
Abstract
The crystal and molecular structures of nicotinohydrazide-derived hydrazones, N′-(2-furfurylidene)pyridine-3-carbohydrazide and N′-(1-(2-thienyl)ethylidene)pyridine-3-carbohydrazide, have been determined. The compounds have been also characterized by the solid state IR and UV–Vis spectroscopy. The hydrazones exist in the keto-amine form. According to the results of molecules quantum–mechanical calculations, performed for isolated molecules, this tautomeric form is energetically unfavorable but in the solid and solution states the observed intermolecular interactions support the presence of this form. The furan derivative crystallizes in two polymorphic forms with two molecules in an asymmetric unit regardless of the crystallization method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hydrazones comprise a class of compounds, which exhibit complex biological properties including antifungal, antitumor, antituberculosis, and anticonvulsant activities [1–4]. These compounds are often used as ligands in coordination chemistry and some of their metal complexes exhibit enhanced biological properties [5–8].
A particular class of hydrazones that has been studied extensively is the nicotinic acid hydrazide derivatives. The nicotinoylhydrazones can coordinate to the metal ions to produce stable metal complexes owing to their facile amide–imidic acid tautomerism. In general, these hydrazones exist in the amide form. The nicotinoylhydrazones molecules acting as a ligand undergo the proton transfer reaction and deprotonation, and in a consequence they coordinate to the central metal ions in the imidol form. Their coordination properties have been utilized for the treatment of Fe overload diseases [9, 10]. The nickel 2,6-diacetylpyridine-bis(nicotinoylhydrazone) has been reported to have antimicrobial and genotoxic activities [5]. The nicotinoylhydrazones also have applications in analytical chemistry because of high sensitivity and selectivity toward metal cations [11, 12].
The origin of the selectivity of these compounds, acting as biologically active molecules or analytical reagents, has not yet been identified. Knowledge of the structural peculiarities such as charge distribution, geometrical parameters, stereoelectronic properties, conformation flexibility is crucial to investigate the coordination properties and for designing new compounds. In this context, the synthesis of new nicotinoylhydrazone derivatives is of interest. Hence, the solid-state characterization of nicotinohydrazide-derived hydrazones: N′-(2-furfurylidene)pyridine-3-carbohydrazide and N′-(1-(2-thienyl)ethylidene)pyridine-3-carbohydrazide, as well as the results of quantum mechanical calculations, are reported here. The hydrazones were derived from the condensation reaction of nicotinohydrazide with furan-2-carbaldehyde and 2-acetylthiophene. These particular carbonyl compounds possessing the donor atoms were chosen to expand the coordination abilities of hydrazones.
Experimental
Synthesis
N′-(2-furfurylidene)pyridine-3-carbohydrazide and N′-(1-(2-thienyl)ethylidene)pyridine-3-carbohydrazide were prepared by one pot synthesis of the carbonyl compound with 3-nicotinohydrazide in methanol. A furan-2-carbaldehyde (5 mmol) was added to a hot solution of 3-nicotinohydrazide (5 mmol) in methanol (30 mL). The reaction mixture was heated under reflux for 2 h. The solution was then reduced by evaporation to half-volume and allowed to cool. After 3 days, yellow crystals of N′-(2-furfurylidene)pyridine-3-carbohydrazide (1A) were formed. Yield: 94%. A part of the product was dissolved in methanol–HCl solution (pH 2) and left to stand at 278 K. After 3 days, yellow crystals of N′-(2-furfurylidene)pyridinium-3-carbohydrazide chloride dihydrate (2) were formed. Since the ionic strength of a solution has effect on condensation reaction process, the reaction was repeated with addition of an inorganic salt and resulting ionic strength of 0.15 mol/L. The resulting product, N′-(2-furfurylidene)pyridine-3-carbohydrazide, was obtained with 95% yield, but hydrazone has crystallized in the polymorphic form (1B). N′-(1-(2-thienyl)ethylidene)pyridine-3-carbohydrazide (3) was synthesized by the same procedure as compound 1A only 2-acetylthiophene was used instead of furan-2-carbaldehyde. After 7 days, bright yellow crystals of 3 were formed. Yield: 91%. In case of 3, the ionic strength does not influence the yield or crystal form. Attempts to grow single crystals of the protonated form of 3 were unsuccessful.
X-ray crystallography
The crystals were mounted in turn on a KM-4-CCD automatic diffractometer equipped with CCD detector, and used for data collection. X-ray intensity data were collected with graphite monochromated MoKα radiation (λ = 0.71073 Å), with ω scan mode. All crystals used for data collection did not change their appearance. Lorentz, polarization, and numerical absorption [13] corrections were applied. The structures were solved by direct methods and subsequently completed by difference Fourier recycling. All the non-hydrogen atoms were refined anisotropically using full-matrix, least-squares technique on F 2. The hydrogen atoms were found by difference Fourier methods and treated as ‘riding’ on their parent non-hydrogen atoms and assigned isotropic displacement parameters equal to 1.5 (methyl groups and water molecules) or 1.2 (other atoms) times the value of equivalent displacement parameters of the parent atoms. The geometry of hydrogen atoms attached to carbon atoms was idealized after each cycle of least-squares refinement. SHELXS97, SHELXL97, and SHELXTL [14] programs were used for all the calculations. Details concerning crystal data and refinement are summarized in Table 1, selected bond lengths and bond angles are given in Table 2.
Theoretical calculations
The gas phase and solution geometry optimization was performed at the B3LYP/6-31++G(d,p) level of theory [15, 16] using the GAUSSIAN03 [17] program package. The geometric parameters were employed from crystal structure data. The optimized geometrical parameters were in agreement with those found from X-ray measurement within three standard deviations. Basis set superposition error (BSSE) corrections were carried out using the counterpoise (CP) method of Boys and Bernardi [18]. The rotation energy barrier was studied by calculations of the total energy value for partially optimized nicotinoylhydrazone molecule with fixed geometry of N1–C6–C1–C5 dihedral angle. The dihedral angles were changed by 20° in range from −180° to 180°, with the angle value from the crystal structure taken as the starting point. The solvent effect on the tautomerization was treated using the polarizable continuum model [19, 20]. The Gibbs free energy for systems in the solution were obtained by applying both, the complete basis set method (CBS-4M [21, 22]) and the computation of frequencies for the geometrically optimized molecules (at the DFT/B3LYP level of theory). Frequencies were used without scaling. The singlet excited-states, starting from the ground-state geometry optimized in the gas phase, were calculated with the time-dependent DFT method (TDDFT method [23–25]). Molecular orbitals were plotted using the Molekel program [26].
Spectroscopic measurements
The IR spectra (400–4000 cm−1) were recorded with samples prepared as KBr disks in a Magma 560 spectrophotometer. The UV–Vis spectra were recorded on a Jasco V-660 spectrophotometer, in transmission mode, for the solid-state samples pressed between two quartz plates.
Result and discussion
The description of the structures
The asymmetric unit of 1A contains two molecules of the nicotinohydrazide-derived hydrazone (A′: C1–C11/N1–N3/O1–O2 and A″: C51–C61/N51–N53/O51–O52). These molecules are joined to pairs by the N51–H51N···O1 and C52–H52···N2 hydrogen bonds (Table 3) forming N2R 22 (10) motif (Fig. 1). The energy of these interactions, with the BSSE correction, is equal to 8.38 kcal/mol, which is in accordance with the energies of intermolecular hydrogen bonds occurring in small molecules [27]. The recrystallization of 1A from a solution of different ionic strength gave polymorphic form 1B. The structural parameters of 1B have been recently reported, but with almost no description and without results of other, than structural, studies [28]. As 1A, this compound crystallizes with two molecules of the nicotinohydrazide-derived hydrazone (B′: C1–C11/N1–N3/O1–O2 and B″: C51–C61/N51–N53/O51–O52) per asymmetric unit (Fig. 2). In this compound, the asymmetric unit molecules interact by two strong N51–H51N···O1 and N51–H51N···N2 hydrogen bonds generating the N2R 21 (5) motif and by one additional, weak C52–H52···O1 hydrogen bond (Table 3). The energy of these attractive electrostatic interactions is equal to 8.03 kcal/mol. Although the number of bonding intermolecular interactions is higher, in comparison to 1A the total binding energy is slightly smaller. This is associated with the bigger directivity of hydrogen bonds and shorter bond lengths in 1A [29].

The molecular structure of 1A showing the atom and ring numbering scheme. The displacement ellipsoids are drawn at 50% probability level and H atoms are shown as spheres of arbitrary radii. Dashed line indicates the intermolecular hydrogen bond

The molecular structure of 1B showing the atom and ring numbering scheme. The displacement ellipsoids are drawn at 50% probability level and H atoms are shown as spheres of arbitrary radii. Dashed line indicates the intermolecular hydrogen bond
The hydrazone molecules of 1A are distorted from planarity: the dihedral angle between the weighted least-squares planes calculated through all non-H atoms of the pyridine and furan rings is 3.88 (13)° in A′ and 15.44 (10)° in A″. These two rings make angles of 13.77 (10)°, 9.89 (13)° and 16.08 (11)°, 0.67 (15)° (for A′ and A″, respectively) with the central aliphatic hydrazone moiety (O1–C6–N1–N2–C7). Two hydrazone molecules of 1A form an interplanar angle of 75.26 (3)°, whereas the molecules of 1B create an interplanar angle of 58.93 (2)° and are non-planar (dihedral angle between the weighted least-squares planes calculated through all non-H atoms of the pyridine and furan rings is 34.31 (7)° in B′ and 17.14 (14)° in B″). The non-H atoms other than the ring atoms are almost planar but the pyridine and furan rings are tilted from the plane comprising of O1, C6, N1, N2 and C7 atoms by 21.13 (9)°, 13.60 (7)° and 19.80 (14)°, 3.64 (18)°, in B′ and B″, respectively.
The hydrazone molecules of both compounds show the configuration E with respect to the azomethine bond. The bonds and angles observed in this structure are normal, but there are differences between the geometrical parameters of these molecules (Table 2). The C6–O1, C56–O51 and C7–N2, C57–N52 bonds show a typical double bond character. The search of the Cambridge Structural Database [30] provided 511 crystal structures containing the benzoyl/nicotinoyl hydrazone moiety. The average C=O, C–N, N–N, and C=N distance from 569 hits is 1.226 (11), 1.351 (13), 1.380 (10) and 1.278 (11) Å, respectively.
The crystal structures of 1A and 1B are stabilized by the hydrogen bonds (Table 3), as well as π···π stacking and C–H···π interactions but, as expected, for each polymorphic form the non-covalent binding mode is different. The hydrazone molecules of 1A are connected by the N–H···O/N and C–H···N hydrogen bonds into a tetramer (Fig. 3). Such an orientation favors the π···π stacking interactions between the five- and six-membered heterocyclic rings of adjacent, symmetry related hydrazone molecules. The stacking molecules of A′ (related by symmetry transformation: −x + 1, −y + 1, −z) are almost parallel [inclined at 3.89 (11)°] and oriented in opposite directions with the distance of 3.791 (2) Å. The tetramers are linked via weak C–H···π contacts (C3–H3···ring#1 (#1: O52, C58–C61) with C···ring#1–centroid distance of 3.625 Å, C–H···ring#1–centroid angle of 135.5°) to form a chain of tetramers propagating along the [001] axis. Additionally, π···π stacking interactions between molecules of A″ and C60–H60···ring#2 (#2: O2, C8-C11) contacts with C···ring#2–centroid distance of 3.831 Å, C–H···ring#2–centroid angle of 148.9° provide some linkage between the tetramers. The stacking molecules of A″ [inclined at 14.45 (12)°] are slightly shifted [symmetry codes: (x − 1, y, z)] with the ring fragments nearly fully overlapping at an interplanar distance of 3.862 (2) Å.

A part of the packing of molecules in 1A. Dashed lines indicate hydrogen bonds
The molecules of second polymorphic form are linked by the N–H···O/N and C–H···N hydrogen bonds to the hydrogen-bonded chain extending along the [001] axis (Fig. 4). The chains are connected by weak π···π stacking interactions (along the [100] axis) between the parallel five- and six-membered heterocyclic rings of adjacent hydrazone molecules B″ oriented in opposite directions [one of the molecules was obtained by symmetry translation: (x − 1, y, z)]. The perpendicular distance between the first ring centroid and that of the second ring is 3.867 (2) Å, and the angle between the vector linking the ring centroids and the normal to the five-membered ring plane is 18.42 (12)°. Moreover, there are weak intermolecular C–H···π contacts: C53–H53···ring#1, C54–H54···ring#1 (#1: O2, C8–C11), C52–H52···ring#2 (#2: O52, C58–C61) with C···ring–centroid distance of 2.877, 3.234, 3.344 Å, respectively, and C–H···ring–centroid angle of 137.9, 119.5, 85.6°, respectively.

A part of the packing of molecules in 1B. Dashed lines indicate hydrogen bonds
In contrast to the structures of the neutral hydrazones, 1A and 1B, the asymmetric unit of 2 consists of one protonated hydrazone molecule, one chloride ion, and two water molecules (Fig. 5). The conformation of the protonated hydrazone is also different. The pyridyl and the imine nitrogen atoms are situated on the same side of the molecule, i.e. they are in a syn position. For a protonated hydrazone molecule, the rotation energy barrier (defined as the difference of energy at the most favorable and the most unfavorable conformation) is equal to 7 kcal/mol (Fig. 6). As expected, energetically the most unfavorable conformation is the one that has the pyridine ring almost perpendicular to the central aliphatic hydrazone moiety. For neutral hydrazone molecule the rotation energy barrier is 4.5 kcal/mol. These relatively small energy values suggest that nicotinohydrazide-derived hydrazones can exist in two conformations depending on the presence of other donor/acceptor atoms.

The molecular structure of 2 showing the atom and ring numbering scheme. The displacement ellipsoids are drawn at 50% probability level and H atoms are shown as spheres of arbitrary radii

The rotation energy barrier diagram for the furan derivative (1) (a) and its protonated form (2) (b)
Owing to the presence of other hydrogen bond receptor/donor species, namely water molecules and chloride ions, the hydrazone chain/tetramer structure is not retained in 2. Cations, anions, and neutral molecules of 2 lying in the [−120] plane are assembled into a chain by extensive hydrogen bonds, involving the carbonyl oxygen atom, the imine nitrogen atom, the chloride ion (acting as acceptors of hydrogen bonds), the aminic group, the pyridinium ring and the water molecule (acting as donors of hydrogen bonds). The chains are interlinked by the additional hydrogen bonds formed between the water molecules, the weak π···π stacking interactions between the furan and pyridinium rings of adjacent protonated organic molecules (with the separation distance of 3.907 Å and the angle between the vector linking the one ring centroid and the normal to the second ring plane of 20.6(2)°), and the C2–H2···π interactions with the furan rings as the π-acceptor (with H···ring–centroid distance of 3.095 Å, C···ring–centroid distance of 3.262 Å, C–H···ring–centroid angle of 92.0°). Noteworthy is the fact that the one water molecule inhabits the position typically occupied by a coordinated metal ion, and, in the presented case, the hydrogen bonds replace the coordination bonds. The furan oxygen atom does not take part in forming hydrogen bonds.
For two polymorphic forms of compound 1 the asymmetric unit contains two molecules regardless of the crystallization method. This can suggest that this form is the stable one. The tendency of –C(O)–NH–N= moiety to form strong bifurcated hydrogen bonds competes with the steric hindrance between the hydrazone molecules containing two rings causing such a twist of the molecules that they can not be related by crystallographic symmetry element, and hence leading to structures where the asymmetric unit contains more than one molecule (i.e., Z′ > 1) [31].
It is worth mentioning that nicotinohydrazide-derived hydrazones display two tautomeric forms. In the pure state they exist mainly in the keto-amine form whereas in the coordination compounds they exist as the enol-imine tautomer. The quantum–mechanical calculations performed for the isolated molecule of 1 show that the compound is less stable in the keto-amine form (found in the crystal structures of 1) (1) than in the enol-imine form (2) (Fig. 7). The proton transfer from form 2 to 1 is an endothermic process with the enthalpy change of only 1.9 kcal/mol. This can explain both the change of tautomeric form during complexation reaction and the existence of two symmetry independent molecules in the crystal structure. The presence of an intramolecular O–H···N hydrogen bond helps stabilize the enol-imine tautomer but it makes forming strong hydrogen bonds by the nitrogen atoms of hydrazone moiety difficult. Thus, the hydrazone molecules exist in the energetically less stable keto-amine form in the solid state because of the possibility of forming the strong hydrogen bonds, which in turn determine the packing.

The tautomeric forms of nicotinohydrazide-derived hydrazone
In the case of 2-thienyl derivative of nicotinohydrazide-derived hydrazone (3) the asymmetric unit contains one molecule (Fig. 8). Owing to the steric hindrance resulting from the presence of the methyl group at the imine carbon atom, the NH group of a hydrazone moiety is able to form strongly limited number of hydrogen bonds. The molecules form chains, along the [010] axis, connected by the N1–H1N···O1, C12–H12A···O1 hydrogen bonds (Fig. 9) and C4–H4···π interactions with the pyridine rings as the π-acceptor (the H···ring–centroid distance is 3.256 Å, the C···ring–centroid distance is 3.729 Å, and the C–H···ring–centroid angle is 106.5°). The strong C12–H12C···π interactions with the furan rings as the π-acceptor (the H···ring–centroid distance is 2.822 Å, the C···ring–centroid distance is 3.593 Å, and the C–H···ring–centroid angle is 138.0°) provide linkage between the chains. There are no π···π stacking interactions in the structure of 3.

The molecular structure of 3 showing the atom and ring numbering scheme. The displacement ellipsoids are drawn at 50% probability level and H atoms are shown as spheres of arbitrary radii

A part of the packing of molecules in 3. Dashed lines indicate hydrogen bonds
The hydrazone molecule of 3 exists as the keto-amine tautomer with the configuration E across the azomethine bond. As for 1, the enol-imine tautomeric form is energetically more favorable. The difference between the energy of the hydrazone molecule in these two forms is equal to 3.04 kcal/mol, with preferred enol-imine form. In this tautomeric form the repulsion between positive charges of hydrogen atoms of methyl, pyridine, and amine groups does not occur. In contrast to the furan derivative of hydrazone, in the thienyl derivative the pyridyl and imine nitrogen atoms are in a syn position. This compound acting as a ligand can coordinate in the tridentate mode.
All above mentioned quantum–mechanical calculations were performed for the isolated molecules in the gas phase. Since solvent has an effect on the tautomeric equilibrium, the energy of each tautomeric form (of 1 and 3) was calculated using the self-consistent reaction field (SCRF) approach, with methanol as a solvent. This polar solvent was used because the obtained nicotinohydrazide-derived hydrazones are insoluble in water, but soluble in organic solvents such as alcohols or DMF. The effect of solute–solvent interactions is bigger in the case of the furan derivative of nicotinohydrazide-derived hydrazone (1) than for thienyl derivative (3), which can result from the possibility of forming stronger O–H···O/N hydrogen bonds in 1. The thienyl derivative of nicotinohydrazide-derived hydrazone can interact with solvent molecules by O–H···O/N hydrogen bonds and by weaker O–H···S hydrogen bonds. Both the total energy and Gibbs free energy values (Table 4) suggest that the keto-amine form of nicotinohydrazide-derived hydrazones is the stable one in the solution and the solid state, in opposition to the gas phase. The methanol molecules assist in stabilizing this tautomeric form what explains the importance of the solute–solvent interactions during crystallization process.
Spectroscopic studies
The vibrational analysis was carried out for both polymorphic forms of 1 and its sulfur-containing derivative (3). Most of the vibrational modes appear to be strongly mixed and only the predominant contributions are indicated in Table 5. The IR spectra of 1A and 1B are similar, the differences are observed only in the hydrogen-stretching region. The spectra contain characteristic bands of the stretching and bending vibrations of the aromatic CC, C=O, C=N and CH groups. An important spectral feature that can be used to distinguish polymorphic forms of 1 is the N–H stretching vibration that typically occurs between 3200 and 3500 cm−1. This frequency displays remarkable bathochromic shift when the N–H group is involved in hydrogen bonding. The N–H stretching vibrations appear as a broad band at 3310–3550 cm−1 with a pronounced shoulder at 3260 cm−1 for 1A, and as a split band at 3210 and 3260 cm−1 for 1B. In both forms, the N–H group acts as a donor of hydrogen bonds, but the observed lowering and splitting of frequency for second polymorphic form is caused by the presence of bifurcated hydrogen bonds formed by this group. The most significant differences between vibrational frequencies of 1 and 3 were observed for the vibrations of five-membered ring. The IR spectrum of 3 showed a strong band at 730 cm−1 originating from the stretching vibrations of C–S bonds whereas the medium intensity band at 910 cm−1 can be attributed to vibrations of C–CH3 group. As for 1A, the vibration of N–H group of 3 appears as the broad band at 3280–3550 cm−1.
The UV/Vis spectra were recorded for the solid-state samples, thus no solvent effects were involved. The spectra of 1A and 1B are almost identical and each exhibits two absorption bands. The bands located at 285 and 360 nm can be attributed to π → π* transitions. The solid-state spectrum of compound 3 is more complex, it contains three partially overlapping absorption maxima. The first maximum located at 250 nm is typical for π → π* transitions in organic molecules. The broad band centered at 290 nm with a shoulder, although significantly less pronounced, at 340 nm is also due to π → π* transitions. Since the π-electron density in such conjugated systems is delocalized, the energy of π → π* transitions is lower, thus the absorption bands are shifted to a longer wavelength.
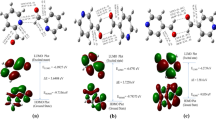
The quantum mechanical calculations were performed for an isolated nicotinohydrazide-derived hydrazone molecule to examine the nature of transitions. Since the experimental spectra were measured for compounds in the solid state, the calculations were carried out for the isolated molecules in the gas phase. The experimental and calculated electronic spectra are compared in Fig. 10. Each calculated excited state was represented by the Lorentzian function with the scale parameter of 5, for 1 and 3. Generally, in the computed UV–Vis spectra the bands are blue shifted. The π → π* transitions observed in the solid-state spectra have decreased energy in comparison to those observed in the calculated spectra, due to creation of non-covalent interactions. Experimental and theoretical data are summarized in Table 6.

The experimental (solid line) and calculated (dashed line) electronic absorption spectra of compounds: 1A (a), 1B (b), 3 (c)
The selected contours of occupied and unoccupied molecular orbitals are presented in Fig. 11. The frontier orbitals of 1 and 3 are similar. The HOMO shows a bonding interaction between the p orbitals from the carbon atoms of the five-membered heterocyclic ring, the imine bond atoms, and it is antibonding with respect to the N1–C6 bond. Energetically lower, the HOMO-4, and HOMO-6 of 1 are mainly localized on the furan and pyridine rings, respectively. The HOMO-1 and HOMO-3 of 3 are similar in shape to HOMOs of 1, they are ring-based with a small contribution from the carbonyl oxygen atom orbitals. The HOMO-4 and HOMO-5 are more delocalized. All LUMOs of 1 and 3 are mainly pyridine ring-based. The LUMO has also contribution from the p orbitals from the C1 atom and imine carbon atom.

Selected orbitals for 1 and 3
Conclusions
The obtained nicotinohydrazide-derived hydrazones exist in the keto-amine form. According to isolated molecules quantum–mechanical calculations results this tautomeric form is energetically unfavorable but in the solid and solution states the observed intermolecular interactions support the presence of this form. The furan derivative crystallizes in the two polymorphic forms with two molecules in the asymmetric unit regardless of the crystallization method, probably due to existence of strong directional hydrogen bonds linking the molecules of both polymorphs. The electronic spectra, calculated by time dependent density functional theory (TD-DFT) method, are blue shifted in comparison to the experimental ones due to lack of non-covalent interactions. The analysis of molecular orbital coefficients showed that the electronic transitions originate mainly from the π → π* transitions.
Supplementary data
CCDC-787761 (1A), 787764 (1B), 787762 (2) and CCDC-787763 (3) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44(0)1223-336033; email: deposit@ccdc.cam.ac.uk].
References
Horiuchi T, Chiba J, Uoto K, Soga T (2009) Bioorg Med Chem Lett 19:305
Chimenti F, Bizzarri B, Bolasco A, Secci D, Chimenti P, Carradori S, Granese A, Rivanera D, Frishberg N, Bordón C, Jones-Brando L (2009) J Med Chem 52:4574
Beraldo H, Gambino D (2004) Mini-Rev Med Chem 4:31
Carvalho SA, da Silva EF, Santa-Rita RM, de Castro SL, Fraga CA (2004) Bioorg Med Chem Lett 14:5967
Bottari B, Maccari R, Monforte F, Ottana R, Vigorita MG, Bruno G, Nicol F, Rotondo A, Rotondo E (2001) Bioorg Med Chem 9:2203
Visbal G, Marchán E, Maldonado A, Simoni Z, Navarro M (2008) J Inorg Biochem 102:547
Singh VP, Katiyar A, Singh S (2008) Biometals 21:491
Avaji PG, Kumar CHV, Patil SA, Shivananda KN, Nagaraju C (2009) Eur J Med Chem 44:3552
Bernhardt PV, Chin P, Sharpe PC, Richardson DR (2007) Dalton Trans 3232
Kalinowski DS, Sharpe PC, Bernhardt PV, Richardson DR (2008) J Med Chem 51:331
Sivaramaiah S, Reddy PR (2005) J Anal Chem 60:828
Szabó K, Marek N (2002) J Biochem Biophys Methods 53:189
Cie STOE (1999) X-RED, version 1.18. STOE & Cie GmbH, Darmstadt, Germany
Sheldrick GM (2008) Acta Cryst A64:112
Becke AD (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B37:785
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision E.01. Gaussian, Inc., Pittsburgh, PA
Boys SF, Bernardi F (1970) Mol Phys 19:553
Miertu S, Scrocco E, Tomasi J (1981) Chem Phys 55:117
Cossi M, Barone V, Cammi R, Tomasi J (1996) Chem Phys Lett 255:327
Ochterski JW, Petersson GA, Montgomery JA Jr (1996) J Chem Phys 104:2598
Montgomery JA Jr, Frisch MJ, Ochterski JW, Petersson GA (2000) J Chem Phys 112:6532
Runge E, Gross EKU (1984) Phys Rev Lett 52:997
Casida ME, Jamorski C, Casida KC, Salahub DR (1998) J Chem Phys 108:4439
Bauernschmitt R, Ahlrichs R (1996) Chem Phys Lett 256:454
Varetto U (2009) MOLEKEL, version 5.4.0.8. Swiss National Supercomputing Centre, Manno, Switzerland
Desiraju GR, Steiner T (1999) The weak hydrogen bond in structural chemistry and biology. Oxford University Press, New York
Qian H, Yin Z, Zhang C, Yao Z (2009) Acta Cryst E65:o2260
Kruszynski R (2008) Central Eur J Chem 6:542
Allen FH (2002) Acta Cryst B58:380
Anderson KM, Goeta AE, Steed JW (2008) Cryst Growth Des 8:2517
Acknowledgments
This work was financed by funds allocated by the Ministry of Science and Higher Education to the Institute of General and Ecological Chemistry, Technical University of Lodz, Poland (Grant no. IP 2010 043770). The GAUSSIAN03 calculations were carried out in the Academic Computer Centre CYFRONET of the AGH University of Science and Technology in Cracow, Poland (Grant No.: MNiSW/SGI3700/PŁódzka/040/2008).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Trzesowska-Kruszynska, A. Exploring the structural, electronic and optical properties of furan-2-carboxaldehyde and 2-acetylthiophene nicotinoylhydrazone. Struct Chem 22, 525–535 (2011). https://doi.org/10.1007/s11224-010-9717-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-010-9717-4