
Abstract
The research was aimed at exploring the biological activities of novel series of β-lactam derivatives against MCF-7 breast cancer cell lines via computer modeling such as quantitative structure-activity relationship (QSAR), designing new compounds and analyzing the drug likeliness of designed compounds. The QSAR model was highly robust as it also conforms to the least minimum requirement for QSAR model from the statistical assessments with a correlation coefficient squared (R2) of 0.8706, correlation coefficient adjusted squared (R2adj) of 0.8411, and cross-validation coefficient (Q2) of 0.7844. The external validation of R2pred was calculated as 0.6083 for model 4. The model parameters (MATS5i and MATS1s) were used in designing new derivative compounds with higher potency against estrogen-positive breast cancer. The pharmacokinetics test on the restructured compounds revealed that all the compounds passed the drug likeness test and they could further proceed to clinical trials. These reveal a breakthrough in medicine, in the research for breast cancer drug with higher effectiveness against the MCF-7 cell line.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer, a dreadful malady, is also referred to as malignant tumors, and it is a heterogeneous tumor that affects almost all parts of the body. Mammary tumor is among the common diseases that brings about morbidity and mortality among the female being [14]. The cases that were encountered with most anti-cancer drugs during the past three decades include drug ineffectiveness, no drug selectivity, growing side effects, and the drug becoming resistant to the tumor [14]. Regardless of the up-to-date diagnostic and therapeutic advancement, mammary tumor is still the common cause of mortality and the primary utmost cancer among women worldwide [2]. Therefore, more effective and safe therapeutic agents are urgently needed with many pathways to increase the positive outcome of the patients clinically [14].
Computational approaches are regularly employed in almost all modern drug discovery effort, and robust success has been attained for computer-aided lead generation and optimization. These techniques are mostly accurate, faster, and cost-efficient. CADD represents more recent applications of software tools in the designing of lead candidate [13]. CADD technique is basically divided into two sections which are the structure-based (SB) and ligand-based (LB) drug discovery. The CADD technique employed depends on the crystal structure (receptor) available. One of the computer-aided tools for drug discovery and design includes quantitative structure-activity relationship (QSAR).
Quantitative structure-activity relationship (QSAR) is a current technique used in optimizing template molecules and re-designing new drug compounds. QSAR calculates the activities, toxicities, and carcinogenicities of molecules obtained from the definition of the molecular parameters from a derived mathematical equation [5]. It is also a known arithmetic relationship connecting molecular compounds and biological activities for a library of molecules quantitatively [3]. An arithmetic equation is generated from the structural info of a well-derived calibration compound and corresponding biological activities, while the model is validated using some validation compounds for which the biological activities are accessible [12].
Malebari et al. [10] reported 43 novel β-lactam derivative compounds as potent inhibitors against estrogen-positive MCF-7 cell line. The purpose of this research is to utilize ligand-based drug design to design new β-lactam derivative compounds based on an established QSAR mathematical model as inhibitors against estrogen-positive breast cancer (MCF-7 cell line) and, furthermore, to test for the pharmacokinetic properties of the designed compounds.
Methodology
QSAR process
Data collection
Forty-four (44) new derivative compounds of β-lactams with thier individual inhibitory concentration (IC50) against breast cancer (MCF-7 cell line) were attained from 14 publications.
Bio-activities
The bio-activities of β-lactam derivative compounds were measured in inhibitory concentration (IC50). The scale of logarithm (pIC50 = − log10 (IC50 × 10−6)) was applied to equalize the IC50 values. The IC50 and pIC50 values of the derivatives are seen in Table 1. It is measured in the concentration of micromolar (μM).
Geometry optimization
This technique was used to get a desirable structure that would be the closest to the initial structural condition [11]. 2D sketching of Chemdraw V (12.0.2) was employed in drawing β-lactam derivatives, and then, it was uploaded on Spartan 14 V (1.1.4) for geometrical optimization. [1]. The template molecule is seen in Fig. 1.

Template molecule of β-lactam derivatives
Model parameters
The model parameters were obtained for the whole derived compounds of β-lactams using Pharmaceutical Data Exploration Laboratory Software V (2.20) [16].
Pretreatment and division of data set
The values from the PADEL V (2.20) were prepped using graphical user interface (GUI) 1.2 (Data Pretreatment software), to remove relentless and undesired descriptor values [1]. Kennard-Stone algorithm [8] was employed in splitting the derivatives into calibration and validation fragments in other to construct the equation (model).
Model building and validation
A mathematical equation was built using the train set as predictor variable, while the pIC50 was used as the predicted variable by using genetic function approximation (GFA) technique of Material Studio Software V 8. The obtained equations were evaluated using Friedman formula [4].
where SEE is the standard estimated error; it is given as
C is the summation of the model definitions, p is the total number of model descriptors, M is the sum of prediction set, and d is a user-defined smoothing parameter [9]. The model is verified externally using the correlation coefficient (R2). The nearer R2 value is to 0.1, the better the regression fitness. R2 is calculated as:
where Yexp and Ypred are means of the actual and calculated activities of the calibration sets [1]. Yminraining indicates the average pIC50 of the train set molecules [7].
R2-based metrics could be a bit misleading, and mean absolute error (MAE)–based metrics are known to be straightforward determinant for predicting errors. MAE values are kept constant, and the effectives of the calculated activity is known if 5% of the residual values are absent [12]. It is expressed as:
Modeling assessment
The mathematical equation (model) produced is made to undertake statistical test like cross-validated test, R2 Fisher’s test, and R2 predicted.
Applicability domain
A model validation should be within the training domain and it is essential for the compounds to be assessed as fitting within the domain to ascertain the model. An applicability domain is evaluated by the leverage value for every molecule. The leverage (L) defines the applicability domain of the generated equation [15]. It is formulated as:
where XT is the matrix transpose of X used in constructing the equation, Xi is the matrix of prediction sets of I, and X is the n x k matrix of train set descriptors. (E*) is the warning leverage; it is a predictive tool that tests for outliers. It is written as:
h stands for the total structural parameters and m is the total compounds of train sets. William’s plot is a plot of standardized values vs. leverage values of both the training (calibration) and test (validation) sets. Molecules that stay within the calculated H* on the graph are the calculated compounds.
Drug likeness analysis
The ADME properties of a molecule are an important determinant of its therapeutic potency. ADME and bio-availability test play an important role in the drug likeness of new drug molecules [17]. In this research, SwissADME was employed in evaluating the physicochemical properties, pharmacokinetics, and drug similarity of the designed compounds. Furthermore, the designed compounds were checked to ensure compliance with five rules of Lipinski [6].
Result and discussion
QSAR investigation
QSAR analysis was used in building a simple mathematical equation in calculating an enhanced biological activity from β-lactam derivatives. The QSAR analysis also correlated the molecular descriptors (model parameters) with the physicochemical properties of the 43 derivative compounds (bio-activities) using statistical methods. Based on the genetic function approximation (GFA) technique employed, 4 mathematical equations were produced to predict the biological activities of β-lactam derivatives. Model 4 (four) passed both internal and external validation with a correlation coefficient squared (R2) of 0.8706, correlation coefficient adjusted squared (R2adj) of 0.8411, and cross-validation coefficient (Q2) of 0.7844. The external validation of R2pred of 0.6083 for model 4 was calculated using the model descriptors from the test set as shown in Tables 2 and 3; the MAE was found to be close to zero which reconfirms the strength of the equation (model) [12]. The effectiveness of the equations was measured by the reliability of the calibration set and calculated pIC50 of the validation set, which agrees with the criteria proposed by Golbraikh and Tropsha (R2pred > 0.6) for a robust equation as seen in Table 6.
The effectiveness of the equation was measured by the value of the calibration set and calculated pIC50 of the validation set. The observed, calculated, and residual values of β-lactam compounds are seen in Table 4. The low residual value from the difference between the biological activities and calculated activities displays the effectiveness of the equation. Both internal and external validations confirm model 4 to be very potent and extremely effective.
Table 5 shows the definition of the descriptors that were used in building the mathematical model; the descriptors were used in validating the model both internally and externally.
The mean effect of the mathematical model was executed statistically to evaluate the contribution of each model parameter individually. From the coefficient of the mean effect values, ALogP2, ATSC0i, MATS5i, and MATS1 had a positive coefficient, meaning that increasing the model parameters would increase the biological activities of the derivatives. Furthermore, ETA_Beta_ns_d having a negative coefficient means that decreasing the model parameter would also increase the biological activities of the derivative compounds as proven in Table 6. Variance inflation factor (VIF) gives a degree of the inter-relationship among the model parameters. The VIF scores were within the approved value of 1–5, indicating that there is no co-linearity between the bio-activities and model parameters of the derived model, as shown in Table 6. The null hypothesis shows no significant connection amid the bio-activity and model parameters of the derived equation at p > 0.05. At a 95% confidence level, the P values of the model parameters were below 0.05. Therefore, the null hypothesis is rejected and the alternative hypothesis is accepted. This indicates that there is no co-linearity between the bio-activity and model parameters of the constructed model, as shown in Table 6.
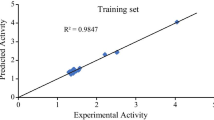
Figure 2 shows a plot of observed activities against the calculated activities of both the test set and the train set of β-lactam derivatives. The graph showed that the predicted activity was in good agreement with its experimental values as shown in Table 2, conforming to the effectiveness and stability of the built model.

Plot of predicted activities versus inhibition concentration
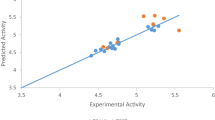
Figure 3 is a graph of standardized against experimental activity; from the plot, it is shown that the values of both test and train set spread on both sides of zero point on the plot, showing no systematic errors between the standardized residual versus the biological activity (experimental activity).

A graph of standardized residual against bio-activities (experimental activities)
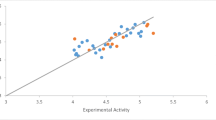
Figure 4 shows a diagram of standardized residual against leverage values also called William’s plot. All the compounds fell within the applicability domain from the calculated leverage of L = 0.6429, thou 1 compound was outside the applicability domain which might be due to a slight change in the molecular structure as compared with the remaining molecules in the data set.

A plot of leverages versus standard residual (William’s plot)
Ligand-based drug design
Six (6) new β-lactam derivative compounds were designed using the ligand-based approach. This approach uses the molecular descriptors obtained from the mathematical QSAR model; adjustments were made on the lead compounds (37 and 43) based on the definition of the molecular descriptors (ATSC0i and MATS55i) as shown in Table 5. The newly designed compounds with their new calculated activities are seen visually in Table 7.
Computational pharmacokinetics of the designed compounds
The physicochemical properties of the designed derivatives were explored for its drug-like properties. Compounds 37 and 43 revealed the characteristics of the effective drug-like template compounds as seen in Table 8. The compounds showed no violation of Lipinski’s rule of five, high GI absorption, 0.55 oral bio-availability score, and zero PAINS alerts + (pain-assay interference structural alerts) indorsing its dependability for further clinical trials. The bio-availability radar is for compounds 37 and 43 and is shown in Fig. 5; it gives a quick glance at the pharmacokinetic properties of the structures.

The bio-availability radar is for compounds 37 and 43
Conclusion
QSAR and pharmacokinetics analysis carried out on the β-lactam derivatives proved the derivative compounds to be standard anti-breast cancer agents against MCF-7 cell line. The effectiveness of the generated QSAR model was assessed using internal and external validation test; the model conformed to the minimum approved values, indicating the equation could be used in designing new β-lactam derivative compounds with enhanced anti-cancer activities. The statistical analysis carried out on the QSAR model showed that ALogP2, ATSC0i, MATS5i, and MATS1s had a positive coefficient, meaning that increasing the model parameters would increase the biological activities of the β-lactam derivative compounds, while ETA_Beta_ns_d having a negative coefficient means decreasing the model parameter would also increase the biological activities of the derivative compounds. Compounds 37 and 43 were chosen as template compounds in designing 6 new derivative compounds because they had higher predicted activity and low residual values. The molecular descriptors (MATS5i and MATS1s) had more significance, and based on their mean effect, adjustments were made on the fragments of the template compounds.
Furthermore, the pharmacokinetic analysis (drug likeliness test) carried out on the newly designed compounds revealed that all the compounds passed the drug likeness test (ADME and other physicochemical properties) and they also had zero violation to Lipinski rule of five: a standard measure used in assessing the drug likeness of molecules. This concludes that the compounds can move on to the next step of pre-clinical trial, proving a tremendous discovery for medicine in finding permanent solutions to estrogen-positive breast cancer (MCF-7 cell line).
References
Abdulrahman HL, Uzairu A, Uba S (2020) Computational pharmacokinetic analysis on some newly designed 2-anilinopyrimidine derivative compounds as anti-triple-negative breast cancer drug compounds. Bull Natl Res Centre 44:1–8
Ahmed EY, Latif NAA, El-Mansy MF, Elserwy WS, Abdelhafez OM (2020) VEGFR-2 inhibiting effect and molecular modeling of newly synthesized coumarin derivatives as anti-breast cancer agents. Bioorg Med Chem 28(5):115328
Dearden JC (2017) The history and development of quantitative structure-activity relationships (QSARs). Oncology: breakthroughs in research and practice. IGI Global, Pennsylvania, pp 67–117
Friedman JH (1991) Multivariate adaptive regression splines. Ann Stat:1–67
Gramatica P (2020) Principles of QSAR modeling: comments and suggestions from personal experience. Int J Quant Structure-Property Relationsh (IJQSPR) 5(3):1–37
Hou Y, Zhu L, Li Z, Shen Q, Xu Q, Li W et al (2019) Design, synthesis and biological evaluation of novel 7-amino-[1, 2, 4] triazolo [4, 3-f] pteridinone, and 7-aminotetrazolo [1, 5-f] pteridinone derivative as potent antitumor agents. Eur J Med Chem 163:690–709
Ibrahim MT, Uzairu A, Uba S, Shallangwa GA (2020) Computational modeling of novel quinazoline derivatives as potent epidermal growth factor receptor inhibitors. Heliyon 6(2):e03289
Kennard RW, Stone LA (1969) Computer aided design of experiments. Technometrics 11:137–148
Khaled KF, Abdel-shafi NS (2011) Quantitative structure and activity relationship modeling study of corrosion inhibitors: genetic function approximation and molecular dynamics simulation methods. Int J Electrochem Sci 6:4077–4094
Malebari AM, Fayne D, Nathwani SM, O’Connell F, Noorani S, Twamley B et al (2020) β-Lactams with antiproliferative and antiapoptotic activity in breast and chemoresistant colon cancer cells. Eur J Med Chem 189:112050
Putri DE, Pranowo HD, Haryadi WINARTO (2019) Study on anti-tumor activity of novel 3-substituted 4 anilino-coumarin derivatives using quantitative structure-activity relationship (QSAR). In: Materials science forum (Vol. 948, pp. 101-108). Trans Tech Publications
Roy K, Das RN, Ambure P, Aher RB (2016) Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom Intell Lab Syst 152:18–33
Sehgal SA, Mirza AH, Tahir RA, Mir A (2018) Quick guideline for computational drug design. Bentham Science Publishers, Sharjah
Tantawy ES, Amer AM, Mohamed EK, Abd Alla MM, Nafie MS (2020) Synthesis, characterization of some pyrazine derivatives as anti-cancer agents: in vitro and in Silico approaches. J Mol Struct 1210:128013
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK (2011) Validation of QSAR models-strategies and importance. Int J Drug Des Discov 3:511–519
Yap CW (2011) PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32:1466–1474
Zhang C, Li Q, Meng L, Ren Y (2020) Design of novel dopamine D2 and serotonin 5-HT2A receptors dual antagonists toward schizophrenia: an integrated study with QSAR, molecular docking, virtual screening and molecular dynamics simulations. J Biomol Struct Dyn 38(3):860–885
Acknowledgments
The authors acknowledge the technical effort of Physical Chemistry Department, Ahmadu Bello University Zaria, for their immense advice and various contributions toward the successful completion of the research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdulrahman, H.L., Uzairu, A. & Uba, S. Computer modeling of some anti-breast cancer compounds. Struct Chem 32, 679–687 (2021). https://doi.org/10.1007/s11224-020-01608-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01608-7