
Abstract
Mitochondrial fatty acid oxidation is an essential pathway for energy production, especially during prolonged fasting and sub-maximal exercise. Long-chain fatty acids are the most abundant fatty acids in the human diet and in body stores, and more than 15 enzymes are involved in long-chain fatty acid oxidation. Pathogenic mutations in genes encoding these enzymes result in a long-chain fatty acid oxidation disorder in which the energy homeostasis is compromised and long-chain acylcarnitines accumulate. Symptoms arise or exacerbate during catabolic situations, such as fasting, illness and (endurance) exercise. The clinical spectrum is very heterogeneous, ranging from hypoketotic hypoglycemia, liver dysfunction, rhabdomyolysis, cardiomyopathy and early demise. With the introduction of several of the long-chain fatty acid oxidation disorders (lcFAOD) in newborn screening panels, also asymptomatic individuals with a lcFAOD are identified. However, despite early diagnosis and dietary therapy, a significant number of patients still develop symptoms emphasizing the need for individualized treatment strategies. This review aims to function as a comprehensive reference for clinical and laboratory findings for clinicians who are confronted with pediatric and adult patients with a possible diagnosis of a lcFAOD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hypoglycemia is a common, and potentially dangerous, sign in medical practice, with an extensive number of etiologies, especially in the neonatal period. When hypoglycemia is accompanied by a low concentration, or even absence, of ketone bodies in plasma and urine, hyperinsulinism and disorders of carnitine transport and mitochondrial fatty acid oxidation, including long-chain fatty acid oxidation disorders (lcFAODs), may be involved as cause. Plasma levels of free fatty acids (FFAs) are low in hyperinsulinism due to a suppression of lipolysis, while FFAs are generally high during hypoglycemia caused by lcFAODs. In lcFAODs, FFAs are mobilized during fasting but cannot be oxidized due to an autosomal recessive inherited deficiency of one of the mitochondrial ß-oxidation enzymes. Hypoglycemia, however, is only one of the signs in lcFAODs. Before lcFAODs were included in newborn screening (NBS) panels, a considerable number of patients was diagnosed in adulthood and these patients typically presented with different clinical presentations compared to patients who presented during childhood. Following the inclusion of lcFAODs in NBS panels in many countries worldwide, the number of patients diagnosed has significantly increased and the clinical spectrum has expanded, necessitating new algorithms for choosing the optimal therapeutic strategy. This review aims to serve as a comprehensive reference for clinical and laboratory findings for those who are confronted with pediatric and adult patients with a possible diagnosis of a mitochondrial lcFAOD.
2 Mitochondrial long-chain fatty acid oxidation
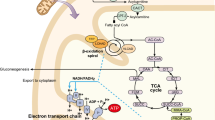
Mitochondrial fatty acid oxidation is an important pathway for maintaining energy homeostasis. Especially during fasting, when glucose and glycogen stores are low, fatty acid oxidation (FAO) is a significant source of energy provision in the form of adenosine triphosphate (ATP). The majority of fatty acids are stored in the body, in particular in adipose tissue, as long-chain triglycerides (LCT). As blood glucose levels drop, triglycerides in adipose tissue are hydrolyzed and free fatty acids (FFAs) are mobilized in the blood stream and become available for other tissues [1, 2]. The FFAs enter the cell via fatty acid transport systems (FAT/CD36) (Fig. 1). Before degradation or elongation can take place, FFAs need to be activated to acyl-coenzyme A (-CoA) esters by one of a variety of different chain-length-specific synthetases localized in distinct subcellular compartments [3]. Medium-chain fatty acids can freely diffuse into mitochondria, whereas long-chain acyl-CoAs cannot. Instead, long-chain fatty acids are esterified with carnitine in the cytoplasm and then are transported by the carnitine shuttle. In this shuttle, the CoA group is first exchanged for carnitine in a reaction catalyzed by carnitine palmitoyltransferase 1 (CPT1), forming acylcarnitine. CPT1 is an important regulator of FAO as it is sensitive for inhibition by malonyl-CoA, formed by carboxylation of the product of fatty acid oxidation, acetyl-CoA. The acylcarnitines are imported into the mitochondrion by carnitine acylcarnitine translocase (CACT) in exchange for a free carnitine molecule. Next, the intramitochondrial acylcarnitines are reconverted to acyl-CoAs by carnitine palmitoyltransferase 2 (CPT2). The mitochondrial acyl-CoAs can subsequently be used for energy production that occurs via degradation by a series of enzymes with different chain-length specificity. The first step concerns the dehydrogenation of acyl-CoAs by different acyl-CoA dehydrogenases. In humans, the key enzymes catalyzing this step for the different chain lengths of fatty acids are very long-chain acyl-CoA dehydrogenase (VLCAD), medium-chain acyl-CoA dehydrogenase (MCAD) and short-chain acyl-CoA dehydrogenase (SCAD) [4]. For long-chain acyl-CoAs (chain length C14-C18), VLCAD is the main enzyme [5]. In rodents, the enzyme long-chain acyl-CoA dehydrogenase (LCAD) can take over part of the VLCAD function, but in humans LCAD makes little contribution to lcFAO [6]. Indeed, nine patients that were originally reported as LCAD-deficient, diagnosed by a reduction in palmitoyl-CoA degradation [7], were reported to have normal LCAD protein levels on western blot [8] and upon subsequent analyses many were in fact VLCAD-deficient [9]. Following the initial dehydrogenation, the resulting long-chain enoyl-CoAs undergo hydration, a second dehydrogenation step, and finally thiolytic cleavage catalyzed by mitochondrial trifunctional protein (MTP) [10]. For degradation of fatty acids with shorter chain length mitochondria have different enzymes which are outside the scope of this review but are described elsewhere [11]. The MTP enzyme complex harbors activity for (a) long-chain enoyl-CoA hydratase (LCEH), (b) long-chain-(S)-3-hydroxyacyl-CoA-dehydrogenase (LCHAD), and (c) long-chain-3-ketoacyl-CoA thiolase (LCKAT). These concerted activities lead to the formation of a molecule of acetyl-CoA and a shortened acyl-CoA with two carbons less. The acetyl-CoA can enter the tricarboxylic acid (TCA) cycle for production of reducing equivalents for oxidative phosphorylation resulting in ATP production, or may enter the ketone body synthesis pathway in the liver. The heart mostly depends on FAO (60–90%), even when glucose levels are high [12]. In contrast, the brain mostly relies on the oxidation of glucose and ketone bodies. However, there is evidence of FAO in astrocytes derived from rat brain and expression of FAO enzymes in other parts of the rat brain as well as in neural cells from human fetuses [13,14,15].

Schematic representation of mitochondrial fatty acid oxidation in humans. Long-chain fatty acids enter the cell from the bloodstream and enter the mitochondria through the carnitine shuttle, followed by a step-wise degradation involving a series of enzymes of the long-chain fatty acid oxidation machinery resulting in the production of acetyl-CoA. Potential treatments to produce acetyl-CoA independent of the fatty acid oxidation enzymes are indicated. These include medium-chain triglycerides, ketone bodies and triheptanoin. Abbreviations: CI-V, Complex I-V; CACT, Carnitine Acylcarnitine Translocase; CD36, Cluster of Differentiation 36; CoA, Coenzyme A; CPT1, Carnitine Palmitoyl Transferase type 1; CPT2, Carnitine Palmitoyl Transferase type 2; FABPpm, plasma membrane-associated Fatty Acid Binding Protein; FATP, Fatty Acid Transport Protein; LCEH, Long-Chain Enoyl-CoA Hydratase; LCHAD, Long-Chain 3-Hydroxyacyl-CoA-Dehydrogenase; LCKAT, Long-Chain Ketoacyl-CoA Thiolase; MCAD, Medium-Chain Acyl-CoA Dehydrogenase; MCKAT, Medium-Chain 3-Ketoacyl-CoA Thiolase; OCTN2, Organic Cation Transporter 2; SCAD, Short-Chain Acyl-CoA Dehydrogenase; SCHAD, Short-Chain 3-hydroxyacyl-CoA Dehydrogenase; T2, acetoacetyl-CoA thiolase
3 Clinical signs and symptoms
Inherited disorders for many of the enzymes of the FAO cascade and carnitine shuttle have been described in the past decades [16,17,18,19]. These disorders give rise to a variety of clinical features especially in organs that rely on energy production by FAO, such as the heart, skeletal muscle and liver. Signs and symptoms can manifest as early as a few hours after birth, but may also only appear during adulthood, with a variation in clinical severity between patients even within the same family [20,21,22,23,24,25,26]. Patients with severe phenotypes can present in infancy with hypoglycemia and lactic acidosis. In rare cases, patients also have low levels of ketones, which might therefore be misinterpreted as respiratory-chain defects [27].
The clinical presentation of lcFAODs is aspecific, but a characteristic feature of the signs and symptoms in lcFAODs is the fact that they can be provoked, or aggravated, by energy requiring states such as fasting, prolonged exercise, illness, fever or a combination of these factors. The most common clinical presentations in lcFAODs are hypoketotic hypoglycemia, cardiomyopathy and myopathy. These presentations occur in combination but also isolated:
-
Hypoketotic hypoglycemia, sometimes resulting in convulsions, coma and brain damage, is most often seen in infancy and the early years of childhood. Hypoketotic hypoglycemia is often observed in combination with hepatomegaly, elevated transaminases, and hyperammonemia. Hepatic dysfunction is described in patients during episodes of metabolic derangement [28] and can be fatal. Chronic liver dysfunction is uncommon [23,24,25].
-
Cardiomyopathy, is most often seen in the neonatal period or early childhood, but can also develop under catabolic conditions like fasting or illness later in life. The outcome can be fatal, especially in infancy. Hypertrophy of the left ventricular wall can be observed initially, which can progress to dilated cardiomyopathy in advanced disease with reduced ejection fraction. Sometimes cardiomyopathy is accompanied by pericardial effusion. Remarkably, the cardiomyopathy can be completely reversed after treatment with medium-chain triglycerides (MCT) or the odd-chain triglyceride triheptanoin [29, 30]. Rhythm disturbances have also been reported in all lcFAODs except CPT1A deficiency, and can occur with, but also without, the presence of cardiomyopathy [20, 31].
-
Myopathy, by which we refer to any muscular sign or symptom, are common in lcFAOD patients. Patients may present with fatigue as myalgia, muscle weakness, exercise intolerance, myoglobinuria and recurrent rhabdomyolysis. Myopathy typically starts in puberty or adulthood, but can be observed at a younger age. Often myopathy is provoked by endurance-type exercise [23,24,25] but can also be seen after anesthesia or viral illness.
Distinctive features of the separate lcFAOD are discussed in Section 5.
4 Diagnostic approach for long-chain fatty acid oxidation disorders
4.1 Laboratory testing
When a patient is suspected of having an lcFAOD based on clinical signs and symptoms, the first laboratory test of choice is measurement of acylcarnitine levels in blood. It is assumed that the level of acylcarnitines in blood mirrors the accumulating acyl-CoA species inside mitochondria which cause the carnitine shuttle to operate in the reverse fashion [32]. CPT2 converts accumulating intramitochondrial acyl-CoAs to acylcarnitines. These are then transported back to the cytoplasm via CACT and out of the cell into the blood (Fig. 1) [33]. With the development of highly sensitive techniques based on mass spectrometry to determine acylcarnitine profiles in blood, urine or dried blood spots (DBS), a fast screening method for lcFAOD has become available [34, 35]. Abnormalities in the acylcarnitine profile are more pronounced during times of metabolic derangement but the acylcarnitine profile can be normal in patients when they are well [36,37,38,39]. This complicates the diagnosis, since false negative results can occur [40]. In addition to analysis in blood and urine, acylcarnitine profiling can also be done in cultured fibroblasts after a fatty acid loading test [41]. Most of the lcFAODs result in a distinctive acylcarnitine profile depending on which of the lcFAO enzymes is deficient (Table 1). Overall lcFAO activity can also be measured in fibroblasts; a whole cell assay, using radiolabeled fatty acids as a substrate [48, 49]. In addition, the activity of specific enzymes can be measured in lymphocytes or in skin fibroblasts, as these cell types express all lcFAO enzymes [5]. After confirmation of the enzyme deficiency, mutation analysis can uncover the underlying molecular defect in the gene coding for the mitochondrial lcFAO protein [50]. With the rapid development of quicker and low-cost sequencing tools, it is tempting to directly proceed to gene panel analysis or targeted sequencing after finding an abnormal acylcarnitine profile. Especially since the enzymatic assays are costly, laborious and only performed in a limited number of centers. It is likely that indeed genetic testing will become the first step in the diagnostic process. However, regularly new mutations and variants of unknown significance (VUS) are identified, requiring functional studies such as enzyme activity measurements or cellular flux analysis for confirmation of the diagnosis and characterization [51]. In addition, prognostic markers are still needed to predict outcome and define personalized treatment strategies [52].
4.2 Newborn screening
LcFAODs are included in NBS panels in many countries, enabling prompt treatment and precautions to avoid catabolism [53, 54]. As described above, all lcFAODs can be identified by specific acylcarnitine profiles in DBS [43, 55] (Table 1). The use of DBS for screening has several advantages over screening in plasma: only a minor amount of blood is required, and it can be easily collected, shipped and stored. Cut-off values for acylcarnitines and free carnitine in DBS vary internationally; reference ranges are collected in the Region 4 Stork (R4S) project [43]. Although DBS is good matrix for detecting most lcFAODs, acylcarnitine levels in CPT2-deficient patients can be in the normal range [56]. The use of ratios of acylcarnitines improves sensitivity [46, 57]. For example for VLCAD deficiency, the ratio of tetradecenoylcarnitine (C14:1) over acetylcarnitine (C2) resulted in less false-negative results when compared to C14:1 as the only marker [46, 57]. A potential disadvantage of a more sensitive screening is that it will likely also result in identification of asymptomatic individuals who may never develop clinical symptoms. The benefit of screening for these patients is not yet clear since their risk of developing symptoms in the future is yet unknown [55, 58]. Since the introduction of NBS, several studies reported follow-up data of VLCAD-deficient patients diagnosed by NBS who were asymptomatic at time of screening and remained so for the follow-up period of the study [21, 59,60,61]. However, despite NBS and subsequent early diagnosis and treatment, some patients still develop severe symptoms, including fatal metabolic derangements [21, 62, 63].
5 Overview of different long-chain fatty acid oxidation disorders
5.1 Disorders of carnitine transport
5.1.1 Primary carnitine deficiency / organic cation transporter 2 (OCTN2) deficiency / carnitine transporter disorder (CTD)
Clinical presentation
Patients with systemic primary carnitine deficiency (also known as OCTN2 deficiency or CTD; OMIM 212140), lose carnitine in urine and as a consequence have low carnitine levels in plasma. Patients can present with hepatic, myopathic and cardiac signs and symptoms as described above (Section 3). Cardiomyopathy and cardiac arrhythmias leading to sudden death are most frequently reported [64]. Some signs and symptoms are only described in a few case reports, and include peripheral neuropathy involving both upper and lower limbs, mild developmental delay, anemia, respiratory distress and proximal muscle weakness [65,66,67]. The disease is particularly prevalent in the Faroe Islands, an archipelago between Scotland and Iceland with little genetic variation. A large population screening revealed a prevalence of 1:297 [68]. After a series of sudden deaths in young Faroese inhabitants who were all homozygous for the c.95A > G (p.N32S) mutation, an association was made between the use of antibiotics containing pivalic acid and development of encephalopathy and sudden cardiac arrest [69]. The following incidences of primary carnitine deficiency in newborns have been reported: 1:40,000 in Japan [70], 1:37,000–1:100,000 in Australia [71], 1:105,000 in Portugal [72], 1:142,000 in the USA [73], 1:250,000 in Germany [74] and 1:297 in the Faroe islands [68].
Acylcarnitine profile
In primary carnitine deficiency the transport of carnitine over the plasma membrane is impaired resulting in low intracellular carnitine concentrations. Carnitine in the blood is wasted via the urine so plasma levels of both free and acylated carnitine are extremely low (free carnitine <5 μM (reference 25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41, 43, 48,49,50,51,52,53,54,55 μM)) and carnitine levels in urine are high [64]. Other plasma acylcarnitines are also low due to impaired formation, but low levels of free carnitine (C0) remains the primary marker for diagnosis and NBS (Table 1) [42]. Transplacental transport of free carnitine from mother to infant complicates the diagnosis of patients by NBS because plasma levels can be higher resulting in false negative screening results. In addition, low levels of C0 found in NBS can also be the result of primary or secondary carnitine deficiency in the mother [75].
Enzymatic activity
Confirmation of primary carnitine deficiency is achieved by measuring carnitine transport in cultured fibroblasts. The severity of clinical symptoms is correlated with the level of residual transport of carnitine [76].
Molecular analysis
The disease is caused by mutations in the SLC22A5 gene (OMIM 603377). Heterogeneity of mutations complicates the formation of a genotype-phenotype correlation, although it appears that the frequency of loss-of-function mutations is higher in patients who present symptomatically compared to asymptomatic women [76].
5.1.2 Carnitine Palmitoyl transferase type 1A (CPT1A) deficiency
Clinical presentation
Patients with CPT1A deficiency (OMIM 255120) present typically with fasting-induced hypoketotic hypoglycemia and encephalopathy without muscular involvement at an early age [77, 78]. A few cases of hepatic encephalopathy without hypoglycemia and renal tubular acidosis have also been reported [79,80,81]. During or after metabolic crisis, elevation of dodecanedioic acid has been observed in urine of patients [82]. Heterozygous female carriers may develop fatty liver of pregnancy when carrying a fetus with this disorder [83]. CPT1A deficiency is particularly prevalent in native populations of Alaska, Canada, Greenland and North-East Siberia [84,85,86]. The common c.1436C > T (p.P479L) variant in these populations causes a partially reduced CPT1A activity and is associated with infant mortality [87,88,89,90]. This mutation also results in a CPT1A protein that is less sensitive for the inhibitory effect of malonyl-CoA on CPT1A under fed conditions, resulting in a 3–4 times higher activity of CPT1A compared to wild type [91,92,93]. Prevalence of CPT1A deficiency in Alaskan infants is 1:780 live births [94], but is very rare among non-Inuit, non-Yupik or non-Hutterite populations. In Taiwan a prevalence of 1:769.230 is reported [95], and combined results of NBS programs in Australia, Germany and USA show an incidence of 1:750,000–1:2000,000 [55]. The prevalence in the Region 4 Stork collaborative, now called Collaborative Laboratory Integrated Reports (CLIR; www.clir-r4s.org), is 1:500,000 to 1:1,000,000.
Acylcarnitine profile
In CPT1A deficiency the conversion of long-chain acyl-CoAs to their corresponding acylcarnitines is impaired, resulting in low long-chain acylcarnitine levels in plasma and DBS. The ratio of C0 over long-chain acylcarnitines in DBS appears to be a good marker to screen for CPT1A deficiency (Table 1) [44, 96]. In addition, free carnitine levels in plasma are usually high [97], but if a child is in a well-fed state false negative results can still occur [37].
Enzymatic testing
Diagnosis should be confirmed by enzymatic and/or molecular testing. To measure CPT1A activity fibroblasts using [U-13C]-palmitoyl-CoA as substrate, and the amount of synthesized labeled palmitoyl-carnitine is measured by mass spectrometry [98]. Most mutations are associated with residual enzyme activity between 0 and 10% of control values.
Molecular analysis
All currently known CPT1-deficient patients have mutations in the CPT1A gene (OMIM 600528). Since the first report of a molecular defect of human CPT1A [99], many different mutations in the CPT1A isoform have been described [92, 100]. Disorders of CPT1B, the muscle isoform, and CPT1C, the neuronal isoform, have not been reported in humans yet. Mutations in the CPT1A gene are usually highly heterogeneous except for the pathogenic variant c.1436C > T (p.P479L) which is observed to have a high prevalence in the Inuit population [101] and the c. 2129G > A (p.G710E) in the North American Hutterites [85].
5.1.3 Carnitine-acylcarnitine translocase (CACT) deficiency
Clinical presentation
Patients with CACT deficiency (OMIM 212138) usually present in the neonatal period with severe hyperammonemia, liver failure, cardiomyopathy and respiratory distress, often with fatal outcome [23, 24, 102, 103]. Less than 60 patients have been reported in literature of whom two were detected by NBS [95, 104].
Acylcarnitine profile
CACT-deficient patients show elevations of C16, C18 and C18:1 carnitines. This profile, however, is not specific for CACT deficiency since CPT2 deficiency has the same profile [41].
Enzyme activity
CACT enzyme activity can be determined in digitonin-permeabilized fibroblasts measuring formation of radiolabeled CO2 from radiolabeled acetylcarnitine as a substrate [105]. There is a correlation between residual capacity of overall lcFAO and milder phenotypes [102, 105].
Molecular analysis
Several, mostly private mutations in the SLC25A20 gene (OMIM 613698) have been identified in patients and most of them express severe phenotypes, which makes genotype-phenotype correlations difficult. The most frequently reported mutations include the splicing mutation c.199-10 T > G that is found in patients from East Asia (Japan, China, Vietnam) and the missense mutation c.713A > G (p.Gln238Arg) in patients of Arabic descent [102].
5.1.4 Carnitine Palmitoyl transferase type 2 (CPT2) deficiency
Clinical presentation
Patients with CPT2 deficiency can present with hypoglycemia, cardiomyopathy and muscular signs and symptoms [23, 24, 106]. Similar to other lcFAODs, muscular symptoms are more prominent in adulthood or adolescence and liver-related signs and symptoms are generally seen in early childhood, but patients can present with both in different stages of life. Yet, CPT2 deficiency is often referred to as either the adult/benign form or the neonatal form, suggesting these are separate identities. In contrast to other lcFAODs, congenital malformations, such as dysmorphic features, renal cysts, intracerebral and intrahepatic calcifications have been reported in several cases that suffered from the lethal neonatal form [107,108,109,110]. However, the majority of patients in literature presents in adolescence or adulthood with myopathy as first complaint [106]. Combined results of NBS programs in Australia, Germany and USA report an incidence of 1:750,000–1:2,000,000 [55], but numbers on prevalence are scarce since CPT2 deficiency has not been introduced in most NBS panels. However, many families have been reported in literature and over 300 patients have been described.
Acylcarnitine profile
CPT2 activity completes the carnitine cycle, by reconverting the acylcarnitines to acyl-CoA esters in mitochondria. The acylcarnitine profile of CPT2-deficient patients is identical to that of CACT patients, i.e. accumulation of C16-, C18- and C18:1-carnitine.
Enzymatic activity
The reaction catalyzed by CPT2 is reversible and can be measured in the forward or backward direction. In the radioisotope exchange assay, the incorporation of labeled carnitine in palmitoyl-carnitine is reflecting (backward) CPT2 activity [111, 112]. Methods to measure the physiological forward reaction have also been developed. Fibroblasts or lymphocytes are incubated with palmitoyl-carnitine, and formed palmitoyl-CoA is measured using (ultra)high performance liquid chromatography ((U)HPLC) in a medium where CPT1 is blocked in the presence of Triton X-100 [5]. For the common c.338C > T (p.S113 L) mutation overall lcFAO flux in fibroblasts can be normal at 37 °C culture conditions whereas at 41 °C the activity was clearly reduced in patient fibroblasts compared to controls [113]. Residual level of long-chain fatty acid oxidation in CPT2-deficient fibroblasts appears to be related to severity of disease [114, 115].
Molecular analysis
Around 60 mutations in the CPT2 gene (OMIM 600650) have been identified, of which most are private mutations. The c.338C > T (p.S113 L) mutation is associated with the more attenuated muscular phenotype and is relatively common with an allele frequency of up to 90% in different cohorts of patients [106, 116, 117].
5.2 Disorders of mitochondrial long-chain fatty acid oxidation
5.2.1 Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency
Clinical presentation
Patients diagnosed with VLCAD deficiency (OMIM 201475) can suffer from hepatic, cardiac and muscular symptoms that can appear in infancy, but also later during adolescence or adulthood. Similar to other lcFAODs, hepatic symptoms are only seen during (early) childhood and patients with onset of symptoms at a later age predominantly experience myopathy-related complaints. Neurological symptoms, such as epilepsy and psychomotor development delay have been reported, but are usually the consequence of cerebral damage due to hypoketotic hypoglycemia. Over the past two decades, many NBS programs have included VLCAD deficiency in their panel. Before inclusion in NBS, many patients died during the first years of life often without any prior signs. One large cohort reports almost 50% deceased patients [23, 25]. Indeed, diagnosis after sudden death of infancy was more common before NBS inclusion [118, 119]. VLCAD deficiency is most frequently diagnosed of all lcFAODs worldwide with a prevalence reported between 1:31.500 to 1:94.569 [55, 120]. The majority of newly diagnosed children by NBS is asymptomatic or mildly affected because of relative high residual enzyme activity [22, 60, 121].
Acylcarnitine profile
Metabolites that are abnormal in VLCAD deficiency patients are usually C12, C14, C14:1, C14:2 and C16 [43], with C14:1 accumulation as most important marker, which is derived from partial breakdown of oleic acid (C18:1w9).
Enzymatic testing
Measurement of VLCAD activity in fibroblasts and lymphocytes can be used to confirm diagnosis and involves the conversion of palmitoyl-CoA in the presence of ferrocenium hexafluorophosphate as electron acceptor. Products (palmitenoyl-CoA and, due to enoyl-CoA hydratase activity, 3-hydroxy-palmitoyl-CoA) are separated and quantified by U(HPLC) [5, 19].
Molecular testing
Disease-causing mutations in the gene encoding VLCAD, i.e. ACADVL (OMIM 609575), are distributed all over the gene, causing a high molecular heterogeneity [122]. A follow-up analysis of 693 unrelated VLCAD-deficient patients detected by NBS underlines the molecular heterogeneity of this disorder, but also shows a relatively high prevalence of the c.848 T > C (p.V283A) pathogenic variant [51, 60].
Phenotype prediction
The detection of patients in the neonatal period before developing symptoms has increased the need for reliable phenotype prediction. To some extent, the phenotype correlates with genotype for VLCAD deficiency [123]. However, the continuing discovery of VUS and new mutations and frequent detection of compound heterozygosity in patients complicates phenotype prediction based on genotype in VLCAD deficiency. In a recent study in which different biochemical assays in patient fibroblasts were correlated to clinical severity, residual oleate fatty acid oxidation flux turned out to be strongly correlated with clinical severity for VLCAD deficiency. Correlation of the clinical severity with either VLCAD activity or C14/C16-acylcarnitine ratio was much less clear [124].
5.2.2 Mitochondrial trifunctional protein (MTP) deficiency/long-chain 3-Hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency/long-chain 3-Ketoacyl-CoA Thiolase (LCKAT) deficiency
Clinical presentation
MTP is a multi-enzyme complex, consisting of three enzymes, in which different enzyme deficiencies can occur. All three enzymes are impaired in generalized MTP deficiency (OMIM 609015). Isolated LCHAD deficiency (OMIM 609016) is most common. Isolated LCKAT deficiency is extremely rare [125]. Patients with isolated LCHAD deficiency or generalized MTP deficiency share most of the clinical signs and symptoms with other lcFAODs, but are the only lcFAOD in which patients can develop peripheral neuropathy and pigmentary retinopathy [23,24,25, 62, 126,127,128]. Even though peripheral neuropathy and pigmentary retinopathy are reported in both LCHAD and MTP deficiency, retinopathy seems to be more prevalent in isolated LCHAD deficiency and peripheral neuropathy is more often seen in MTP deficiency [127,128,129,130]. There is a broad clinical spectrum, but the proportion of patients with a severe clinical presentation and who are refractory to treatment - despite early detection by NBS - is larger than seen in VLCAD deficiency [62, 131]. Some cases of hypoparathyroidism are reported for both isolated LCHAD as well as generalized MTP deficiency [132,133,134]. Typical for LCHAD deficiency is the increased risk for female heterozygous carriers to develop hemolysis, elevated liver enzymes, low platelets (HELLP) syndrome and acute fatty liver of pregnancy (AFLP) [135, 136]. The pathophysiology is incompletely understood, but decreased FAO of the placenta and fetus are likely to cause accumulation of long-chain 3-hydroxyacyl fatty acid metabolites in the maternal circulation. This could, in combination with increased lipase activity in the last trimester of pregnancy, have lipotoxic impact on the liver of the mother [137,138,139]. A meta-analysis showed an estimated clinical detection prevalence of 0.41 per 100,000 for the combination of LCHAD/MTP deficiency, this was mainly based on studies in Western populations [140]. Combined results of NBS programs in Australia, Germany and USA showed an incidence of 1:250,000 for isolated LCHAD deficiency and 1:750,000 for MTP deficiency [55].
Acylcarnitine profile
LCHAD, LCKAT and MTP deficiency are all characterized by accumulation of long-chain 3-hydroxyacylcarnitine species in DBS and plasma and cannot be separated from each other based on acylcarnitine profile [128]. In two separate cohort studies of LCHAD-deficient patients, the level of long-chain 3-hydroxyacylcarnitines accumulation does not increase with age, but in general, concentrations are lower after the diagnosis is known and patients are put on a long-chain fatty acid restricted diet [62, 63].
Enzymatic testing
Because there is no specific substrate for LCHAD, its activity is determined in the backward reaction by measuring 3-ketopalmitoyl-CoA dehydrogenase activity (LCHAD+SCHAD activity) in the presence and absence of a powerful LCHAD inhibitor (N-ethylmaleimide). The difference between the two measurements is taken to represent specific LCHAD activity [5]. When activity is reduced, mutation analysis should be performed. The thiolase activity of MTP (LCKAT) can be measured by using 3-ketopalmitoyl-CoA as substrate [5]. In case of elevated 3-hydroxyacylcarnitine levels in plasma of DBS, both the LCHAD and the LCKAT activity of MTP should be measured.
Molecular testing
The diagnosis is confirmed by mutation analysis of the two genes (HADHA (OMIM 600890) and HADHB (OMIM 143450)) that encode the α and ß subunits of the enzyme MTP. A mutation in one of both genes can lead to a generalized MTP deficiency due to instability of the complex [141]. In addition mutation in HADHA can lead to isolated LCHAD deficiency and mutations in the HADHB gene to isolated LCKAT deficiency. The majority of LCHAD-deficient patients carries the c.1528G > A (p.E510Q) mutation in at least one allele of the HADHA gene [142].
6 Management of lcFAODs
6.1 Acute management
Patients with lcFAODs are unable, or insufficiently able, to utilize fatty acids when the available glucose pool becomes depleted. Therefore, in case of metabolic crisis, emergency treatment should be aimed at supplying sufficient glucose to prevent cell damage, especially in muscles. The amount of glucose infusion depends on various factors such as residual enzyme activity, age and the level of stress due to anxiety or fever. Recommendations vary widely and there is no clear consensus.
Unfortunately, normoglycemia is not a sign that a catabolic crisis is averted. Patients may still develop rhabdomyolysis without hypoglycemia and should still receive glucose. Hyperglycemia should be treated with insulin, not with a decreasing glucose intake. Sodium and potassium should be monitored and supplemented if necessary. In case of fever, anti-pyrogenic medication should be prescribed [143]. Creatine kinase in plasma is the recommended marker to monitor rhabdomyolysis, but symptoms usually occur several hours before any rise in plasma CK levels is noticed.
6.2 Diet and MCT
Although the understanding of the pathophysiology of lcFAODs has increased significantly in the past decades, treatment options are still limited [144]. Traditionally treatment of lcFAODs consists of prevention of catabolism, LCT restriction, and supplementation of MCT [143, 145, 146]. Intake of LCT and MCT is dependent on the specific type of lcFAOD, the residual enzyme activity, and age of the patient. In case of LCHAD/MTP deficiency and symptomatic VLCAD deficiency, MCT at 20–25% of energy intake, and total fat at 25–35% of energy intake is advised [143]. Breastfeeding is not contraindicated during LCT restriction, but should be complemented with MCT-containing formula [52, 143] . More patients have been diagnosed since the inclusion of lcFAODs in NBS panels, often before development of symptoms and it is debated whether dietary restrictions should be alleviated in certain patient groups [147, 148]. Therefore, early phenotype prediction is warranted, but this is complicated as most lcFAODs lack a clear genotype-phenotype correlation. Patients are often compound heterozygote with a combination that includes at least one novel mutations or VUS. For VLCAD deficiency, we recently proposed a dietary treatment strategy based on a functional assay that measures the overall lcFAO flux in skin fibroblasts [52]. Based on clinical and nutritional data of VLCAD-deficient patients diagnosed before introduction of VLCAD deficiency in NBS [52], LCT restriction and MCT supplementation can be alleviated under normal circumstances in patients with >10% lcFAO as long as they follow strict dietary measures during illness and a maximum feeding pause in line with the guidelines that specify maximal fasting periods per age group [143].
6.3 Carnitine
Supplementation of carnitine is the primary therapy for OCTN2 deficiency in which very low levels of free and acylated carnitine (<10 μmol/L) in plasma occur due to abnormally high urinary loss. Complete recovery of cardiac, muscular and hepatic symptoms has been described for OCTN2-deficient patients after supplementation [149, 150]. Next to its essential role in the import of acid into the mitochondria, carnitine can also remove acyl-CoAs out of the mitochondria by facilitating their conversion to acylcarnitines which can be excreted in urine [151]. The use of carnitine in the other lcFAODs is controversial and not advised during acute management of a metabolic crisis [21, 143]. Even though supplementation can aid carnitine transport and subsequently improve FAO in theory, evidence for the efficacy of carnitine supplementation to treat secondary carnitine deficiency in lcFAODs is lacking [152]. Furthermore, concerns have been raised about the safety of carnitine supplementation in lcFAODs because it could also induce the accumulation of toxic intramitochondrial long-chain acyl-carnitines and -CoAs, and supplementation has been associated with ventricular fibrillation and rhabdomyolysis [153,154,155].
6.4 Follow-up diagnostics
The frequency and choice of diagnostic follow-up for lcFAOD patients depends on the type of disorder, severity of the disease and accessibility. The monitoring of lcFAOD patients is mainly based on expert opinion (grade 4 evidence), as case-control studies are lacking [156]. In 2009, treatment recommendations have been published for lcFAODs, based on clinical data of 18 metabolic centers [21, 143]. In most centers, patients were annually seen by a metabolic specialist. For lcFAOD the specific long-chain acylcarnitines and CK levels should be measured in plasma to evaluate status of disease. Because of the risk of cardiac disease, electrocardiography and echocardiography should be performed, especially at a young age. Cardiac monitoring is usually done once every one-to-two years. This interval can be prolonged for the asymptomatic and mild patients detected by NBS, for whom the risk of cardiac disease is much smaller [22, 23, 157]. LCHAD deficiency and MTP deficiency have long-term complications not seen in other lcFAODs: progressive pigmentary retinopathy and a progressive peripheral neuropathy, which should be monitored. Furthermore anthropometric measurements should be performed during outpatient clinic visits in all lcFAOD patients, to make sure the child is thriving well, and also to detect excess weight gain at an early stage. Due to extra calories and prevention of fasting, lcFAOD patients have an extra risk to gain weight, and losing weight can be risky because this often involves fasting and reliance on lcFAO [158].
6.5 Therapies under investigation
6.5.1 Anaplerotics
Anaplerotic therapy for lcFAOD is based on the hypothesis that a deficit in TCA cycle intermediates in these disorders hampers ATP production from substrates such as glucose, in conditions of impaired capability to produce ATP from FAO. This deficit has been shown in LCAD knockout mice, in which myocardial TCA cycle intermediates were depleted, indicating an increased but unmet need for anaplerosis [159]. Triheptanoin, a triglyceride with odd numbered fatty acids (C7), is an anaplerotic compound [160]. A recent double-blind randomized controlled trial included 32 subjects with different lcFAODs and compared the effects of a 4-month diet containing 20% of total energy from triglycerides containing either C8 or C7 fatty acids. Results seemed promising for cardiac symptoms as the left ventricle wall mass decreased and an increase in ejection fraction was observed in patients in the C7 group, but it should be noted that all the included patients had normal cardiac function [161]. Beneficial effects of triheptanoin for cardiac symptoms have been described before in a few case studies [30, 162, 163]. However, triheptanoin gave no decline in rhabdomyolysis episodes in a double-blind placebo-controlled trial [161], emphasizing the need for new therapeutic options.
6.5.2 Resveratrol and bezafibrate
Residual lcFAO can be increased in fibroblasts from VLCAD- and CPT2-deficient patients carrying missense mutations by incubation with the hypolipidemic drug bezafibrate or the polyphenol resveratrol [164, 165]. Resveratrol increases mitochondrial biogenesis through the activation of sirtuin 1 (SIRT1) and the downstream effector peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α). It is likely that through this mechanism the enzyme activity can be increased if the mutated enzyme has some residual enzyme activity [166]. Bezafibrate acts as an agonist of peroxisome proliferator activator receptors (PPAR). A complete restoration of palmitate oxidation accompanied with increased protein and mRNA levels can be achieved in VLCAD-deficient fibroblasts and CPT2-deficient myoblasts of patients with considerable residual enzyme activity [167, 168]. These promising findings have led to the investigation of bezafibrate in clinical trials with CPT2-deficient patients. A clinical trial reported improved exercise tolerance and reduction of rhabdomyolysis episodes under bezafibrate treatment in all six CPT2-deficient patients tested [169]. Additionally, palmitate oxidation increased in skeletal muscle. However, another trial showed no effect of bezafibrate in CPT2-deficient patients in terms of lcFAO or exercise capacity [170, 171].
6.5.3 Ketone therapy
Ketogenic diets are often used in patients with inborn errors of metabolism, but are not suitable for lcFAOD patients, since they are unable to synthesize ketone bodies. Synthetic ketone bodies on the other hand are hypothesized to serve as an efficient alternative energy substrate that will generate sufficient acetyl-CoA for the TCA cycle to prevent symptoms. Ketone-salts have been described as therapy in multiple acyl-CoA dehydrogenase deficiency (MADD), a mitochondrial disorder that also involves impaired lcFAO, but with a more severe phenotype than lcFAODs [172]. A disadvantage of ketone salts is the undesirable salt loading, especially in patients with cardiac disease, which is why ketone-esters have more potential to serve as a new lcFAOD therapy [173, 174]. A recent study has tested the therapeutic potential of an ester of (D)-ß-hydroxybutyrate and 1,3-butanediol in VLCAD-deficient patients and found improved muscular energy balance and plasma acylcarnitine profiles after ingestion of a single dose of the ketone ester [175]. Whether daily use of this ketone ester results in alleviation of symptoms and reduces episodes of rhabdomyolysis in lcFAOD patients still needs to be investigated in a long-term follow-up study.
7 Conclusions and future perspectives
With the inclusion of lcFAODs in many newborn screening programs and improvement of diagnostic techniques more patients are diagnosed but the clinical severity varies between patients. Consequently the need for prognostic markers to predict outcome is rising. In terms of treatment, up until now, dietary strategies are the best therapeutic option, but there are several other potential treatments under investigation. Nevertheless, confirmation of effectiveness of new treatments for lcFAOD is challenging, since outcome parameters to measure functional improvement are hard to find, and because it is challenging or even impossible to collect large cohorts of patients for all lcFAODs.
References
Lopaschuk GD, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–58.
Bartlett K, Eaton S. Mitochondrial β-oxidation. Eur J Biochem. 2004;271(3):462–9.
Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Annu Rev Nutr. 2014;34:1–30.
Swigonova Z, Mohsen AW, Vockley J. Acyl-CoA dehydrogenases: dynamic history of protein family evolution. J Mol Evol. 2009;69(2):176–93.
Wanders RJ, et al. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J Inherit Metab Dis. 2010;33(5):479–94.
Chegary M, et al. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta. 2009;1791(8):806–15.
Hale DE, Batshaw ML, Coates PM, Frerman FE, Goodman SI, Singh I, et al. Long-chain acyl coenzyme a dehydrogenase deficiency: an inherited cause of nonketotic hypoglycemia. Pediatr Res. 1985;19(7):666–71.
Indo Y, Coates PM, Hale DE, Tanaka K. Immunochemical characterization of variant long-chain acyl-CoA dehydrogenase in cultured fibroblasts from nine patients with long-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 1991;30(3):211–5.
Yamaguchi S, Indo Y, Coates PM, Hashimoto T, Tanaka K. Identification of very-long-chain acyl-CoA dehydrogenase deficiency in three patients previously diagnosed with long-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 1993;34(1):111–3.
Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33(5):469–77.
Houten SM, Violante S, Ventura FV, Wanders RJA. The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu Rev Physiol. 2016;78:23–44.
Schulz H. Regulation of fatty acid oxidation in heart. J Nutr. 1994;124(2):165–71.
Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res. 1987;18(4):551–61.
Jernberg JN, Bowman CE, Wolfgang MJ, Scafidi S. Developmental regulation and localization of carnitine palmitoyltransferases (CPTs) in rat brain. J Neurochem. 2017;142(3):407–19.
Oey NA, et al. Long-chain fatty acid oxidation during early human development. Pediatr Res. 2005;57(6):755–9.
DiMauro S, DiMauro PMM. Muscle carnitine Palmityltransferase deficiency and Myoglobinuria. Science. 1973;182(4115):929–31.
Izai K, Uchida Y, Orii T, Yamamoto S, Hashimoto T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. I. Purification and properties of very-long-chain acyl-coenzyme a dehydrogenase. J Biol Chem. 1992;267(2):1027–33.
Karpati G, et al. The syndrome of systemic carnitine deficiency. Clinical, morphologic, biochemical, and pathophysiologic features. Neurology. 1975;25(1):16–24.
Wanders RJ, et al. Human trifunctional protein deficiency: a new disorder of mitochondrial fatty acid beta-oxidation. Biochem Biophys Res Commun. 1992;188(3):1139–45.
Bonnet D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100(22):2248–53.
Spiekerkoetter U, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32(4):488–97.
Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33(5):527–32.
Baruteau J, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2013;36(5):795–803.
Baruteau J, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study from 187 patients. Complementary data. J Inherit Metab Dis. 2014;37(1):137–9.
Saudubray JM, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999;22(4):488–502.
Vianey-Saban C, et al. Mitochondrial very-long-chain acyl-coenzyme a dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta. 1998;269(1):43–62.
Das AM, et al. Secondary respiratory chain defect in a boy with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: possible diagnostic pitfalls. Eur J Pediatr. 2000;159(4):243–6.
Glasgow AM, et al. Hypoglycemia, hepatic dysfunction, muscle weakness, cardiomyopathy, free carnitine deficiency and long-chain acylcarnitine excess responsive to medium chain triglyceride diet. Pediatr Res. 1983;17(5):319–26.
Cox GF, et al. Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long-chain acyl-coenzyme a dehydrogenase deficiency. J Pediatr. 1998;133(2):247–53.
Sharef SW, Al-Senaidi K, Joshi SN. Successful treatment of cardiomyopathy due to very long-chain acyl-CoA dehydrogenase deficiency: first case report from Oman with literature review. Oman Med J. 2013;28(5):354–6.
Kluge S, Kühnelt P, Block A, Merkel M, Gocht A, Lukacs Z, et al. A young woman with persistent hypoglycemia, rhabdomyolysis, and coma: recognizing fatty acid oxidation defects in adults. Crit Care Med. 2003;31(4):1273–6.
Schaefer J, et al. Characterisation of carnitine palmitoyltransferases in patients with a carnitine palmitoyltransferase deficiency: implications for diagnosis and therapy. J Neurol Neurosurg Psychiatry. 1997;62(2):169–76.
Violante S, et al. Carnitine palmitoyltransferase 2: new insights on the substrate specificity and implications for acylcarnitine profiling. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2010;1802(9):728–32.
Millington DS, et al. Application of fast atom bombardment with tandem mass spectrometry and liquid chromatography/mass spectrometry to the analysis of acylcarnitines in human urine, blood, and tissue. Anal Biochem. 1989;180(2):331–9.
Rashed MS, et al. Diagnosis of inborn errors of metabolism from blood spots by acylcarnitines and amino acids profiling using automated electrospray tandem mass spectrometry. Pediatr Res. 1995;38(3):324–31.
Topcu Y, et al. Importance of acylcarnitine profile analysis for disorders of lipid metabolism in adolescent patients with recurrent rhabdomyolysis: report of two cases. Ann Indian Acad Neurol. 2014;17(4):437–40.
Borch L, et al. Normal levels of plasma free carnitine and Acylcarnitines in follow-up samples from a Presymptomatic case of carnitine Palmitoyl transferase 1 (CPT1) deficiency detected through newborn screening in Denmark. JIMD Rep. 2012;3:11–5.
Browning MF, et al. Normal acylcarnitine levels during confirmation of abnormal newborn screening in long-chain fatty acid oxidation defects. J Inherit Metab Dis. 2005;28(4):545–50.
Burrage LC, et al. Elevations of C14:1 and C14:2 plasma Acylcarnitines in fasted children: a diagnostic dilemma. J Pediatr. 2016;169:208–213.e2.
Spiekerkoetter U, et al. Lethal undiagnosed very long-chain acyl-CoA dehydrogenase deficiency with mild C14-Acylcarnitine abnormalities on newborn screening. JIMD Rep. 2012;6:113–5.
Ventura FV, et al. Quantitative acylcarnitine profiling in fibroblasts using [U-13C] palmitic acid: an improved tool for the diagnosis of fatty acid oxidation defects. Clin Chim Acta. 1999;281(1–2):1–17.
Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta. 2016;1863(10):2422–35.
McHugh D, et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med. 2011;13(3):230–54.
Fingerhut R, et al. Hepatic carnitine palmitoyltransferase I deficiency: acylcarnitine profiles in blood spots are highly specific. Clin Chem. 2001;47(10):1763–8.
Gempel K, et al. Screening for carnitine Palmitoyltransferase II deficiency by tandem mass spectrometry. J Inherit Metab Dis. 2002;25(1):17–27.
Diekman E, et al. The newborn screening paradox: sensitivity vs. Overdiagnosis in VLCAD deficiency. JIMD Rep. 2016;27:101–6.
Sander J, et al. Neonatal screening for defects of the mitochondrial trifunctional protein. Mol Genet Metab. 2005;85(2):108–14.
Manning NJ, et al. A comparison of [9,10-3H]palmitic and [9,10-3H]myristic acids for the detection of defects of fatty acid oxidation in intact cultured fibroblasts. J Inherit Metab Dis. 1990;13(1):58–68.
Olpin SE, et al. Improved detection of long-chain fatty acid oxidation defects in intact cells using [9,10-3H]oleic acid. J Inherit Metab Dis. 1997;20(3):415–9.
Gregersen N, et al. Mutation analysis in mitochondrial fatty acid oxidation defects: exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat. 2001;18(3):169–89.
Miller MJ, et al. Recurrent ACADVL molecular findings in individuals with a positive newborn screen for very long chain acyl-coA dehydrogenase (VLCAD) deficiency in the United States. Mol Genet Metab. 2015;116(3):139–45.
Bleeker JC, Kok IL, Ferdinandusse S, et al. Proposal for an individualized dietary strategy in patients with very long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 2018. https://doi.org/10.1007/s10545-018-0164-5.
Pena LD, et al. Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol Genet Metab. 2016;118:272–81.
Loeber JG, et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J Inherit Metab Dis. 2012;35(4):603–11.
Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33(5):521–6.
de Sain-van der Velden MG, et al. Differences between acylcarnitine profiles in plasma and bloodspots. Mol Genet Metab. 2013;110(1–2):116–21.
Hall PL, et al. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet Med. 2014;16(12):889–95.
Lindner M, et al. Efficacy and outcome of expanded newborn screening for metabolic diseases--report of 10 years from south-West Germany. Orphanet J Rare Dis. 2011;6:44.
Merritt Ii JL, et al. Infants suspected to have very-long chain acyl-CoA dehydrogenase deficiency from newborn screening. Mol Genet Metab. 2014;111(4):484–92.
Evans M, et al. VLCAD deficiency: follow-up and outcome of patients diagnosed through newborn screening in Victoria. Mol Genet Metab. 2016;118(4):282–7.
Ryder B, et al. The natural history of elevated tetradecenoyl-L-carnitine detected by newborn screening in New Zealand: implications for very long chain acyl-CoA dehydrogenase deficiency screening and treatment. J Inherit Metab Dis. 2016;39(3):409–14.
Karall D, et al. Clinical outcome, biochemical and therapeutic follow-up in 14 Austrian patients with long-chain 3-Hydroxy acyl CoA dehydrogenase deficiency (LCHADD). Orphanet J Rare Dis. 2015;10:21.
De Biase I, et al. Diagnosis, treatment, and clinical outcome of patients with mitochondrial trifunctional protein/long-chain 3-Hydroxy acyl-CoA dehydrogenase deficiency. JIMD Rep. 2017;31:63–71.
Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142c(2):77–85.
Makhseed N, et al. Carnitine transporter defect due to a novel mutation in the SLC22A5 gene presenting with peripheral neuropathy. J Inherit Metab Dis. 2004;27(6):778–80.
Erguven M, Yılmaz O, Koc S, Cakı S, Ayhan Y, Donmez M, et al. A case of early diagnosed carnitine deficiency presenting with respiratory symptoms. Ann Nutr Metab. 2007;51(4):331–4.
Wang Y, et al. Phenotype and genotype variation in primary carnitine deficiency. Genet Med. 2001;3(6):387–92.
Rasmussen J, et al. Carnitine levels in 26,462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands. J Inherit Metab Dis. 2014;37(2):215–22.
Rasmussen J, Nielsen OW, Lund AM, Køber L, Djurhuus H. Primary carnitine deficiency and pivalic acid exposure causing encephalopathy and fatal cardiac events. J Inherit Metab Dis. 2013;36(1):35–41.
Koizumi A, et al. Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum Mol Genet. 1999;8(12):2247–54.
Wilcken B, et al. Carnitine transporter defect diagnosed by newborn screening with electrospray tandem mass spectrometry. J Pediatr. 2001;138(4):581–4.
Vilarinho L, et al. Four years of expanded newborn screening in Portugal with tandem mass spectrometry. J Inherit Metab Dis. 2010;33(Suppl 3):S133–8.
Therrell BL Jr, Lloyd-Puryear MA, Camp KM, Mann MY. Inborn errors of metabolism identified via newborn screening: ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol Genet Metab. 2014;113(1–2):14–26.
Schulze A, et al. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111(6 Pt 1):1399–406.
El-Hattab AW, et al. Maternal systemic primary carnitine deficiency uncovered by newborn screening: clinical, biochemical, and molecular aspects. Genet Med. 2010;12(1):19–24.
Rose EC, et al. Genotype-phenotype correlation in primary carnitine deficiency. Hum Mutat. 2012;33(1):118–23.
Bougneres PF, et al. Fasting hypoglycemia resulting from hepatic carnitine palmitoyl transferase deficiency. J Pediatr. 1981;98(5):742–6.
Demaugre F, et al. Hepatic and muscular presentations of carnitine palmitoyl transferase deficiency: two distinct entities. Pediatr Res. 1988;24(3):308–11.
Vianey-Saban C, et al. Carnitine palmitoyl transferase I deficiency presenting as a Reye-like syndrome without hypoglycaemia. Eur J Pediatr. 1993;152(4):334–8.
Falik-Borenstein ZC, et al. Brief report: renal tubular acidosis in carnitine palmitoyltransferase type 1 deficiency. N Engl J Med. 1992;327(1):24–7.
Bergman AJ, et al. Rate-dependent distal renal tubular acidosis and carnitine palmitoyltransferase I deficiency. Pediatr Res. 1994;36(5):582–8.
Korman SH, et al. Novel metabolic and molecular findings in hepatic carnitine palmitoyltransferase I deficiency. Mol Genet Metab. 2005;86(3):337–43.
Innes AM, et al. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr Res. 2000;47(1):43–5.
Skotte L, et al. CPT1A missense mutation associated with fatty acid metabolism and reduced height in Greenlanders. Circ Cardiovasc Genet. 2017;10(3):e001618.
Prasad C, et al. Hepatic carnitine palmitoyl transferase 1 (CPT1 a) deficiency in north American Hutterites (Canadian and American): evidence for a founder effect and results of a pilot study on a DNA-based newborn screening program. Mol Genet Metab. 2001;73(1):55–63.
Fohner AE, et al. Carnitine palmitoyltransferase 1A P479L and infant death: policy implications of emerging data. Genet Med. 2017;19(8):851–7.
Collins SA, et al. Causes and risk factors for infant mortality in Nunavut, Canada 1999-2011. BMC Pediatr. 2012;12:190.
Sinclair GB, et al. Carnitine palmitoyltransferase I and sudden unexpected infant death in British Columbia first nations. Pediatrics. 2012;130(5):e1162–9.
Gessner BD, et al. Evidence for an association between infant mortality and homozygosity for the arctic variant of carnitine palmitoyltransferase 1A. Genet Med. 2016;18(9):933–9.
Gessner BD, et al. Evidence for an association between infant mortality and a carnitine palmitoyltransferase 1A genetic variant. Pediatrics. 2010;126(5):945–51.
Morillas M, et al. Structural model of a malonyl-CoA-binding site of carnitine octanoyltransferase and carnitine palmitoyltransferase I: mutational analysis of a malonyl-CoA affinity domain. J Biol Chem. 2002;277(13):11473–80.
Brown NF, et al. Molecular characterization of L-CPT I deficiency in six patients: insights into function of the native enzyme. J Lipid Res. 2001;42(7):1134–42.
Morillas M, et al. Identification of conserved amino acid residues in rat liver carnitine palmitoyltransferase I critical for malonyl-CoA inhibition. Mutation of methionine 593 abolishes malonyl-CoA inhibition. J Biol Chem. 2003;278(11):9058–63.
Butler JC, McLaughlin J. Carnitine palmitoyl transferase-1A deficiency rates in alaska. State of alaska bulletin 2006;19. Retrieved from http://www.epi.Alaska.gov. Accessed on 12 January 2018.
Chien YH, et al. Fatty acid oxidation disorders in a chinese population in Taiwan. JIMD Rep. 2013;11:165–72.
Sim KG, et al. Carnitine palmitoyltransferase I deficiency in neonate identified by dried blood spot free carnitine and acylcarnitine profile. J Inherit Metab Dis. 2001;24(1):51–9.
Stanley CA, et al. Elevated plasma carnitine in the hepatic form of carnitine palmitoyltransferase-1 deficiency. J Inherit Metab Dis. 1992;15(5):785–9.
van Vlies N, et al. An improved enzyme assay for carnitine palmitoyl transferase I in fibroblasts using tandem mass spectrometry. Mol Genet Metab. 2007;90(1):24–9.
IJlst L, et al. Molecular basis of hepatic carnitine palmitoyltransferase I deficiency. J Clin Invest. 1998;102(3):527–31.
Gobin S, et al. Functional and structural basis of carnitine palmitoyltransferase 1A deficiency. J Biol Chem. 2003;278(50):50428–34.
Gessner BD, et al. Prevalence and distribution of the c.1436C-->T sequence variant of carnitine palmitoyltransferase 1A among Alaska Native infants. J Pediatr. 2011;158(1):124–9.
Vitoria I, et al. Carnitine-acylcarnitine translocase deficiency: experience with four cases in Spain and review of the literature. JIMD Rep. 2015;20:11–20.
Rubio-Gozalbo ME, et al. Carnitine-acylcarnitine translocase deficiency: case report and review of the literature. Acta Paediatr. 2003;92(4):501–4.
Wang GL, et al. Expanded molecular features of carnitine acyl-carnitine translocase (CACT) deficiency by comprehensive molecular analysis. Mol Genet Metab. 2011;103(4):349–57.
IJlst L, et al. Functional analysis of mutant human carnitine acylcarnitine translocases in yeast. Biochem Biophys Res Commun. 2001;280(3):700–6.
Bonnefont JP, et al. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Asp Med. 2004;25(5–6):495–520.
Albers S, et al. Detection of neonatal carnitine palmitoyltransferase II deficiency by expanded newborn screening with tandem mass spectrometry. Pediatrics. 2001;107(6):E103.
North KN, et al. Lethal neonatal deficiency of carnitine palmitoyltransferase II associated with dysgenesis of the brain and kidneys. J Pediatr. 1995;127(3):414–20.
Zinn AB, et al. Carnitine Palmitoyltransferase-B (Cpt B) deficiency - a heritable cause of neonatal cardiomyopathy and dysgenesis of the kidney. Pediatr Res. 1991;29(4):A73.
Witt DR, et al. Carnitine Palmitoyl transferase-type 2 deficiency - 2 new cases and successful prenatal-diagnosis. Am J Hum Genet. 1991;49(4):109–9.
Norum KR. Palmityl-Coa:carnitine palmityltransferase. purification from calf-liver mitochondria and some properties of the enzyme. Biochim Biophys Acta. 1964;89:95–108.
Zierz S, Engel AG. Regulatory properties of a mutant carnitine palmitoyltransferase in human skeletal muscle. Eur J Biochem. 1985;149(1):207–14.
Olpin SE, et al. Mutation and biochemical analysis in carnitine palmitoyltransferase type II (CPT II) deficiency. J Inherit Metab Dis. 2003;26(6):543–57.
Thuillier L, et al. Correlation between genotype, metabolic data, and clinical presentation in carnitine palmitoyltransferase 2 (CPT2) deficiency. Hum Mutat. 2003;21(5):493–501.
Bonnefont JP, et al. Carnitine palmitoyltransferase deficiencies. Mol Genet Metab. 1999;68(4):424–40.
Joshi PR, Deschauer M, Zierz S. Carnitine palmitoyltransferase II (CPT II) deficiency: genotype-phenotype analysis of 50 patients. J Neurol Sci. 2014;338(1–2):107–11.
Isackson PJ, Bennett MJ, Vladutiu GD. Identification of 16 new disease-causing mutations in the CPT2 gene resulting in carnitine palmitoyltransferase II deficiency. Mol Genet Metab. 2006;89(4):323–31.
Ficicioglu C, et al. Very long-chain acyl-CoA dehydrogenase deficiency in a patient with normal newborn screening by tandem mass spectrometry. J Pediatr. 2010;156(3):492–4.
Pryce JW, et al. Changing patterns of infant death over the last 100 years: autopsy experience from a specialist children's hospital. J R Soc Med. 2012;105(3):123–30.
Boneh A, et al. VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab. 2006;88(2):166–70.
Pena LD, et al. Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol Genet Metab. 2016;118(4):272–81.
Mathur A, et al. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999;99(10):1337–43.
Andresen BS, et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64(2):479–94.
Diekman EF, et al. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet Med. 2015;17(12):989–94.
Das AM, et al. Isolated Mitochondrial Long-Chain Ketoacyl-CoA Thiolase Deficiency Resulting from Mutations in the HADHB Gene. Clin Chem. 2006;52(3):530–4.
Tyni T, et al. Pathology of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation. Pediatr Pathol Lab Med. 1997;17(3):427–47.
den Boer ME, et al. Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J Pediatr. 2003;142(6):684–9.
den Boer ME, et al. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics. 2002;109(1):99–104.
Tyni T, Pihko H, Kivela T. Ophthalmic pathology in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation. Curr Eye Res. 1998;17(6):551–9.
Boese EA, et al. Characterization of Chorioretinopathy associated with mitochondrial trifunctional protein disorders: long-term follow-up of 21 cases. Ophthalmology. 2016;123(10):2183–95.
Diekman EF, et al. Necrotizing enterocolitis and respiratory distress syndrome as first clinical presentation of mitochondrial trifunctional protein deficiency. JIMD Rep. 2013;7:1–6.
Dionisi-Vici C, et al. Hypoparathyroidism in mitochondrial trifunctional protein deficiency. J Pediatr. 1996;129(1):159–62.
Tyni T, et al. Hypoparathyroidism in a patient with long-chain 3-hydroxyacyl-coenzyme a dehydrogenase deficiency caused by the G1528C mutation. J Pediatr. 1997;131(5):766–8.
Labarthe F, Benoist JF, Brivet M, Vianey-Saban C, Despert F, Ogier de Baulny H. Partial hypoparathyroidism associated with mitochondrial trifunctional protein deficiency. Eur J Pediatr. 2006;165(6):389–91.
Wilcken B, et al. Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme a dehydrogenase deficiency. Lancet. 1993;341(8842):407–8.
Tyni T, Ekholm E, Pihko H. Pregnancy complications are frequent in long-chain 3-hydroxyacyl-coenzyme a dehydrogenase deficiency. Am J Obstet Gynecol. 1998;178(3):603–8.
Liu J, Ghaziani TT, Wolf JL. Acute fatty liver disease of pregnancy: updates in pathogenesis, diagnosis, and management. Am J Gastroenterol. 2017;112(6):838–46.
Ibdah JA. Acute fatty liver of pregnancy: an update on pathogenesis and clinical implications. World J Gastroenterol. 2006;12(46):7397–404.
Bartha JL, et al. Decreased mitochondrial fatty acid oxidation in placentas from women with preeclampsia. Placenta. 2012;33(2):132–4.
Moorthie S, et al. Systematic review and meta-analysis to estimate the birth prevalence of five inherited metabolic diseases. J Inherit Metab Dis. 2014;37(6):889–98.
Spiekerkoetter U, et al. General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatr Res. 2004;55(2):190–6.
IJlst L, et al. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest. 1996;98(4):1028–33.
Spiekerkoetter U, et al. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009;32(4):498–505.
Spiekerkoetter U, Mayatepek E. Update on mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33(5):467–8.
Arnold GL, et al. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96(3):85–90.
Bach AC, Babayan VK. Medium-chain triglycerides: an update. Am J Clin Nutr. 1982;36(5):950–62.
Hoffmann L, et al. VLCAD enzyme activity determinations in newborns identified by screening: a valuable tool for risk assessment. J Inherit Metab Dis. 2012;35(2):269–77.
Spiekerkoetter U, et al. Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33(5):555–61.
Roussel J, et al. Carnitine deficiency induces a short QT syndrome. Heart Rhythm. 2016;13(1):165–74.
Rijlaarsdam RS, et al. Ventricular fibrillation without overt cardiomyopathy as first presentation of organic cation transporter 2-deficiency in adolescence. Pacing Clin Electrophysiol. 2004;27(5):675–6.
Chalmers RA, et al. Urinary excretion of l-carnitine and Acylcarnitines by patients with disorders of organic acid metabolism: evidence for secondary insufficiency of l-carnitine. Pediatr Res. 1984;18:1325–8.
Nasser, M., et al., Carnitine supplementation for inborn errors of metabolism. Cochrane Database Syst Rev, 2012(2): p. Cd006659.
Corr PB, Heathers GP, Yamada KA. Mechanisms contributing to the arrhythmogenic influences of alpha 1-adrenergic stimulation in the ischemic heart. Am J Med. 1989;87(2A):19S–25S.
Exil VJ, Gardner CD, Rottman JN, Sims H, Bartelds B, Khuchua Z, et al. Abnormal mitochondrial bioenergetics and heart rate dysfunction in mice lacking very-long-chain acyl-CoA dehydrogenase. Am J Physiol Heart Circ Physiol. 2006;290(3):H1289–97.
Watanabe K, et al. Two siblings with very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency suffered from rhabdomyolysis after l-carnitine supplementation. Mol Genet Metab Rep. 2018;15:121–3.
Angelini C, et al. Task force guidelines handbook: EFNS guidelines on diagnosis and management of fatty acid mitochondrial disorders. Eur J Neurol. 2006;13(9):923–9.
Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis. 2010;33(5):501–6.
Lund AM, et al. Clinical and biochemical monitoring of patients with fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33(5):495–500.
Bakermans AJ, et al. Myocardial energy shortage and unmet anaplerotic needs in the fasted long-chain acyl-CoA dehydrogenase knockout mouse. Cardiovasc Res. 2013;100(3):441–9.
Roe CR, Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis. 2006;29(2–3):332–40.
Gillingham MB, et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40(6):831–43.
Vockley J, et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol Genet Metab. 2016;119(3):223–31.
Roe CR, et al. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 2002;110(2):259–69.
Aires V, et al. Stilbenes and resveratrol metabolites improve mitochondrial fatty acid oxidation defects in human fibroblasts. Orphanet J Rare Dis. 2014;9:79.
Bastin J, Lopes-Costa A, Djouadi F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum Mol Genet. 2011;20(10):2048–57.
Bleeker JC, Houtkooper RH. Sirtuin activation as a therapeutic approach against inborn errors of metabolism. J Inherit Metab Dis. 2016;39:565–72.
Djouadi F, et al. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum Mol Genet. 2005;14(18):2695–703.
Djouadi F, et al. Peroxisome proliferator activated receptor delta (PPARdelta) agonist but not PPARalpha corrects carnitine palmitoyl transferase 2 deficiency in human muscle cells. J Clin Endocrinol Metab. 2005;90(3):1791–7.
Bonnefont JP, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. 2010;88(1):101–8.
Orngreen MC, et al. Bezafibrate in skeletal muscle fatty acid oxidation disorders: a randomized clinical trial. Neurology. 2014;82(7):607–13.
Orngreen MC, Vissing J, Laforet P. No effect of bezafibrate in patients with CPTII and VLCAD deficiencies. J Inherit Metab Dis. 2015;38(2):373–4.
Van Hove JL, et al. D,L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD). Lancet. 2003;361(9367):1433–5.
Wüst RC, et al. Ketones and inborn errors of metabolism: old friends revisited. J Inherit Metab Dis. 2017;40(1):3–4.
Cox PJ, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2016;24(2):256–68.
Bleeker, J.C., et al., Ketonesters as a Possible Novel Therapeutic Option for Patients With Fatty Acid Oxidation Disorders [abstract]. Abstracts presented at the 13th international congress of inborn errors of metabolism - ICIEM 2017. JIEMS abstract nr. 508, 2017. 5: p. 2326409817722292.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest related to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Knottnerus, S.J.G., Bleeker, J.C., Wüst, R.C.I. et al. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord 19, 93–106 (2018). https://doi.org/10.1007/s11154-018-9448-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-018-9448-1