OH)2]−
As seen, k obs2,OH ≫ k obs1,OH, which demonstrates substantially higher reactivity of the dihydroxo-complex. Looking at Figs. 10 and 11, a remarkable difference in k obs,OH versus [OH−] functions is observed. The rate constants for [Cr(gly)2(O–gly)(OH)]− are [OH−] independent (Eq. 7), whereas those for [Cr(gly)2(OH)2]− linearly depend on [OH−] (Eq. 8).
The OH− catalysed reaction path for [Cr(gly)2(OH)2]− species, represented by the b 2,OH parameter (Eq. 8), may be rationalized in terms of deprotonation of one hydroxo ligand and formation of a much more reactive conjugate base, the oxo-form. Lack of an analogous reaction path for the [Cr(gly)2(O–gly)(OH)]− complex can result from formation of the intramolecular hydrogen bond [Scheme 2 (a)], which protects the OH− ligand against deprotonation. This type of intermolecular hydrogen bond cannot be formed by both OH− ligands in [Cr(gly)2(OH)2]− [Scheme 2 (b)].

Structure of [Cr(gly)2(O–gly)(OH)]− (a) and [Cr(gly)2(OH)2]− (b) complexes
Based on the obtained results, the following reaction sequence for the dissociation of the first (Scheme 3, stage I) and the second glycine ligand (Scheme 2, stage II) can be proposed:

Mechanism of glycine dissociation from [Cr(gly)2(O–gly)(OH)]− (stage I) and from [Cr(gly)2(OH)2]− (stage II) in NaOH solution
Liberation of glycine from [Cr(gly)2(O–gly)(OH)]− (S) is a result of the Cr–O–gly bond breaking, which is weakened by the coordinated OH− ligand. This process generates the aqua-intermediate (P1′), which, under the conditions applied, is rapidly deprotonated to give the dihydroxo-product (P1 –OH). The overall process is described by one kinetic parameter, k 1,OH, representing the rate-limiting step.
In stage II, two reaction paths are observed: spontaneous aquation of the dihydroxo-complex (k 2,OH) and an OH−-catalysed process proceeding via the conjugate base, (P1 –O). Three parameters should be used to describe the glycine substitution: the deprotonation constant (K OH) and the first-order rate constants (k 2,OH and k 2,O). Since under the conditions applied, the equilibrium leading to the oxo-complex is shifted to the left, the rate law of stage II is of the form:
Assumption that at stage II the Cr–N bond breaking is the rate-determining step was additionally supported by stopped flow measurements for the first 100 s of the reaction. As the character of the spectroscopic changes did not indicate formation of a monodentate bonded intermediate, we conclude that the collected data are consistent with the chelate-ring opening via the Cr–N bond breaking (Scheme 3, stage II) being the rate-limiting step. The monoglycinato-complex is very labile in alkaline media and is rapidly transformed into “so-called” chromates(III).
Comparing appropriate experimental and derived rate expressions (7 with 9 and 8 with 10) gives the meaning of parameters a OH and b OH:
Thus, a OH are first-order rate constants for the spontaneous aquation and b OH is a pseudo second-order rate constant, being the product of the rate constant of the conjugate base (the oxo-complex) and the deprotonation constant, k 2,O K OH. These two quantities cannot be separated from linear dependence of the k obs2,OH on [OH−]. Determination of the kinetic parameters at different temperatures allows evaluation of the activation parameters for spontaneous and OH− -catalysed reaction paths (Table 7) .
Activation parameters for the spontaneous process were determined with sufficient accuracy only for [Cr(gly)2(O–gly)(OH)]−, whereas for [Cr(gly)2(OH)2]− the OH− -dependent reaction path substantially predominates and errors for the intercepts (Fig. 11) are large, leading to large errors in ∆H ≠ and ∆S ≠ (Table 7). The activation process for the one-end bonded glycine liberation from [Cr(gly)2(O–gly)(OH)]− is characterized by substantially negative entropy of activation attributed to higher hydration of dissociated small glycinate anion than the much larger starting complex (the intermediate formed after dissociation of gly is electrically neutral). The OH−-catalysed reaction path is characterized by the pseudo ∆H ≠ and pseudo ∆S ≠ (Table 7) because they were calculated from the product k 2,O K OH (Eq. 10).
Comparison of the first-order rate constants for the spontaneous processes (k 1,OH and k 2,OH at 298 K, Table 7) demonstrates ca. 4 times higher reactivity of [Cr(gly)2(OH)2]− species than [Cr(gly)2(O–gly)(OH)]−. Reactivity of the former complex is additionally increased by the catalytic reaction path. At 1 M NaOH, the base hydrolysis of [Cr(gly)2(OH)2]− is ca. 30 times faster than that of [Cr(gly)2(O–gly)(OH)]−, mainly due to formation of the oxo-complex.
Comparing some kinetic results for acidic and alkaline media, the distinct labilizing influence of coordinated OH− ligands on glycine liberation is clearly seen, especially in the case of the diglycinato-complex, which in acidic solution, in the diaqua-form, does not undergo glycine substitution even during elongated heating in 1 M HClO4.
In the case of tris-complex with one monodentate O-bonded glycine the rate constants for spontaneous processes of the aqua- and the hydroxo-forms can be compared at 313 K, [Table 5 (stage II) and 7 (complex A)]. The ratio of k 1OH/k 0′ is ca. 140, which is consistent with typical values for hydroxo and aqua chromium(III) complexes [23]. This reactivity difference is obviously a result of labilizing effect of hydroxo ligands. Generally, the more OH− ligands in the coordination sphere, the higher the reactivity of the complex.
Conclusions
Much higher solubility of [Cr(gly)3] than the dimer makes it a better candidate for chromium bioavailability studies. These kinetic studies indicate that, under physiological conditions, tris-glycinatochromium(III) is converted into the intermediate with one monodentate gly, which is able to form Cr(III)-peptide bonds or interact with peptides via the terminal amine group.
References
Mertz W (1993) J Nutr 123:626
Anderson RA (2000) Diabetes Metab 26:22
Slesinski RS, Clarke JJ, San RHC, Gudi R (2005) Mutation Res 585:86
Vincent JB, Bennet R (2007) Potential and purported roles for chromium in insulin signaling: The search for holy grail. In: Vincent JB (ed) Nutritional biochemistry of Chromium(III). Elsevier, Amsterdam, pp 139–160
Vincent JB (2010) Dalton Trans 39:3787
Kita E, Marai H, Jasiński M, Drewa T (2008) Transit Met Chem 33:585
Kita E, Marai H, Iglewski Ł (2009) Transit Met Chem 34:75
Kita E, Marai H (2009) Transit Met Chem 34:585
Kita E, Marai H, Jach K, Orłowska A (2009) Transit Met Chem 34:217
Kita E, Marai H, Lisiak M, Jasiński M, Drewa T (2010) Transit Met Chem 35:177
Ernshaw A, Lewis J (1961) J Chem Soc 1961:396
Bryan RF, Greene PT, Stokely PF, Wilson EW Jr (1971) Inorg Chem 10:1468
Gillard RD, Laurie SH, Price DC, Phipps DA, Weick CF (1974) J Chem Soc Dalton Trans 1974:1385
Oki H, Otsuka K (1976) Bull Chem Soc Jpn 49:1841
Veal TJ, Hatfield WE, Jeter DY, Hempel JC, Hodgson DJ (1973) Inorg Chem 12:342
Cooper JA, Blacwell LF, Buckley PD (1984) Inorg Chim Acta 92:23
Bailey JL (1962) Techniques in protein chemistry. Elsevier, Amsterdam, p 73
CrysAlis RED, CrysAlis CCD (2000) Oxford Diffraction Ltd., Abingdon, Oxfordshire, England (1)
Sheldrick GM (2008) Acta Crystallogr Sect A 64:112
Farrugia LJ (1997) J Appl Crystallogr 30:565
Rao L, Zhang Z, Friese JI, Ritherdon B, Clark SB, Hess NJ, Rai D (2002) J Chem Soc Dalton Trans 267
Kallen TW, Hamm RE (1977) Inorg Chem 16:1147
Richens DT (2005) Chem Rev 105:1961
Acknowledgments
Hasan Marai wishes to thank the Libyan Government for financial support of his Ph.D. studies in Poland.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kita, E., Marai, H., Muzioł, T. et al. Kinetic studies on chromium-glycinato complexes in acidic and alkaline media. Transition Met Chem 36, 35–44 (2011). https://doi.org/10.1007/s11243-010-9431-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-010-9431-5