
Abstract
Theoretical density functional theory (B3LYP/6-31G**) was used to study the intra- and intermolecular interactions of nitrobenzene, aniline, and meta and para nitroaniline in various solvation models. The studied molecules were solvated by one or two water molecules in the presence of continuum solvation (the PCM model) or without it. Finally, the studied molecules were surrounded by a cluster of water molecules. For comparison, calculations were also made for separated molecules. Geometries, energies, hydrogen bonding between solutes and solvent molecules, atomic charges, and aromaticity were examined. The analysis was based on the Atoms in Molecules methodology and the Harmonic Oscillator Model of Aromaticity (HOMA) index. As a result, an extensive description of the solvation of nitro and amino groups and the effect of solvation on mutual interactions between these groups in meta and para nitroanilines is provided. It was found that in general, the PCM description of the hydration effect on the electronic structure of the studied systems (substituents) is consistent with the approach taking into account all individual interactions (cluster model).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Intramolecular interactions in para and meta nitroanilines belong to the category of substituent effects (SE) in widely studied systems of a general structure X-R-Y, where Y is a fixed functional group (reaction site), X is a variable substituent, and R is a transmitter. At the beginning, this kind of systems has been subject of the application of the Hammett-type approaches [1,2,3,4,5] with original substituent constants σ [6] or one of their modifications [7] as explanatory descriptors.
Introduction of quantum chemistry-based descriptors of substituent electron donating/accepting properties opened a new chapter in this field of investigations. Introduced by Taft et al. [8], energies of homodesmotic reactions as (1)
allowed to quantify the substituent effect. When ΔE = {E(X-R) + E(Y-R)} − {E{(X-R-Y-Y) + E(R)} is greater than 0, then the interactions between X and Y in X-R-Y system are stabilizing. The ΔE term was later named as substituent effect stabilization energy (SESE) [9] and revealed well correlations with substituent constants [9,10,11].
Almost 30 years later, Sadlej-Sosnowska presented a new descriptor of the substituent effect based on quantum chemistry: cSAR(X) (abbreviation from charge of the substituent active region) [12, 13] defined as a sum of atomic charges of the substituent X and the substituted carbon atom, Cipso {cSAR(X) = q(X) + q (Cipso)}. Unlike atomic charges at substituents X, cSAR(X) nicely correlated with substituent constants in series of monosubstituted benzenes. Moreover in para substituted derivatives of nitrosobenzene and N,N-di-methylaniline, cSAR(X) described changes in geometries of amine and nitroso groups even slightly better than the Hammett substituent constants [14]. For the same reaction series, it was shown that the charge transferred from substituents to the NO groups, expressed by the difference cSAR (NO) − cSAR(X), well correlates with the Hammett substituent constants, whereas this difference for charges at X and NO completely failed [15]. It is important to stress that cSAR(X) values estimated by various atomic charge assessments lead to well-correlated regression lines, whereas for charges at substituents, these relations failed [16].
From the beginning of application Hammett-like relationships, it was well known that sensitivity of the substituent effect expressed by the so-called reaction constants ρ is strongly dependent on the environment in which the physicochemical processes are carried out [3]. The acid-base equilibria of substituted benzoic acids are very sensitive to solvent: The reaction constant (slope) for the Hammett relation is for water equal 1.00 (by definition), but for dimethyl sulfoxide is 2.48 [17], whereas for the gas phase measurements is 5.6 [18], and for wider review, see [19]. Recently it was shown [20] that the substituent effect in olefinic (cyclohexa-1,3-diene) and aromatic (benzene) derivatives exhibits a substantially greater sensitivity in water represented by polarizable continuum model (PCM model) [21] than in the gas phase (GP). For saturated 2,2,2-bicyclooctane derivatives, the effect of solvent was significantly weaker [20].
The purpose of this report is to confront results obtained in the gas phase and polarizable continuum model with the approach based on a model that takes into account chemical interactions of solute (aniline, nitrobenzene, and meta and para nitroaniline) molecules with explicit water molecule(s). The solvation of studied molecules was simulated by surrounding them by the 50 water molecules and full geometry optimization of the whole system at the DFT level, see the “Methods” section for details. This model (abbreviated here as H2Osolv) was successfully used for the investigation of aromaticity of the benzene molecule in water [22]. For better understanding what happens when the single molecule is fully solvated by discrete solvent molecules, we added also some more models representing intermediate stages between the gas phase and the full solvation. They are the first model in which each substituent (–NO2 or –NH2) is solvated by one water molecule (the H2O model) and in the second model where the just mentioned model, the PCM environment is added (the H2O+PCM model). In cases of meta nitroaniline and para nitroaniline, models with solvated only one functional group, NH2 (H2Oamino) or NO2 (H2Onitro), are also included. Corresponding models with continuum solvation added (H2Oamino+PCM and H2Onitro+PCM) are studied too. The electronic properties of substituents in this study are described by the cSAR model estimated by the use of Bader [23] methods for the atomic charge assessments. The aniline, nitrobenzene, meta nitroaniline, and para nitroaniline molecules were chosen since nitro and amino groups are well known as very strong electron-attracting [24] and donating [25] substituents, respectively. Thus, intramolecular interactions between them have been the subject of numerous studies [26,27,28,29,30]. Hence, they may be an excellent probe for studying the environmental impact on their mutual interactions. Another aspect of the effect of solvation is its influence on the aromaticity of benzene ring in the studied systems which will be studied by the use of aromaticity index HOMA [31, 32].
Methods
Full geometry optimizations for studied molecules and their different water clusters were performed using the Gaussian’03 package [33]. A standard optimization procedure implemented in the Gaussian package was used in all cases. The nature of optimized structures was checked by frequency calculations. No one imaginary frequency was detected. Thus, all calculated structures are minima. Three different minima were optimized for water clusters (H2Osolv model) with each studied compound. The B3LYP [34] DFT functional and 6-31G basis set [35] enriched with a polarization function [36] for all the atoms (6-31G**) were used. Complexation energies of one or two water molecules to studied compounds were calculated for the gas phase according to the supramolecular approach (energy of complexation reaction, i.e., the energy difference of the product and the reactants) and corrected by the basis set superposition error values estimated by the counterpoise method [37].
Atoms in Molecules (AIM) [23] charges were calculated by means of the AIMAll software [38], which was also used to detect hydrogen bonds between a solute and water molecule(s).
Harmonic Oscillator Model of Aromaticity (HOMA) is an aromaticity index based on the geometry of studied systems (bond lengths of the ring under investigation) [31, 32]. It is calculated using the HOMA = 1 − [α/N Ʃ (Ropt − Ri)2] formula, where Ropt and Ri are optimal bond lengths and bond lengths in the real system, respectively. N is the number of bonds in the studied system and empirical factor α sets the HOMA value equal to 0 for the Kekule structure of benzene (reference non-aromatic system) and 1 for the real benzene structure (reference aromatic system).
Results and discussion
Series of three subsequent figures present visualization of studied compounds: aniline, nitrobenzene, and meta and para nitroanilines in their complexes with water in various interactions with one or two water molecule(s), and these complexes in PCM model and finally when solute molecules interact with a cluster of 50 water molecules, see Figs. 1, 2, and 3. The lengths of the observed H-bonds are shown in Table 1. In addition, in the same table, energies connected with the complexation of one or two water molecules in the gas phase are presented.

Structures of aniline and nitrobenzene complexes with one water molecule
To begin with, let us compare the strength of hydrogen bonds formed by the studied molecules with water in the gas phase. The amino group can act as both a proton donor and a proton acceptor. In the case of aniline, both H-bonds are formed in the gas phase: (i) between water oxygen and hydrogen of the amino group, denoted H2O(H···O), and (ii) between hydrogen of a water molecule and the amino’s nitrogen, denoted H2O(N···H). The first H-bond is shorter than the second (2.054 Å and 2.067 Å, respectively), but the second is slightly stronger (by 2.12 kJ/mol). The presence of a nitro group in the molecule significantly strengthens the H2O(H···O) H-bond (dH···O = 1.975 and 2.008 Å, with energies − 23.57 and − 20.22 kJ/mol, respectively for para and meta nitroaniline) and eliminates the second hydrogen bond (H2O(N···H)). The nitro group acts only as a proton acceptor, but both oxygen atoms can participate in hydrogen bonding. This is observed in the gas phase for nitrobenzene or nitroaniline complexes with one molecule of water. The addition of an amino group to nitrobenzene does not cause such significant changes in characteristics of the H-bonds between the nitro group and water molecule as in the case of the abovementioned analogous amino group interactions, see Table 1. In addition, the simultaneous complexation of both groups by water molecules in para and meta nitroanilines enhances intermolecular interactions (by 3.64 and 2.55 kJ/mol, respectively); this strengthening is also documented by H-bond shortening. This means that intermolecular hydrogen bonds also cause changes in intramolecular interactions, i.e., the electronic structure of the remaining part of nitroaniline (solute).
Figure 1 shows also nitrobenzene and aniline molecules interacting with a water molecule in the PCM environment. As mentioned above, there is only one binding mode of a single water molecule to the nitro group of nitrobenzene. In the gas phase, the water connects to nitrobenzene using its both hydrogen atoms, see Fig. 1. It is quite curious that this seemingly logical and symmetrical structure changes in conditions of continuous solvation simulated by the PCM model, where only one hydrogen bond is formed. The reason why only one hydrogen bond is formed in the presence of solvation (here with the PCM solvation, but the same is observed in case of discrete solvation in the H2Osolv model) is that hydrogen bonds between water and the nitro group in the gas phase are relatively weak. The single hydrogen bond in the asymmetrical structure observed for solvated nitrobenzene is stronger (hydrogen bond in asymmetrical structure is shorter by about 0.2 Å, see Table 1). At the same time, the second hydrogen of the coordinated water molecule can interact with the environment.
On the contrary to the nitrobenzene-water complex, there are two modes of water interactions with the amino group of the aniline molecule. Water can interact through one of its hydrogens (the N...H hydrogen bond is formed) or by its oxygen atom (in this case, the H...O hydrogen bond appears). The results of performed calculations reveal that the energetic preference of these H-bonds depends on the environment. In the case of the aniline-water complex without the PCM environment, the structure with the H...O bond has energy lower by 3.84 kJ/mol. On the other hand, the introduction of the PCM environment changes this so that the complex with the N...H hydrogen bond becomes more stable by 5.61 kJ/mol.
The nitro group types of interaction in meta and para nitroanilines follow rules observed in the case of nitrobenzene, see Fig. 2. The nitro group without continuum solvation interact with water by two hydrogen bonds between oxygen atoms of the nitro group and hydrogen atoms from water. This changes after using the PCM model, and the nitro group is interacting with water through one hydrogen bond only for both nitroaniline molecules.

Structures of meta and para nitroanilines with one and two water molecule(s)
The behavior of the solvated amino group in meta and para nitroanilines is more complicated. Not always two binding modes of the amino group with a water molecule are possible. In the case of both compounds, meta nitroaniline and para nitroaniline, geometries started with suggested N...H hydrogen bond easily transform during optimization procedure to complexes with the H...O hydrogen bond, if only additional continuum solvation is not present. Thus, such structures are not present in Fig. 2 and in all the following tables. In structures in which we can compare relative stability between complexes with N...H and H...O motifs (i.e., H2Oamino+PCM(H...O) vs. H2Oamino+PCM(N...H), and H2O+PCM(H...O) vs. H2O+PCM(N...H)), energies of structures containing the H...O bond are lower by about 11 and 2 kJ/mol, for para and meta derivatives respectively. All these data suggest, as expected, that the N...H hydrogen bond is more difficult to form than the H...O one.
Typical modes of studied compound interactions with solvent molecules in water clusters are presented in Fig. 3. The nitrobenzene molecule interacts with the solvent sometimes through one, sometimes through two oxygen(s), which formed always two hydrogen bonds. But always a particular water molecule interacts with the nitro group by one hydrogen bond. Structure in which one of the water molecules in cluster forms two hydrogen bonds with the NO2 group (the structure preferred in the gas phase) is not observed.

Examples of studied compound complexes with 50 water molecules
The amino group of aniline interacts strongly with at least three water molecules. Both possible types of hydrogen bonds (N...H and H...O) are observed. Sometimes a hydrogen atom of the amino group takes part in two hydrogen bonds, and then aniline interacts directly with four water molecules (see the structure of aniline-b, Fig. 3). Thus, the amino group interacts with water in a more differentiated way than the nitro ones. This is the reason why aniline is slightly soluble in water while nitrobenzene is considered water insoluble compound.
Different modes of solvations are observed also in cases of para and meta nitroanilines. The nitro group directly interacts always with two water molecules but in two modes: either by two H...O bonds between each of oxygen atoms in the NO2 group and two hydrogen atoms of different water molecules (see structures p-nitroaniline-b and m-nitroaniline-b, Fig. 3), or by two H...O from different water molecules to the same oxygen atom of the nitro group (structures p-nitroaniline-a and p-nitroaniline-c, Fig. 3). Sometimes, both these types of interactions are present, see the structure m-nitroaniline-a, where the meta nitroaniline molecule interacts with three water molecules.
The nitro group of the meta nitroaniline is hydrated stronger than this group in the para nitroaniline (see Table 1 and Fig. 3). In the para nitroaniline, always two water molecules strongly interact with the solute molecule. In the case of meta derivative, three or two water molecules are binded to the nitro group, and the structure with three water molecules is more frequent. Also an amino group of meta nitroaniline is hydrated by more water molecules than the para derivative. Always three water molecules form hydrogen bonds with the amino group of meta nitroaniline (two N...H and one H...O hydrogen bonds) while the amino group of para nitroaniline is involved in strong interactions with three (the same type of interactions as in case of the meta nitroaniline) or two water molecules. In the case of the p-nitroaniline-c structure, the nitrogen atom of the amino group has not formed any hydrogen bond (as it was shown above, the N...H hydrogen bond is not as easy to form as the H...O one). These all observations suggest that para nitroaniline effectively interacts with a lower number of water molecules than its meta counterpart. This effect is probably responsible for relative water solubility of these compounds—meta nitroaniline (0.090 g/100 g H2O, 25 °C) is slightly more soluble than para nitroaniline (0.057 g/100 g H2O, 25 °C) [39].
The main topic of this paper is to study the influence of solvent (water molecules) on electronic structure and the substituent effects in studied compounds by means of the analysis provided in the cSAR approach. As it was presented above, studied molecules can be solvated in different ways. With no doubts, this can affect effective cSAR values evaluated in our calculations. This may be some inconvenience but anyway, performed calculations for many different water clusters surrounding studied compounds allow us to understand better the nature of interactions and their impact on the electronic structure of functional groups as well as the interactions between them. However, such calculations are time and computer resources consuming. Thus, we must base our analysis on a limited number of cases.
The obtained cSAR (NH2), cSAR (NO2), and charges at both groups q (NH2) and q (NO2) are gathered in Tables 2 and 3. To facilitate a quantitative view, the above data are expressed also as percentage quantities in relation to the values estimated in the gas phase for NO2 and NH2 groups (which in all cases correspond to 100%).
Changes of cSAR (NH2) and cSAR (NO2) values in studied molecules depend significantly on the hydration and the differences observed are due to the type of interactions of NH2 group with water molecules and the applied model of hydration. To show this, let us consider first results for aniline and nitrobenzene.
For NH...O (water) interactions, an increase of cSAR (NH2) is very significant—by 68%, whereas for N...H (water), a great decrease, by 45%, is observed (Table 2). This is fully understandable: In the first case, the hydrogen bond NH...O results in drawing back proton from the NH bond towards oxygen atom in water molecule leading to an increase of a negative charge at the nitrogen atom and hence on the whole NH2 group. The opposite is in another case: the H-bond in the N...H (water) system where hydrogen atom of water molecule attracts pi-electron pair of the amino group leading to a decrease of a charge at the whole group and hence decreasing cSAR (NH2) values. The application of the PCM approach leads also to significant changes. Direct comparison of cSAR (NH2) values in the gas phase and in PCM results in an increase of 29%. When the PCM model is applied to interactions NH...O (water) and N...H (water), then PCM computation leads to a significant increase of cSAR (NH2) by 56% and only by 12%, respectively. It is well known that NH in the amino group is a better proton-donating system than the lone pair in this group, a proton-accepting moiety [40, 41]. Thus, the PCM model, which describes the medium by means of its dielectric constant, supports the interactions with a stronger charge separation i.e., stronger H-bonding, whereas the weaker does decrease less significantly. In the case of interactions with many water molecules, N...H interactions are even strengthened: The decrease of cSAR (NH2) values are lower than in the gas phase by 54% and 75% which may be related to the additional interactions with neighboring water molecules stabilizing the proton transfer process.
In the case of nitrobenzene, it is shown that cSAR (NO2) values are much less dependent on interactions with water molecules and on the choice of the hydration model applied. In all cases, an increase of cSAR (NO2) values is observed. The greatest changes are observed for PCM applied to the nitrobenzene alone, by 19%, and for NO2 interacting with a water molecule, by 21%, whereas for nitrobenzene interacting with a water molecule in the gas phase, the change is only 7%.
Commenting these two cases, it may be said that the stronger H-bond type interactions the higher changes in cSAR(X) and the greater influence of dielectric constant of the medium, as observed via the PCM approach. Both cases of nitroanilines reveal one more interesting problem: How far different intramolecular electron interactions between the nitro group and the amino group differentiate the influence of hydration on cSAR values of both the nitro and amino groups. It is well known that resonance effect (or intramolecular charge transfer effect) is much stronger for para substituted species than for the meta ones [42]. In order to facilitate description of changes in cSAR values for all cases considered, apart of original values of cSAR(X) (X = NH2 or NO2), their relative changes are given in a percentage scale, taking as 100% the values for cSAR(X) for the individual molecules in the gas phase.
The first observation is that cSAR (NH2) values for both para and meta nitroaniline molecules are dramatically higher than for the mono substituted species. Even the gas phase data are considerably greater for amino group in para and meta nitroaniline than for individual aniline by 124.9 and 101.2%, respectively. In contrast, significantly weaker is the effect revealed for the nitro group. When cSAR (NO2) in nitrobenzene is compared with the values of para and meta nitroanilines, the increase is only by 11.4 and 1.0%, respectively. The same tendencies are observed when the mean values of the overall effect of variously realized hydration are concerned (H2Osolv model). The averaged cSAR (NH2) value for these cases for aniline complexes, compared with aniline gas phase data, gives an increase of approximately 15% and for para and meta nitroaniline values higher by 295% and 154%, respectively. These changes are very significant but in a relative agreement with those observed for aniline and nitroanilines without intermolecular interactions, which caused only a minor increase. In the case of cSAR (NO2), the changes due to hydration are again decisively much weaker in comparison with those estimated for cSAR (NH2). The appropriate cSAR (NO2) values expressed in percentage scale in relation to the gas phase in nitrobenzene are as follows: in nitrobenzene, an increase by 31%, while in para and meta nitroanilines, an increase by about 63% and 31%, respectively. Tables 2 and 3 present all details of relative percentage values, whereas some summaries of the above discussion are shown in Table 4.
Table 5 shows explicitly how various interactions with water molecules influence the intramolecular charge transfer measured by values of the Charge Flow Index (CFI) [43] between amino and nitro groups (CFI = cSAR (NH2) − cSAR (NO2)) estimated by AIM atomic charge assessment. The obtained CFI data (Table 5) reveals that for para nitroaniline and its H-bonded complexes, CFI values are always higher than for the meta ones. The last right column of Table 5 shows the increase in CFI values in para isomers in comparison with the meta ones, presented in a percentage scale. Taking into account individual interactions, the differences are between 15 and 30%, indicating numerically stronger charge transfer for para nitroanilines than for the meta ones. Usually, PCM approach reveals greater changes of CFI in both cases, inside the series of meta or para isomers as well as in the relation between para isomer and the meta one.
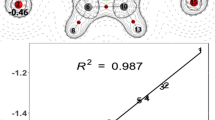
When the CFI values for meta derivative are plotted against the para ones, then the slope is 0.71, with correlation coefficient 0.93. Once again, the transmission of charge via resonance effect for meta substituted nitroaniline is significantly weaker than for the para substituted one.
The CFI index informs about the transfer of electrons between the substituents through the ring. One more information about this effect can be found by analyzing changes in aromaticity of the ring in studied systems. Table 6 presents these data.
In the gas phase, aromaticity of the ring, estimated by the HOMA index [31, 32], for nitrobenzene is equal to 0.988, whereas for aniline, it is 0.966. In PCM, these values are 0.985 and 0.965, respectively. Because the substituent effect of amino and nitro groups is similar, in their moduli of substituent constant values (− 0.66 and 0.78 for the Hammett σp, respectively) [44], it allows concluding that benzene ring is more sensitive for accepting pi-electrons than for withdrawing them. Neither aniline nor nitrobenzene does not demonstrate any substantial influence of solvation, independently of its mode; for aniline, the HOMA values are between 0.948 and 0.979 (incidentally 0.915 for one case of H2Osolv) and for nitrobenzene between 0.972 and 0.988. The case of meta nitroaniline does not differ from these data, with the range between 0.946 and 0.978. The case of para nitroaniline is significantly different, and the range of HOMA index is greater, between 0.848 and 0.945 (incidentally 0.840 and 0.841 for two cases of H2Osolv). This may be attributed to strong NH···OH2 interactions (see Table 1), which in the para position of NH2 group in nitroaniline causes pi-electron pair at the amino group looser bound and easier to be involved in intramolecular charge transfer. In consequence, a quinoid-like structure is induced associated with a partial decrease of the aromatic character of the ring [45].
Conclusions
A quantum chemical study, carried out for variously solvated molecules of aniline, nitrobenzene, and meta and para nitroaniline, enabled us to make several conclusions. It is well known that both NO2 and NH2 groups are able to interact with water molecules. However, the type and number of hydrogen bonds formed are different in the gas and solvated phases. It was found that, in general, the PCM description of the hydration effect on the electronic structure of studied systems (substituents) is in line with the approach taking into account all individual interactions (cluster model). It was detected that interactions of the amino group with water lead to much larger changes in its electronic structure than those observed for the nitro group. Obtained data indicate that the intramolecular charge transfer from the amino group to the nitro group is significantly stronger for the para nitroaniline than for the meta nitroaniline. Moreover, it is much more affected by interactions with water molecules. A comparison of the CFI values for variously hydrated NH2 and NO2 groups reveals the changes in the range between 15 and 30%. Direct correlation between the CFI values and meta and para nitroanilines reveals a slope of 0.71. Such a result again indicates much weaker charge transfer in meta substituted species. It is documented that the effect of solvation on the ring aromaticity for nitrobenzene, aniline, and meta nitroaniline is also very weak. Oppositely to that, this effect is significantly greater for para nitroaniline. This is due to the NH bond involved in H-bonding with a water molecule and hence, lone pair at nitrogen may be involved in charge transfer, leading to a quinoid-like structure with a low aromatic character.
References
Hammett LP (1940) Physical organic chemistry. McGraw – Hill, New York
Jaffe HH (1953) A reëxamination of the Hammett equation. Chem Rev 53:191–261
Exner O (1972) In: Chapman NB, Shorter J (eds) Advances in linear free energy relationships. London, Plenum Press
Johnson CD (1973) The Hammett equation. Cambridge University Press, Cambridge
Shorter J (1991) In: Zalewski RI, Krygowski TM, Shorter J (eds) Similarity models in organic chemistry, biochemistry and related fields. Amsterdam, Elsevier
Hammett LP (1937) The effect of structure upon the reactions of organic compounds. Benzene derivatives. J Am Chem Soc 59:96–103
Hansch C, Leo A, Taft RW (1991) A survey of Hammett substituent constants and resonance and field parameters. Chem Rev 91:165–195
Pross A, Radom L, Taft WR (1980) Theoretical approach to substituent effects. Phenols and phenoxide ions. J Org Chem 45:818–826
Siodla T, Oziminski WP, Hoffmann M, Koroniak H, Krygowski TM (2014) Toward a physical interpretation of substituent effects: the case of fluorine and trifluoromethyl groups. J Org Chem 79:7321–7331
Varaksin KS, Szatyłowicz H, Krygowski TM (2017) Towards a physical interpretation of substituent effect: quantum chemical interpretation of Hammett substituent constants. J Mol Struct 1137:581–588
Szatyłowicz H, Jezuita A, Siodla T, Varaksin KS, Domanski MA, Ejsmont K, Krygowski TM (2017) Toward the physical interpretation of inductive and resonance substituent effects and reexamination based on quantum chemical modeling. ACS Omega 2:7163–7171
Sadlej-Sosnowska N (2007) On the way to physical interpretation of Hammett constants: how substituent active space impacts on acidity and electron distribution in p-substituted benzoic acid molecules. Polish J Chem 81:1123–1134
Sadlej-Sosnowska N (2007) Substituent active region–a gate for communication of substituent charge with the rest of a molecule: monosubstituted benzenes. Chem Phys Lett 447:192–196
Sadlej-Sosnowska N, Krygowski TM (2009) Substituent effect on geometry of the NO and N (CH3)2 groups in para substituted derivatives of nitrosobenzene and N,N-dimethylaniline. Chem Phys Lett 476:191–195
Krygowski TM, Sadlej-Sosnowska N (2011) Towards physical interpretation of Hammett constants: charge transferred between active regions of substituents and functional group. Struct Chem 22:17–22
Stasyuk OA, Szatylowicz H, Guerra CF, Krygowski TM (2015) Theoretical study on electron-attracting ability of the nitro group: classical and reverse substituent effects. Struct Chem 26:905–913
Kalfus K, Vecera M (1972) Dissociation constants of substituted benzoic acids in anhydrous dimethyl sulphoxide. Coll Czech Chem Commun 37:3607–3609
Yamdagni R, McMahon TB, Kebarle P (1974) Substituent effects on the intrinsic acidities of benzoic acids determined by gas phase proton transfer equilibria measurements. J Am Chem Soc 52:4035–4037
Krygowski TM, Fawcett WR (1975) Separations of the enthalpic and entropic contributions to substituent effects for the equilibrium constants of aromatic acids. Can J Chem 53:3622–3633
Szatyłowicz H, Jezuita A, Siodła T, Varaksin KS, Ejsmont K, Madura ID, Krygowski TM (2018) Dependence of the substituent effect on solvent properties. J Phys Chem A 122:1896–1904
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105:2999–3093
Zborowski KK (2014) Aromaticity of benzene in condensed phases, a case of a benzene-water system. Chem Phys Lett 602:1–3
Bader RWF (1990) Atoms in molecules: a quantum theory. Clarendon Press, Oxford
Exner O, Krygowski TM (1996) The nitro group as substituent. Chem Soc Rev 25:71–75
Patai S (1968) The chemistry of the aminogroups. Interscience Publishers, London
Hutley MC, Jacobs DJ (1969) The resonance Raman effect in p-nitroaniline. Chem Phys Lett 3:711–714
Prakash R, Kalia RK, Verma RS (1976) Polarographic reduction of nitrophenols and nitroanilines in aqueous tetrahydrofuran. Proc Ind Acad Sci A 84:64–70
Krygowski TM, Maurin J (1989) Crystallographic studies of intra- and inter-molecular interactions. Crystal and molecular structure of N,N-dimethyl-4-nitro-3,5-xylidine. Structural evidence against the classical through-resonance concept in p-nitroaniline and derivatives. J Chem Soc Perkin Trans II 695–698
Krawiec M, Krygowski TM (1991) Crystallographic studies of intra- and inter-molecular interactions Part III. Refinement of the crystal and molecular structure of N,N-dimethyl-m-nitroaniline: additivity of substituent effects on geometrical parameters of the ring. J Mol Struct 246:113–122
Obi-Egbedi NO, Targema M, Adeoye MD, Gbangban ST (2015) Calculation of electronic properties of some 4-nitroaniline derivatives: molecular structure and solvent effects. Int Res J Pure Appl Chem 8:165–174
Kruszewski J, Krygowski TM (1972) Definition of aromaticity basing on the harmonic oscillator model. Tetr Lett 13:3839–3842
Krygowski TM (1993) Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of -electron systems. J Chem Inf Comput Sci 33:70–78
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian‘09 revision D.01. Gaussian Inc, Wallingford
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Ditchfield R, Hehre WJ, Pople JA (1971) Self-consistent molecular-orbital methods. IX An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J Chem Phys 54:724–728
Hariharan PC, Pople JA (1973) The influence of polarization functions on molecular-orbital hydrogenation energies. Theor Chem Accounts 28:213–222
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Keith TA (2011) AIMAll version 11.10.16. TK Gristmill Software, Overland Park
Bańkowski K (1974) Poradnik fizykochemiczny. (in polish, Physicochemical Handbook). WNT, Warsaw
Szatyłowicz H (2008) Structural aspects of the intermolecular hydrogen bond strength: H-bonded complexes of aniline, phenol and pyridine derivatives. J Phys Org Chem 21:897–914
Szatyłowicz H, Krygowski TM, Zachara-Horeglad JE (2007) Long-distance structural consequences of H-bonding. How H-bonding affects aromaticity of the ring in variously substituted aniline/anilinium/anilide complexes with bases and acids. J Chem Inf Model 47:875–886
Krygowski TM, Palusiak M, Plonka A, Zachara-Horeglad JE (2007) Relationship between substituent effect and aromaticity – Part III: naphthalene as a transmitting moiety for substituent effect. J Phys Org Chem 20:297–306
Szatyłowicz H, Jezuita A, Siodla T, Varaksin KS, Ejsmont K, Shahamirian M, Krygowski TM (2018) How far the substituent effects in disubstituted cyclohexa-1,3-diene derivatives differ from those in bicyclo[2.2.2] octane and benzene? Struct Chem 29:1201–1212
Exner O (1988) In: Chapman NB, Shorter J (eds) Correlation analysis of chemical data. New York, Plenum Press
Krygowski TM (1984) Crystallographic studies and physicochemical properties of pi-electron systems. 5. Substituent effect on the geometry of the benzene-ring in benzene derivatives. J Chem Res (S) 238–239
Funding
The authors received funding support for the computer equipment used in this work from the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.22.01.00-12-023/08). HS also received financial support from the Warsaw University of Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zborowski, K.K., Szatyłowicz, H. & Krygowski, T.M. Solvent influence on intramolecular interactions and aromaticity in meta and para nitroanilines. Struct Chem 31, 1717–1728 (2020). https://doi.org/10.1007/s11224-020-01582-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01582-0