
Abstract
The effect of H-bonding and metal complexation (probed by HF, F−, Li+, Na+, and K+) on structural and π-electron delocalization changes in four most stable guanine tautomers and their structural subunits has been studied in the gas phase using the B3LYP/6−311++G(2d,2p) computational level. In both cases, i.e., H-bonding and metal complexation, the strongest interactions are found in bifurcated complexes of the keto guanine tautomers. Interactions in which the functional groups participate (NH2 or C=O) regularly lead to the greatest geometric and aromaticity changes. As a consequence, aromaticity of substituted six-membered rings is decisive for aromaticity of whole ring system in guanine tautomers. Aromaticity of guanine tautomers and their structural subunits changes in the same way (increase or decrease) depending on particular type of interactions.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
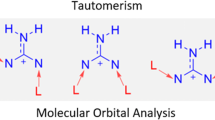
Guanine is one of the two purine nucleobases consisting of fused imidazole and pyrimidine (with two functional groups: oxo/hydroxo and amino/imino) rings. Among all nucleic acid bases, guanine has the largest number of tautomers—36, including rotamers [1, 2]. According to theoretical and experimental results, two keto-amino forms (7H and 9H) predominate in the gas phase [3–6]. In polar solvent and for hydrated polycrystalline guanine, the 9H keto-amino tautomer is the most favored species [7, 8]. However, two enol forms of 9H guanine with cis and trans orientation of OH group are very close energetically to the 9H keto one, and all four tautomers were detected experimentally [5, 9].
In Watson–Crick pair guanine interacts with cytosine via three H-bonds [10] that results in proper double-stranded structure of DNA. However, quite often in RNA guanine can form pair with uracil (wobble base pair [11]), which has comparable thermodynamic stability to Watson–Crick base pairs [12]. Distortion of a proper DNA structure is also induced by stabilization of rare guanine tautomers caused by metal cations [13, 14] and formation of non-Watson–Crick base pairs [15, 16]. Metal ions can also weaken [17, 18], and in some cases even disrupt [19], one or more hydrogen bonds in the base pairs and stabilize non-canonical structures of nucleic acids [20]. Such interactions between metal cations and nucleobases can be direct or solvent-mediated [21]. However, in the case of alkali metals, the X-ray investigations [22, 23] and MD simulations [24] support mostly direct interactions between nucleobases and partially dehydrated metal ions [25]. Interactions with active centers of guanine located in major groove of DNA (N7 and O at C6 atom) have been discussed for years [26–31]. More specific interactions with other centers of guanine were also described [32–34]. However, despite the extensive literature on this topic, insufficient attention to effects of intermolecular interactions with different active centers of guanine tautomers on their electronic structure has been paid.
The aim of the present work is to investigate π-electron delocalization of the most stable guanine tautomers and their complexation via H-bonding (with F−/HF) and with alkali metal cations (Li+, Na+, K+), as well as to analyze consequences of the intermolecular interactions on geometry and aromaticity of the studied systems. In this work, seven or nine active sites for the H-bonding and two or three possible sites for the metal binding were taken into account, depending on the tautomer under consideration. The sodium and potassium cations are the common ions in biological systems. A choice of the lithium cation is motivated by an opportunity to show the greatest possible changes in the electronic structure due to the strongest intermolecular interactions. To study the effect of fusion of two aromatic rings into one guanine molecule, we also performed a comparative analysis of guanine tautomers and subunits of which they are composed, in particular imidazole and substituted pyrimidine rings.
Methodology
Calculations were carried out at B3LYP/6−311++G(2d,2p) level using the Gaussian 09 program [35]. Justification of the theoretical method choice, which remains unchanged throughout our research on the effects of H-bonding and complexation with metal ions on structural and electronic properties of the nucleobases [36–39], is given in our previous work [36].
For studied systems, optimization without any symmetry constraints was performed. Based on harmonic frequency analysis, we confirmed that all equilibrium structures correspond to true ground-state stationary points.
Two types of aromaticity parameters have been used as quantitative measures of π-electron delocalization: (1) structural [40] and (2) magnetic indices [41].
The first is HOMA [42, 43], the geometry-based aromaticity index, which is defined as:
where n is the number of bonds taken into the summation and α i is a normalization constant (for CC and CO bonds α CC = 257.7 and α CO = 157.38) fixed to give HOMA = 0 for a model non-aromatic system and HOMA = 1 for a system with all bonds equal to the optimal value R opt,i assumed to be realized for fully aromatic systems (for CC and CO bonds R opt,CC = 1.388 and R opt,CO = 1.265 Å), where as R j denotes bond lengths taken into calculation.
The second index is NICS, which was calculated: (1) in the center of the ring [44], NICS(0), (2) 1 Å above the center [45], NICS(1), and (3) the component of the tensor perpendicular to the molecular plane [46, 47], NICS(1)zz.
To gain insight into changes in electron density distribution induced by fusion of two subunits into guanine tautomers, the atomic charges were analyzed using NBO method by NBO 5.G program [48].
To elucidate the modification of the π-electron delocalization in studied systems, we approached different types of partners to obtain three types of complexes: (1) neutral (with HF), (2) anionic (with F−), and (3) cationic (with M+, M = Li, Na, K). The same procedure was applied to guanine tautomers and their subunits.
The total interaction energy was decomposed into deformation (E def) and interaction (E int) components. The first term represents the amount of energy required to deform the geometries of individual fragments (E 0A and E 0B ) into their geometries in the complex (E A and E B):
Interaction energy, corrected by BSSE [49, 50], was calculated as described elsewhere [51] and corresponds to the actual energy change when the deformed fragments are combined to form the complex.
Results and discussion
Discussion of the results will be presented in three subsections dealing with: (1) non-interacting (free) guanine tautomers and their structural subunits (substituted pyrimidine and unsubstituted imidazole rings), (2) H-bonded complexes, and (3) complexes with alkali metal cations of the studied tautomers.
Electronic structure of free guanine tautomers
Four of the most stable guanine tautomers are presented in Fig. 1. Their relative energies, E rel, are within 2 kcal/mol (Table 1S), and all of them were observed experimentally [5, 9]. In agreement with previous theoretical studies [2, 52–54], the most stable is 7H keto-amino form, g2 in Fig. 1. This form was also experimentally found in crystal structure of anhydrous guanine [55].

Structures of the most stable guanine tautomers
As mentioned above, the guanine consists of two aromatic units: pyrimidine with two functional groups (oxo/hydroxo and amino/imino) and imidazole fused into one molecule. In the case of benzenoid hydrocarbons, such fusion leads to a substantial decrease in aromatic character [56, 57]. In particular, HOMA values for ring in benzene and naphthalene are 0.996 and 0.803, respectively. Thus, the decrease in aromaticity is about 0.2 HOMA unit. To compare the data for guanine case, we have introduced quantities: ΔHOMA6 and ΔHOMA5, which are defined as differences of HOMA for the appropriate fused ring and its monocyclic form. These data are presented in Table 1. As it is clearly seen, fusion of the individual structural units into guanine moiety leads to increase in aromaticity in both rings only in the case of g2 tautomer. In other cases, the decrease in HOMA index is observed; however, here this is manifested to a lesser degree than in the case of benzene/naphthalene systems. It is worth mentioning that changes in aromaticity are equally well described by HOMA as well as by three kinds of NICS index: NICS(0), NICS(1), and NICS(1)zz, as presented in Fig. 1S. All details about aromaticity of studied structures are given in Tables 1S and 2S.
In order to analyze the effect of fusion of two aromatic subunits into guanine tautomers on their electronic structure in detail, the distribution of NBO atomic charges was also studied. Even a cursory look at the atomic charges in guanine tautomers and their individual components allows us to conclude that the greatest changes occur only at the atoms of C4C5 bond shared between two fused rings, see Figs. 2S and 3S. In all tautomers, the NBO charge at C4 is always strongly positive (from 0.353 to 0.391 a.u.), whereas the charge at C5 is weakly negative (from −0.035 to −0.054 a.u.), compensating charges at neighboring atoms. For the non-fused pyrimidine rings different from the above charge distribution was found, the C4 charge is positive (from 0.091 to 0.116), and the C5 charge is strongly negative (from −0.366 to −0.403 a.u.). Moreover, if we look at the same atoms in imidazole unit, we find that both atoms, C4 and C5, are weakly negative (−0.068 and −0.088) and fusion with six-membered ring leads to their significant changes: The C4 atomic charge increases up to 0.391 a.u., and the C5 one slightly goes down to −0.035 a.u. All differences in NBO charges at other atoms in guanine tautomers and their subunits are much smaller and usually not more than 0.02 a.u. This indicates that only atoms involved in the common C4C5 bonds are the subject of substantial perturbation due to the fusion of the rings. As a result, the shared C4C5 bonds are longer in guanine tautomers than appropriate bonds in non-fused structural subunits (red numbers in Figs. 2S and 3S). The C4C5 bond lengths in guanine tautomers are between 1.390 and 1.401 Å, whereas in pyrimidine derivatives, they are between 1.361 and 1.384 Å and amounts to 1.368 Å in imidazole.
The above-described differences of the charge distributions concern fused and individual rings of the free systems. Therefore, the question arises whether they can result in different characteristic (behavior) of guanine tautomers and their structural subunits in complexes with intermolecular interactions.
Electronic structure of guanine tautomers involved in H-bonding
First, we studied the fusion effect on hydrogen bond strength, comparing similar H-bonds formed by guanine tautomers and their structural subunits (Table 2). Characteristics of H-bonded complexes are presented in Tables 3S and 4S for subunits and guanine tautomer complexes, respectively.
In the case of imidazole, two types of H-bond may be formed: neutral N···HF and charge-assisted NH···F−. However, when fluoride approaches the NH moiety, the proton transfer occurs and complex with N−···HF interaction is created. The H-bond energy in such complex is equal to −28.9 kcal/mol and may be compared to strength of similar interactions with F− observed in guanine tautomers, where −23.0 ≤ E HB ≤ −23.9 kcal/mol (details in Tables 3S and 4S). Thus, in individual imidazole, this type of H-bonding is stronger than in guanine five-membered subunits by ~5 kcal/mol. It can be rationalized by a greater negative charge at the nitrogen atom in imidazole anion than in guanine one, −0.621 and −0.598 a.u., respectively. The picture is slightly different for interactions of N···HF type, where E HB(imidazole) = −13.2 kcal/mol, whereas for subunits embedded in guanine moiety, E HB is between −11.3 and −14.7 kcal/mol. In this way, for neutral H-bonds no significant differences are observed between individual five-membered ring and fused ones.
Due to two functional groups attached to the six-membered ring, a greater variety of H-bonds is observed in pyrimidine derivative complexes (Table 3S). The following intermolecular interactions are possible: bifurcated H-bonds, N(ring)···HF, O···HF or OH···F−, and N···HF or NH···F− of the amino group. Interactions with F− usually lead to proton transfer, except cases where F− participates in bifurcated H-bonding. The latter is realized in complexes of the subunit for g1 and g2 tautomers where F− is located between H13 (hydrogen atom of the amino group) and H14 (attached to the ring) atoms. The total interaction energy for this complex is two times greater than obtained for complex of the same subunit with single charge-assisted H-bond, −50 versus −25 kcal/mol (Table 3S). Moreover, the fusion with five-membered ring does not influence energetic characteristics of the same type interactions observed in complexes of g1 and g2 tautomers (Fig. 4S). A variety of H-bonds considered in guanine tautomers is shown in Fig. 2. Furthermore, the above conclusion covers also the other H-bonds in pyrimidine derivatives and guanine tautomer complexes (Table 2).

Possible H-bonded complexes for the g1 tautomer
According to expectations, energy of bifurcated H-bonds is the greatest among all studied complexes. For the remainder H-bonds realized in complexes of subunits and guanine tautomers, the sequence of their strength is as follows: N−(amino)···HF > N−(ring5)···HF > O−···HF > N(ring)···HF ≈ O(keto)···HF > N(amino)···HF > O(hydroxy)···HF. Thus, charge-assisted H-bonds are stronger than the neutral ones. In the case of similar types of intermolecular interactions with a different proton acceptor atom, e.g., N−(amino)···HF and O−···HF, H-bonds with the oxygen atom are always weaker than interactions with the nitrogen atom, in line with other studies [58, 59].
Results of general comparative analysis of H-bonds formed with guanine tautomers and their structural subunits demonstrate that effect of fusion of two aromatic rings into guanine moiety is weakly pronounced and does not change significantly the strength of the H-bonds (Table 2). Data for H-bond energies agree well with results obtained recently for cytosine tautomers [38].
Next, we investigated the effect of fusion on aromatic character of H-bonded complexes. As presented in Table 1, the fusion of two components into guanine moiety slightly decreases aromaticity of the particular rings, except for g2 tautomer, where increase in aromaticity is observed. Another question that arises is how much guanine tautomers behave differently in comparison with their subunits when both systems are involved in H-bonding. Tables 3S and 4S contain geometric and energetic characteristics of considered H-bonded complexes, as well as their aromaticity described by HOMA index. Due to structural complexity of studied systems, HOMA index was estimated for five- and six-membered rings, as well as for the whole molecules, separately. To make data more clear, Table 3 represents only values related to changes in aromaticity occurring in systems involved in H-bonding, whereas HOMA trends for particular H-bonded systems are presented in Table 2.
The data for five-membered rings in guanine tautomers indicate that their aromaticity is rather insensitive to H-bonding. The differences of HOMA index between H-bonded and free species, Δ(5), are very small, unlike imidazole itself where aromaticity of the ring slightly arises due to H-bonding. The same observation has been found for six-membered rings of the most stable tautomers—g1 and g2. In the six-membered subunit alone, aromaticity increases greater than in the rings embedded in guanine moiety. In turn, for two less stable tautomers, g3 and g4, H-bonding promotes a similar decrease in aromaticity (by ~0.05 HOMA unit) for both six-membered rings (free and fused with imidazole ring). When we compare a variation of HOMA values due to the H-bond formation in guanine tautomers, we find that it is almost two times greater for six-membered rings (0.114–0.160) than for five-membered ones (0.069–0.090). This fact indicates a greater sensitivity of six-membered rings to perturbation of π-electron structure caused by H-bonding.
Furthermore, in most cases, trends in aromaticity changes found for pyrimidine part of complexes define the aromaticity changes observed in total systems. For this reason, in detailed analysis of the effect caused by different types of H-bond, we use only HOMA6 data, except the interactions, which occur only in five-membered ring, i.e., N−(ring)···HF. In all cases, trends in HOMA index for particular types of H-bond are the same for guanine tautomers and their subunits (Table 2). The weakest interactions, O(hydroxy)···HF, in g3 and g4 tautomers as well as pyrimidine subunits almost do not influence the aromaticity of complexes. However, similar interactions with the oxygen atom of the keto form in g1 and g2 tautomers have quite pronounced effect, increasing aromaticity of six-membered ring and total system due to elongation of CO bond length and disturbance of partly quinoid structure. Completely opposite effect was found for O−···HF interactions. Moreover, it has been established that greater changes in aromaticity are induced by such H-bond interactions in which functional groups such as NH2 or C=O participate. The same trend was found in H-bonded complexes of thymine and cytosine tautomers [37, 38].
Electronic structure of guanine tautomers involved in complex with metal cation
Comparison of energetic characteristics and aromaticity changes for metal complexes of guanine tautomers and particular subunits is presented in Table 4. The strengths of particular interactions are shown in Tables 5S and 6S, whereas aromaticity of individual rings is gathered in Tables 8S and 9S.
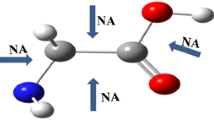
For all guanine tautomers, mainly the bifurcated binding of alkali metal cations takes place, i.e., two neighboring atoms (N,N or N,O) are involved in the interaction (Fig. 3). Only for g2 tautomer, a singular coordination at the oxygen atom has been obtained. The complexes with bifurcated interactions N,O···M+ are always more stable than those with M+···N,N ones, in line with previously found results [32, 38]. It should be noted that the most stable and the least stable complexes have been found for g1 tautomer (Table 5S). In the former, the cation is located between N7 and O atoms, whereas in the latter one between N3 and N10 atoms. The same is observed for interaction energies between guanine and metal cations: The strongest and the weakest interactions correspond to the complexes of g1 tautomer. The magnitude of total energy, consisting of interaction and deformation components, depends on cationic radius and site of interaction. The overall energy ranges are: (1) −41 to −74 kcal/mol for Li+ complexes, (2) −23 to −56 kcal/mol for Na+ complexes, and (3) −13 to −43 kcal/mol for K+ ones (see Table 7S). As mentioned above, the weakest interactions are observed for bifurcated complexes where metal is located between the nitrogen atoms of the six-membered guanine ring (N1 or N3) and of the amino group (N10). Interestingly, these interactions are even weaker than singular O···M+ one (observed in g2 complexes), see Table 4. For the strongest interactions, i.e., N,O···M+ ones, energy depends on the form of the oxygen atom; in complexes with the keto O atom, these interactions are stronger by about 25 % than those with the O from the hydroxyl group. Complexes of particular structural subunits with cations are characterized by interaction energies similar to those found for guanine tautomers with two exceptions: stronger O···M+ interactions and weaker N,O···M+ ones (Table 4).

Possible sites of metal coordination in the g1 tautomer (M = Li, Na, K)
The greatest effect of the complexation with metal cations on aromaticity of the ring takes place for the strongest and weakest interactions. In the former case corresponding to N7,O···M+ complexes of g1, an increase in the six-membered ring aromaticity in comparison with non-interacting tautomer by 0.18, 0.16, and 0.15 HOMA unit is observed for interactions with Li+, Na+, and K+, respectively (Table 8S). This fact can be ascribed to the prevalence of the resonance structure with separated charges (Fig. 5S) which contributes to more aromatic character of the six-membered rings [31]. On the other hand, for the g1 and g2 complexes with the weakest interactions (N3,N10···M+), the opposite changes, i.e., a decrease aromaticity of the six-membered ring by more than 0.09 HOMA unit, are found. This type of interactions also decrease the π-electron delocalization of the g1, g2 subunit ring by 0.20, 0.15, and 0.12 HOMA unit in complexes with Li+, Na+, and K+, respectively (Table 9S). Monotonic changes in aromaticity in line with the increase in metal ionic radii can be also observed.
Considering aromaticity changes caused by the formation of complexes with metal cations, it has been found that for less aromatic tautomers, g1 and g2, the greatest changes (in both directions: increase and decrease) are observed in six-membered rings, and they are responsible for total changes in aromatic character of tautomers (Table 8S). In turn, for more aromatic tautomers, g3 and g4, the complexation does not cause significant changes in aromaticity with maximum values ΔHOMA6 = 0.035 and ΔHOMA5 = 0.029. The interactions with oxygen atom of the keto tautomers (g1 and g2) lead to increase in aromaticity of the six-membered ring and the total ring system. The opposite happens in the case of interactions with O atom of the enol tautomers.
Conclusions
-
1.
Effect of fusion of two heterocyclic structural subunits into guanine moiety on aromaticity of six- and five-membered rings is less pronounced than in the case of fusion of two rings into purine and naphthalene. However, similarly as in the case of benzene/naphthalene pair, the fusion of imidazole and pyrimidine rings with two functional groups (amino and oxo/hydroxo) into guanine leads to the elongation of the common C4C5 bond by 0.01–0.03 Å in all studied tautomers.
-
2.
Aromaticity of six-membered rings and, as a consequence, aromaticity of whole guanine tautomers strongly depend on the presence of the C=O group. Tautomers with hydroxyl group (g3 and g4) are significantly more aromatic than their keto analogs (g1 and g2).
-
3.
The strongest intermolecular interactions have been found in complexes of keto tautomers. In both cases, i.e., H-bonding and metal complexation, these interactions are bifurcated.
-
4.
Larger changes of π-electron delocalization caused by intermolecular interactions are observed in the six-membered rings, which are also responsible for total aromaticity changes in tautomers. In all cases, trends in aromaticity changes caused by particular type of interactions are the same for guanine tautomers and their subunits.
-
5.
The greatest aromaticity changes are always caused by interactions in which functional groups NH2 or C=O participate. These changes are realized in both directions—an increase and a decrease in the π-electron delocalization, expressed by HOMA index.
References
Sabio M, Topiol S, Lumma WC (1990) J Phys Chem 94:1366–1372
Liang W, Li H, Hu X, Han S (2006) Chem Phys 328:93–102
Chung G, Oh H, Lee D (2005) J Mol Struct (THEOCHEM) 730:241–249
Zhou J, Kostko O, Nicolas C, Tang X, Belau L, De Vries MS, Ahmed M (2009) J Phys Chem A 113:4829–4832
Choi MY, Miller RE (2006) J Am Chem Soc 128:7320–7328
Seefeld K, Brause R, Häber T, Kleinermanns K (2007) J Phys Chem A 111:6217–6221
Lopes RP, Marques MPM, Valero R, Tomkinson J, De Carvalho LAEB (2012) Spectrosc Int J 27:273–292
Pluhařová E, Jungwirth P, Bradforth SE, Slavíček P (2011) J Phys Chem B 115:1294–1305
Alonso JL, Peña I, López JC, Vaquero V (2009) Angew Chem Int Ed 48:6141–6143
Watson JD, Crick FH (1953) Nature 171:737–738
Crick FHC (1966) J Mol Biol 19:548–555
Varani G, McClain WH (2000) EMBO Rep 1:18–23
Pedersen DB, Simard B, Martinez A, Moussatova A (2003) J Phys Chem A 107:6464–6469
Lippert B, Gupta D (2009) Dalton Trans 4619–4634
Eichhorn GL, Butzow JJ, Shin YA (1985) J Biosci 8:527–535
Müller J (2010) Metallomics 2:318–327
Noguera M, Bertran J, Sodupe M (2004) J Phys Chem A 108:333–341
Zhang Y, Huang K (2007) J Mol Struct (THEOCHEM) 812:51–62
Pelmenschikov A, Zilberberg I, Leszczynski J, Famulari A, Sironi M, Raimondi M (1999) Chem Phys Lett 314:496–500
Biver T (2013) Coord Chem Rev 257:2765–2783
Pyle AM (2002) J Biol Inorg Chem 7:679–690
Tereshko V, Minasov G, Egli M (1999) J Am Chem Soc 121:3590–3595
Timsit Y, Bombard S (2007) RNA 13:2098–2107
Varnai P, Zakrzewska K (2004) Nucleic Acids Res 32:4269–4280
Egli M (2002) Chem Biol 9:277–286
Burda JV, Sponer J, Hobza P (1996) J Phys Chem 100:7250–7255
Sychrovsky V, Sponer J, Hobza P (2004) J Am Chem Soc 126:663–672
Sponer JE, Sychrovsky V, Hobza P, Sponer J (2004) Phys Chem Chem Phys 6:2772–2780
Robertazzi A, Platts JA (2005) J Biol Inorg Chem 10:854–866
Baker ES, Manard MJ, Gidden J, Bowers MT (2005) J Phys Chem B 109:4808–4810
Poater J, Sodupe M, Bertran J, Solà M (2005) Mol Phys 103:163–173
Kabelac M, Hobza P (2006) J Phys Chem B 110:14515–14523
Bagchi S, Mandal D, Ghosh D, Das AK (2012) Chem Phys 400:108–117
Yu CY, Yu Y, Gong LD, Yang ZZ (2012) Theor Chem Acc 13:1098
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam MJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) GAUSSIAN 09 (Revision B.01) Gaussian Inc, Wallingford CT
Stasyuk OA, Szatylowicz H, Krygowski TM (2012) J Org Chem 77:4035–4045
Stasyuk OA, Szatylowicz H, Krygowski TM (2014) Org Biomol Chem 12:456–466
Stasyuk OA, Szatylowicz H, Krygowski TM (2014) Croat Chem Acta 87:335–342
Stasyuk OA, Szatylowicz H, Krygowski TM (2014) Org Biomol Chem 12:6476–6483
Krygowski TM, Szatylowicz H, Stasyuk OA, Dominikowska J, Palusiak M (2014) Chem Rev 114:6383–6422
Chen Z, Wannere CS, Corminboeuf C, Puchta R, Schleyer PVR (2005) Chem Rev 105:3842–3888
Kruszewski J, Krygowski TM (1972) Tetrahedron Lett 13:3839–3842
Krygowski TM (1993) J Chem Inf Comput Sci 33:70–78
Schleyer PVR, Maerker C, Dransfeld A, Jiao H, Hommes NJRVE (1996) J Am Chem Soc 118:6317–6318
Schleyer PVR, Manoharan M, Wang ZX, Kiran B, Jiao H, Puchta R, Hommes NJRVE (2001) Org Lett 3:2465–2468
Corminboeuf C, Heine T, Seifert G, Schleyer PVR, Weber J (2004) Phys Chem Chem Phys 6:273–276
Fallah-Bagher-Shaidaei H, Wannere CS, Corminboeuf C, Puchta R, Schleyer PVR (2006) Org Lett 8:863–866
Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F (2004) NBO 5.G Theoretical Chemistry Institute, University of Wisconsin, Madison WI
Boys SF, Bernardi F (1970) Mol Phys 19:553–566
Simon S, Duran M, Dannenberg JJ (1996) J Chem Phys 105:11024–11031
Szatylowicz H (2008) J Phys Org Chem 21:897–914
Gorb L, Kaczmarek A, Gorb A, Sadlej AJ, Leszczynski J (2005) J Phys Chem B 109:13770–13776
Shukla MK, Leszczynski J (2006) Chem Phys Lett 429:261–265
Yu LJ, Pang R, Tao S, Yang HT, Wu DY, Tian ZQ (2013) J Phys Chem A 117:4286–4296
Guille K, Clegg V (2006) Acta Cryst C62:o515–o517
Cyranski MK, Stepien BT, Krygowski TM (2000) Tetrahedron 56:9663–9667
Ciesielski A, Krygowski TM, Cyranski MK (2008) J Chem Inf Model 48:1358–1366
Steiner T (1998) J Phys Chem A 102:7041–7052
Gilli P, Pretto L, Bertolasi V, Gilli G (2009) Acc Chem Res 42:33–44
Acknowledgments
We thank the Foundation for Polish Science for supporting this work under MPD/2010/4 project “Towards Advanced Functional Materials and Novel Devices—Joint UW and WUT International PhD Programme” and the Interdisciplinary Center for Mathematical and Computational Modeling (Warsaw, Poland) for providing computer time and facilities.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to the memory of Professor Oleg V. Shishkin (1966–2014).
Electronic supplementary material
Below is the link to the electronic supplementary material.
11224_2015_605_MOESM1_ESM.pdf
Tables: Main characteristics for free guanine tautomers and their subunits (1S); NICS indices for guanine tautomers and their subunits (2S); Characteristics for H-bonded complexes of guanine tautomers and subunits (3S and 4S); Energetic characteristics for metal complexes of guanine tautomers and subunits (5S and 6S); Comparison of total interaction energies for studied complexes with metal ions (7S); HOMA indices for metal complexes of guanine tautomers (8S and 9S). Figures: Correlations between HOMA and NICS indices (1S), NBO charge distributions for guanine tautomers and subunits (2S and 3S), Structures of the strongest H-bonded complexes of guanine (4S), Resonance structure for N7,O∙∙∙M+ complex of g1 tautomer (5S) (PDF 694 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Stasyuk, O.A., Szatylowicz, H. & Krygowski, T.M. Aromaticity of H-bonded and metal complexes of guanine tautomers. Struct Chem 27, 111–118 (2016). https://doi.org/10.1007/s11224-015-0605-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-015-0605-9