
Abstract
This article presents a combined experimental and computational study of Ru(II) complex containing 2,5-pyridinedicarboxylic acid ligand. The novel complex [Ru(py-2,5-COOH)2(PPh3)2]·3H2O has been obtained in the reaction of [RuCl2(PPh3)3] with 2,5-pyridinedicarboxylic acid in methanol and has been studied by IR, 1H, 31P NMR, UV–Vis spectroscopy, and X-ray crystallography. The electronic structure of [Ru(py-2,5-COOH)2(PPh3)2] has been calculated with the density functional theory (DFT) method. The spin-allowed electronic transitions of the complex have been calculated with the time-dependent DFT method, and the UV–Vis spectrum has been discussed on this basis and rationalized by determination of ligand field splitting (10Dq) and Racah’s parameters from the experimental spectrum. The luminescence property of the complex has been examined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The coordination chemistry of ruthenium is a field of current growing interest from various viewpoints. The attention of scientists concentrates on synthetic aspects, structural, physicochemical properties, and reactivity. The pyridine derivative ligands have energetically low lying π-antibonding orbitals, which can accept electrons from filled d orbitals of metal atoms. In consequence, they can exhibit charge transfer bands with interesting spectroscopic properties in the visible region [1]. Ligands containing pyridine ring are wide studied and their π-donor properties are interesting. Their combination with other donor atoms should in principle afford complexes with tunable spectroscopic properties [2]. Furthermore, phosphine ruthenium(II) complexes with N-donor ligands are still of interest for their potential applications as well biological activity [3–9].
The azine ligands have energetically low lying π-antibonding orbitals, which can accept electrons from filled metal d orbitals. In consequence, they can exhibit charge transfer bands with interesting spectroscopic properties in the visible region [1]. Ligands containing pyridine ring are wide studied and their π-donor properties are interesting. Its combination with other donor atoms should in principle afford complexes with tunable spectroscopic properties [2, 10, 11].
Hence, synthesis and spectral characterization of new ruthenium complexes containing triphenylphosphine are of great importance. Earlier was published the complex with 2,3-pyridine dicarboxylic acid ligand in which triphenylphosphine ligands were in trans position [12].
In this communication, the 2,5-pyridinedicarboxylic acid was used as ligand, and in the obtained complex, ligands (PPh3, py-2,5-COOH) were in cis positions. The quantum chemical study included a characterization of the molecular and electronic structures of the complex by analysis of optimized molecular geometries, electronic populations using the natural bond orbitals scheme. The latter was used to identify the nature of the interactions between the ligands and the central ion. The calculated density-of-states (DOS) showed the interactions and influences the orbital composition in the frontier electronic structure. The time-dependent density functional theory (TD-DFT) was finally used to calculate the electronic absorption spectra. Based on a molecular orbital scheme, these results allowed the interpretation of the UV–Vis spectra obtained at an experimental level. The complex reported in this article combines the interest in ruthenium phosphine coordination compounds and complexes containing pyridine derivative ligands [13–19].
Experimental
All other reagents were commercially available and were used without further purification.
Synthesis of the [Ru(py-2,5-COOH)2(PPh3)2]·3H2O complex
[RuCl2(PPh3)3] (0.19 g, 0.2 mmol) and 2,5-pyridinedicarboxylic acid (0.06 g, 0.4 mmol) in CH3OH (80 cm3) were refluxed for 3 h. The starting material gradually dissolved and the color of the reaction solution became red-brown. The reaction solution was filtered, and the single crystals were obtained by slow evaporation of solvent. Yield 69%.
IR: 3455 νOH; 3058 νPhH; 1715 νCOOH; 1594 νCOO; 1562 νCN; 1481 δ(C–CH in the plane); 1433 νPh(P–Ph); 1352, 1286, 1274 νCOO; 1090 δ(C–CH in the plane); 799; 747 δ(C–C out of the plane); 691 δ(C–C in the plane); 521, 488 νP–Ph, Ru–N.
1H NMR (ν, CDCl3): 7.820–7.476 (m, PPh3/pyridine). 31P NMR (δ, CDCl3): 29.175 (s, PPh3), 22.183 (s, PPh3).
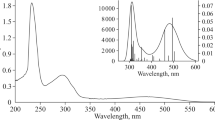
UV–Vis (methanol, λ [nm] (log ε)): 444 (1.14), 332 (1.97), 305 (sh), 265 (2.74), 212 (4.98).
Physical measurements
Infrared spectrum was Perkin Elmer FT-IR spectrophotometer in the spectral range 4000–450 cm−1 with the sample in the form of KBr pellet. Electronic spectrum was measured on a Lab Alliance UV–Vis 8500 spectrophotometer in the range of 600–180 nm in methanol solution. 1H and 31P NMR spectra were obtained at room temperature in CDCl3 using a Bruker 400 spectrometer. Luminescence measurement was made in methanolic solutions on an F-2500 FL spectrophotometer at room temperature.
DFT calculations
The calculations were carried out using the Gaussian09 [20] program. The DFT/B3LYP [21] method was used for the geometry optimization and electronic structure determination, and electronic spectra were calculated by the TD-DFT [22] method. The calculations were performed using a DZVP basis set [23] with f functions having exponents of 1.94722036 and 0.748930908 for ruthenium atom, and polarization functions for all other atoms: 6–31 g**—carbon, nitrogen and 6–31 g—hydrogen. The PCM (polarizable continuum model) solvent model [24] was used in the Gaussian calculations with methanol as the solvent. The contribution of a group to a molecular orbital was calculated using Mulliken population analysis. GaussSum 2.2 [25] was used to calculate group contributions to molecular orbitals and to prepare partial DOS spectra. The DOS spectra were created by convoluting the molecular orbital information with Gaussian curves of unit height and FWHM (Full Width at Half Maximum) of 0.3 eV.
Crystal structure determination and refinement
X-ray intensity data were collected with graphite monochromated Mo Kα radiation at temperature of 150.0(2)K, with ω scan mode. Lorentz, polarization, and empirical absorption correction using spherical harmonics implemented in SCALE3 ABSPACK scaling algorithm [CrysAlis RED, Oxford Diffraction Ltd., Version 1.171.29.2] were applied. The structure was solved by the Patterson method and subsequently completed by the difference Fourier recycling. All the non-hydrogen atoms were refined anisotropically using full-matrix, least-squares technique. All the hydrogen atoms were found from difference Fourier synthesis after four cycles of anisotropic refinement, and refined as “riding” on the adjacent carbon atom with individual isotropic temperature factor equal 1.2 times the value of equivalent temperature factor of the parent atom. The Olex2 [26] and SHELXS97, SHELXL97 [27] programs were used for all the calculations. Details concerning crystal data and refinement are gathered in Table 1.
Results and discussion
The complex was synthesized by a simple reaction between [RuCl2(PPh3)3] and chemically twofold quantities of the ligand in refluxed methanolic solution. In the IR spectrum of the complex, the ν(C–O) bands are found decreased scientifically compared to the free ligand and appeared in the 1594 and 1352–1274 cm−1 regions corresponding to the νas(C–O) and νs(CO) modes of the coordinated carboxylate moiety. This considerable difference between νas and νs is indicative of strong coordination of the carboxylate oxygen to the ruthenium(II) acceptor center. The presence of a band at the 1715 cm−1 indicates the carboxylic group not involved in coordination.
The complex crystallises in monoclinic P21/n space group in solvated form by three water molecules. Figure 1 presents the molecular structure of the complex, and the selected bond distances and angles are collected in Table 2. The structure can be can be considered as a distorted octahedral with the largest deviation from the expected 90° bond angles coming from the bite angle of 2,5-pyridinedicarboxylic acid. It equals to 78.16(9)° and 78.44(9)° for N(2)–Ru(1)–O(5) and N(1)–Ru(1)–O(1) angles, respectively. The angles are practically the same indicating identical binding of the two pyridine derivative ligands. The P(1)–Ru(1)–P(2) angle is greater than 90° (96.46(3)) which may be attributed to the steric interactions between bulky PPh3 ligands. The mutually cis position of triphenylphosphine ligands confirms two signals of the 31P NMR spectrum at 29.175 and 22.183 ppm. The bond length is normal and comparable with ruthenium(II) complexes with N-carboxylate-donor ligands. The C=O bonds of the coordinated moiety (1.243(4) Å) is slightly longer (~0.04 Å) and the C–O bonds (1.276(3) Å) is shorter (~0.02 Å) than the corresponding bonds length of the free carboxyl. In the structure, several weak inter- and intramolecular hydrogen bonds exist [28] collected in Table 3. The crystal packing with some of hydrogen bonds are presented on the Fig. 2. In the structure of the complex, some electronic interactions (π–π stacking) between PPh3 phenyl and pyridine dicarboxylic acid ring is visible. Figure 3 presents the alignment of centroids formed by pyridine and phosphine phenyl rings. The plane-to-plane distance between the #Ph(P) centroid, determined by C(33) to C(38) carbons, and pyridine ring is equal to 3.345 Å, and the angle between the between normal to #Ph(P) and #py is 9.97° indicating π–π stacking interaction.

Molecular structure of [Ru(py-2,5-COOH)2(PPh3)2]

The crystal packing of [Ru(py-2,5-COOH)2(PPh3)2]·3H2O complex

The π-stacking interactions in the [Ru(py-2,5-COOH)2(PPh3)2]·3H2O molecule
To obtain an insight in the electronic structures and bonding properties of the complex, calculations using the density functional theory (DFT) method with the B3LYP functional of GAUSSIAN-09 were carried out. Before the calculations, their geometries were optimized in singlet states using the DFT method with the B3LYP functional. In general, the predicted bond lengths and angles are in a good agreement with the values based on the X-ray crystal structure data, and the general trends observed in the experimental data are well reproduced in the calculations. The stabilization energies calculated in NBO [29] analysis have shown that the 2,5-pyridinedicarboxylic ligands donate the charge to ruthenium, and the stabilization energy is 353.70 kcal/mol, while the back donation is (Ru → py-2,5-COOH) 76.02 kcal/mol. The data suggest that the donation from ligands to d Ru orbitals plays a role in the electronic structure of the complex which can be seen in the natural atomic charge on the ruthenium central ion in the complex is −0.025.
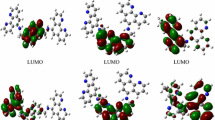
In the frontier region, neighboring orbitals may be a quasi-degeneracy of the energetic levels, and taking into consideration only the HOMO and LUMO may not yield a realistic description of the frontier orbitals. For this reason, the DOS and overlap population density-of-states (OPDOS) in terms of Mulliken population analysis were calculated using the GaussSum program. They provide a pictorial representation of MOs compositions and their contributions to chemical bonding. The DOS and OPDOS diagrams are shown in Fig. 4, and they may enable us to ascertain the bonding, nonbonding, and antibonding characteristics with respect to the particular fragments. A positive value in OPDOS plots means a bonding interaction, while a negative value represents antibonding interaction, and a near zero value indicates a non-bonding interaction. From the DOS plot of the complex, one can see that the d Ru orbitals play significant role in frontier HOMO orbitals with contribution of pyridine derivatives ligands. The frontier LUMOs are localized on the py-2,5-COOH ligands and in the higher virtual orbitals (LUMO + 4 to LUMO + 7) participate the d orbitals of ruthenium central ion. The interaction of ruthenium d orbitals with py-2,5-COOH ligands causes noticeable increases in energy levels of highest HOMO orbitals, which is reflected in the fact that the presence of PPh3 is visible at lower occupied orbitals (HOMO-4). Additionally, the change in energy levels of molecular orbitals refers to luminescent properties of the complex. On the OPDOS plot (inset in Fig. 4), the antibonding interaction between py-2,5-COOH ligands and ruthenium(II) central ion in the frontier HOMO and LUMO orbitals is visible. The values of the interaction and mentioned earlier stabilization energies indicate the ligand as a strong σ-donor and medium π-acceptor.

The density-of-states (DOS) and overlap DOSs (inset) diagrams for [Ru(py-2,5-COOH)2(PPh3)2] complex
Based on the pseudo-octahedral geometry of the studied complexes and taking into account the d–d transitions assigned to 1A1 → 1T1 and 1A1 → 1T2 in octahedron (or 1A1 → 1A2/B1/E in lower symmetry fields), the ligand field parameter 10Dq can be estimated to 23592 cm−1 for the complex. Adequately, Racah’s parameters are B = 475 cm−1; C = 1891 cm−1 and the nepheloauxetic parameter has value β55 = 0.66. The values of Racah parameters are in consistence with the calculated Mayer bond orders [30] pointing covalent character of the bonds between ruthenium and 2,5-pyridinedicarboxylic acid (Ru–N 0.8, Ru–O 0.9, Ru–P 1.6).
The electronic spectrum of the complex was calculated with the TDDFT method with methanol as solvent in the PCM. The longest wavelength experimental band (444 nm) originates in the H−1 → L+1 (92%) and H−2 → LUMO (97%) transitions. As the HOMO-1 an HOMO-2 are delocalized on central ion and py-2,5-COOH ligands, whereas the LUMO and LUMO + 1 are formed of π*-bonding orbitals of pyridine dicarboxylic ligand the transitions can be seen as a delocalized MLLCT (Metal–Ligand-to-Ligand CT) transitions. The same character can be assigned to the experimental absorption at 332 nm (d Ru → π *N,O-ligand H−3/−4 → LUMO (68%), H−2 → L+3 (91%) and d Ru → π *Ph H−2 → L+4 (41%), H−2 → L+5 (23%)). The experimental absorption band at 265 nm can be attributed to Metal–Ligand Charge Transfer transitions occurring from the d ruthenium orbitals to py-2,5-COOH and PPh3 ligands (H−1 → L+11 (23%), HOMO → L+10 (66%); H−2 → L+8 (81%); H−2 → L+9 (97%)). The highest experimental bands close to 212 nm may result from transitions in the PPh3 ligands and from π → π* excitations in the pyridine carboxylic ligands.
Emission property of the complex has been examined in the methanol solution (with concentration of 1·10−3 mol/dm3) at room temperature. Figure 5 presents the fluorescence 2D map and the plot of emission spectrum.

Fluorescence 2D map and spectrum of [Ru(py-2,5-COOH)2(PPh3)2] complex in methanolic solution
The excitations at 340 and 439 nm gave two emission peaks with maxima at 430 and 510 nm. The excitation at shorter wavelength gave much stronger emission (I 430/I 510 = 5.5:1). The emissions originating from the lowest energy metal-to-ligand charge transfer (MLCT) state, derived from the excitation involving a d π → π *ligand transition are observed. The assignment is supported by the analysis of the frontier orbitals of the complex showing a partial contribution of ligands nature. Moreover, the differences in the intensity of the fluorescence maxima can be associated with the higher share of the ligands (py-2,5-COOH and PPh3) the molecular orbitals involved in the transitions at 340 nm.
Summarizing, new ruthenium(II) complex with pyridine dicarboxylic acid ligands has been synthesized. The molecular structure of the complex is determined by X-ray, and the spectroscopic properties as infrared, 1H, 31P NMR spectra were studied. Based on the crystal structures, the computational studies were carried out in order to determine the electronic structure. The electronic spectrum was calculated with the use of TD-DFT method, and the transitions character was commented in connection with structure of molecular orbitals. Emission property of the complex has been examined. Emissions originating from the lowest energy metal-to-ligand charge transfer (MLCT) state, derived from the excitation involving a d π → π *ligand transition are observed. The assignment is supported by the analysis of the frontier orbitals of the corresponding complex showing a partial contribution of ligands nature.
Supplementary data
Supplementary data for C50H38N2O8P2Ru·3(H2O) is available from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK on request, quoting the deposition number 817684.
References
Chandra M, Sahay AN, Pandey DS, Puerta MC, Valerga P (2002) J Organomet Chem 648:39
Singh AK, Kumar P, Yadav M, Pandey DS (2010) J Organomet Chem 695:567
Kannan S, Sivagamasundari M, Ramesh R, Liu Yu (2008) J Organomet Chem 693:2251
Sivagamasundari M, Ramesh R (2007) Spectrochim Acta A 67:256
Sivagamasundari M, Ramesh R (2007) Spectrochim Acta A 66:427
Kaveri MV, Prabhakaran R, Karvembu R, Natarajan K (2005) Spectrochim Acta A 61:2915
Ulaganatha Raja M, Gowri N, Ramesh R (2010) Polyhedron 29:1175
Basu S, Halder S, Pal I, Samanta S, Karmakar P, Drew MGB, Bhattacharya S (2008) Polyhedron 27:2943
Al-Noaimi M, El-Barghouthi MI, El-khateeb M, Abdel-Rahman OS, Görls H, Crutchley RJ (2008) Polyhedron 27:2698
Arias M, Concepción J, Crivelli I, Delgadillo A, Díaz R, Francois A, Gajardo F, López R, Leiva AM, Loeb L (2006) Chem Phys 326:54
Henry W, Browne WR, Ronayne KL, O’Boyle NM, Vos JG, McGarvey JJ (2005) J Mol Struct 735–736:123
Sengupta P, Ghosh S, Mak TCW (2001) Polyhedron 20:975
Małecki JG, Kusz J (2007) J Coord Chem 60:2461
Małecki JG (2007) J Coord Chem 60:1949
Kruszyński R, Małecki JG (2007) Acta Cryst E 63:m3052
Małecki JG, Kruszyński R (2008) Struct Chem 19:63
Małecki JG, Kruszyński R, Kusz J, Tabak D, Mazurak Z (2008) Struct Chem 19:257
Małecki JG, Kruszyński R (2010) Polyhedron 29:1023
Małecki JG (2010) Polyhedron 29:1973
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.1. Gaussian, Inc., Wallingford, CT
Becke AD (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Eichkorn K, Weigend F, Treutler O, Ahlrichs R (1997) Theor Chim Acc 97:119
Tomasi J, Mennucci B, Cammi R (2005) Chem Rev 105:2999
O’Boyle NM, Tenderholt AL, Langner KM (2008) J Comput Chem 29:839
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Cryst 42:339
Sheldrick GM (2008) Acta Cryst 64:112
Desiraju GR, Steiner T (1999) The weak hydrogen bond in structural chemistry and biology. Oxford University Press, Oxford
Glendening ED, Reed AE, Carpenter JE, Weinhold F. NBO (version 3.1)
Tenderholt AL. QMForge, Version 2.1. Stanford University, Stanford, CA
Acknowledgments
The GAUSSIAN-09 calculations were carried out in the Wrocław Centre for Networking and Supercomputing, WCSS, Wrocław, Poland, http://www.wcss.wroc.pl, under calculational Grant No. 18.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Małecki, J.G. Synthesis, characterization, and molecular structure of Ru(II) complex containing 2,5-pyridinedicarboxylic acid. Struct Chem 23, 71–77 (2012). https://doi.org/10.1007/s11224-011-9842-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-011-9842-8