
Abstract
Over the years, the mitochondrial fatty acid β-oxidation (FAO) pathway has been characterised at the biochemical level as well as the molecular biological level. FAO plays a pivotal role in energy homoeostasis, but it competes with glucose as the primary oxidative substrate. The mechanisms behind this so-called glucose–fatty acid cycle operate at the hormonal, transcriptional and biochemical levels. Inherited defects for most of the FAO enzymes have been identified and characterised and are currently included in neonatal screening programmes. Symptoms range from hypoketotic hypoglycaemia to skeletal and cardiac myopathies. The pathophysiology of these diseases is still not completely understood, hampering optimal treatment. Studies of patients and mouse models will contribute to our understanding of the pathogenesis and will ultimately lead to better treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucose, fatty acids and amino acids are the three substrates an organism can use to maintain metabolic homoeostasis. They are required for the generation of energy, but also as building blocks for the biosynthesis of (macro)molecules. The prime pathway for the degradation of fatty acids is mitochondrial fatty acid β-oxidation (FAO) (Bartlett and Eaton 2004). FAO is a key metabolic pathway for energy homoeostasis in organs such as the liver, heart and skeletal muscle. During fasting, when glucose supply becomes limited, FAO is of particular importance. Under this condition, most tissues, except the brain, can use fatty acids directly to generate energy. Furthermore, the liver converts fatty acids into ketone bodies, a process for which FAO is indispensable. Ketone bodies serve as an additional energy source that is used by all tissues including the brain. In this review, we sketch a historical perspective of FAO research and introduce different aspects of the topic.
Historical perspective
Georg Franz Knoop discovered fatty acid β-oxidation. In 1904, he published his classical experiments using odd and even chain ω-phenyl fatty acids such as ω-phenylvaleric acid and ω-phenylbutyric acid (Knoop 1904). Knoop fed these compounds to dogs and analysed their urine. In dogs that had been fed the odd chain fatty acids, he found hippuric acid (conjugate of benzoic acid and glycine), whereas, the dogs that had been fed even chain fatty acids excreted phenaceturic acid (conjugate of phenylacetic acid and glycine). From this he concluded that the metabolism of fatty acids proceeds by the successive removal of two carbon fragments. The remaining fatty chain had to contain a carboxylic acid. He postulated that oxidation took place on the β carbon atom, an oxidation unknown to organic chemistry. Henry Drysdale Dakin confirmed and further developed the β-oxidation theory of Knoop, also by using phenyl derivatives of fatty acids (Dakin 1908). Another landmark was Irving B. Fritz’s discovery of the stimulation of FAO by carnitine (Fritz 1955; Fritz and McEwen 1959). The first inherited defects in the FAO pathway were identified in the 1970s, carnitine palmitoyltransferase 2 (CPT2) deficiency in 1973, primary carnitine deficiency in 1975 and medium chain acyl-coenzyme A (CoA) dehydrogenase (MCAD) deficiency in 1976 (DiMauro and DiMauro 1973; Karpati et al. 1975; Gregersen et al. 1976). Most FAO enzymes were purified in the 1980s (Furuta et al. 1981), followed by the cloning of the individual genes and the subsequent identification of disease-causing mutations in patients (Kelly et al. 1990; Yokota et al. 1990; Matsubara et al. 1990).
Biochemical basis of fatty acid oxidation
Transport across the plasma membrane and fatty acid activation
Mitochondria, as well as peroxisomes, harbour all enzymes necessary for FAO. Mitochondria are the main site for the oxidation of plasma free fatty acids or lipoprotein-associated triglycerides. This implies that several transport steps are necessary before fatty acids are oxidised. Triglycerides are first hydrolysed by the action of endothelium-bound lipoprotein lipase. The uptake of fatty acids seems to be largely mediated by membrane proteins, although passive uptake probably also occurs.
Fatty acid transport proteins (FATPs) are integral transmembrane proteins that enhance the uptake of long chain and very long chain fatty acids into cells. In humans, FATPs comprise a family of six highly homologous proteins, FATP1–FATP6, which are found in all fatty acid-utilising tissues of the body (Doege and Stahl 2006). FATPs have acyl-CoA synthetase activity, suggesting that fatty acids are rapidly converted to acyl-CoAs after translocation across the plasma membrane, a process that may drive the transport. Skeletal muscle expresses FATP1 (SCL27A1) and FATP4 (SCL27A4), whereas heart specifically expresses FATP6 (SCL27A6) as well as FATP1 (Gimeno et al. 2003). In liver, FATP5 (SCL27A5) plays a crucial role in the hepatocellular uptake of fatty acids (Doege et al. 2006). Interestingly, there is a second protein sub-family with long chain acyl-CoA synthetase activity, the long-chain acyl-CoA synthetases (ACSL) (Soupene and Kuypers 2008), illustrating that fatty acid activation is a complex process. Besides FATPs, plasma membrane fatty acid-binding protein (FABPpm, which is identical to mitochondrial aspartate aminotransferase, gene name GOT2), and fatty acid translocase (FAT, CD36) are involved in the uptake of fatty acids (Glatz et al. 2006; Kiens 2006).
In order to maintain high rates of FAO in liver, heart and skeletal muscle, cytoplasmic fatty acid-binding proteins (FABPs) are required (Schaap et al. 1999; Binas et al. 2003; Erol et al. 2004). There are many tissue-specific FABPs. Liver FABP is encoded by the FABP1 gene. Heart and skeletal muscle express heart type FABP (FABP3).
The carnitine shuttle
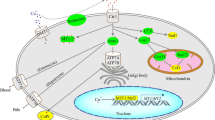
The mitochondrial membrane is impermeable to acyl-CoAs. For their import into mitochondria, acyl-CoAs use the carnitine shuttle (Fig. 1). The first step of this shuttle is performed by CPT1, which converts an acyl-CoA into an acylcarnitine. There are two isoforms that are important for FAO. CPT1A (gene CPT1A), also called liver CPT1, is not only expressed in the liver, but also in the brain, kidney, lung, spleen, intestine, pancreas, ovary and fibroblasts. CPT1B (gene CPT1B) is the muscle isoform that is highly expressed in heart, skeletal muscle and testis. Both proteins are present at the outer mitochondrial membrane and are sensitive to inhibition by malonyl-CoA. Carnitine acylcarnitine translocase (CACT, SLC25A20) exchanges acylcarnitines for a free carnitine molecule from the inside. Once the acylcarnitines have entered the mitochondria, CPT2 (gene CPT2), located at the mitochondrial inner membrane, reconverts the acylcarnitines into their CoA esters, which can then undergo FAO (van der Leij et al. 2000; Ramsay et al. 2001; Bonnefont et al. 2004). CPT1C is a brain-specific CPT with a currently unknown function (Price et al. 2002). Both CPT1 and CPT2 are primarily involved in the import of (dietary) long-chain acyl-CoAs, such as palmitoyl-CoA, oleoyl-CoA, and linoleoyl-CoA.

Mitochondrial fatty acid β-oxidation in humans a and in the mouse b. After transport across the plasma membrane, fatty acids are activated to acyl-CoAs at the cytosolic site. CPT1 converts the acyl-CoA into an acylcarnitine, which is subsequently transported across the mitochondrial membrane by CACT. CPT2 converts the acylcarnitine back into an acyl-CoA. Long chain acyl-CoAs are metabolised by the membrane bound enzymes, very long chain acyl-CoA dehydrogenase (VLCAD) and mitochondrial trifunctional protein (MTP), which has hydratase, long chain hydroxyacyl-CoA dehydrogenase (LCHAD) and thiolase activity. Short and medium chain acyl-CoAs are metabolised in the mitochondrial matrix by medium chain acyl-CoA dehydrogenase (MCAD), short chain acyl-CoA dehydrogenase (SCAD), crotonase, medium and short chain hydroxyacyl-CoA dehydrogenase (M/SCHAD) and medium chain 3-ketoacyl-CoA thiolase (MCKAT). The oxidation of unsaturated fatty acids such as oleic acid requires the action of an isomerase [dodecenoyl-CoA delta isomerase (DCI)]. C18 denotes an acyl-CoA with a chain length of 18 carbon atoms, and so forth
Alternatively, carnitine can be converted in the mitochondrial matrix into an acylcarnitine by the action of CPT2 or carnitine acetyl transferase (CAT, gene CRAT). These acylcarnitines can cross the mitochondrial membrane also in the opposite direction via CACT, resulting in the transport of these acylcarnitines into the cytosol. Acylcarnitines can also cross the plasma membrane, but the mechanism is currently unknown. After crossing the plasma membrane, acylcarnitines are excreted from the body via either urine or bile. This detoxification mechanism is especially important when acyl-CoAs accumulate; for example, in disorders of mitochondrial FAO. Recently this pathway has been proposed to play a role in the pathogenesis of type 2 diabetes and insulin resistance. These conditions are characterised by a metabolic overload of mitochondria with fatty acids, which is reflected by increased levels of plasma acylcarnitines and decreased free carnitine levels. The elevation of free carnitine and, thereby, promotion of the export of acylcarnitines is proposed as a novel therapeutic approach (Koves et al. 2008; Noland et al. 2009; Mynatt 2009; Adams et al. 2009). For the maintenance of carnitine homoeostasis, plasma carnitine can be taken up by tissues via the carnitine transporter, organic cation transporter 2 (gene OCTN2).
The β-oxidation cycle
Once inside the mitochondria, acyl-CoAs are degraded into acetyl-CoA units via the classic series of four enzyme reactions called β-oxidation (Fig. 1). The β-oxidation pathway is a cyclic process in which acyl-CoAs are shortened, whereby the two carboxy-terminal carbon atoms are released as acetyl-CoA units each time a cycle is fully completed. First, an acyl-CoA-ester is dehydrogenated to yield a trans-2-enoyl-CoA. This is followed by hydration of the double bond. In the third step the resulting L-3-hydroxy-acyl-CoA is dehydrogenated to 3-keto-acyl-CoA. Finally, thiolytic cleavage of the 3-keto-acyl-CoA produces a two-carbon chain-shortened acyl-CoA plus acetyl-CoA. Each cycle yields an acyl-CoA shortened by two carbon atoms, an acetyl-CoA, and one nicotinamide adenine dinucleotide (NADH) and one flavin adenine dinucleotide (FADH2) as electron carriers (or reducing equivalents). The resulting acyl-CoA enters another cycle of FAO; the acetyl-CoA can enter the citric acid cycle and the electron carriers deliver the electrons to the electron transport chain.
Directly after import, the long chain acyl-CoAs are metabolised first by the membrane-bound enzymes, very long chain acyl-CoA dehydrogenase (VLCAD, encoded by ACADVL) and mitochondrial trifunctional protein (MTP, encoded by HADHA and HADHB), which has hydratase, long chain hydroxyacyl-CoA dehydrogenase (LCHAD) and thiolase activity. After 2–3 FAO cycles using these enzymes, the resulting medium acyl-CoAs are metabolised in the mitochondrial matrix. The first step is performed by MCAD (encoded by ACADM) and, after another 3–4 FAO cycles, short chain acyl-CoA dehydrogenase (SCAD, encoded by ACADS) participates in the final 1–2 cycles. For the second, third and fourth steps of FAO of medium and short chain acyl-CoAs, there is one set of three distinct matrix-soluble enzymes: the enoyl-CoA hydratase called crotonase (encoded by ECHS1), the medium and short chain hydroxyacyl-CoA dehydrogenase (M/SCHAD, encoded by HADH) and medium chain 3-ketoacyl-CoA thiolase (MCKAT, encoded by ACAA2). All have broad substrate specificity towards CoA-esters with chain lengths of up to ten carbon atoms. The oxidation of mono-unsaturated fatty acids such as oleic acid requires the action of dodecenoyl-CoA delta isomerase (3,2 trans-enoyl-CoA isomerase, encoded by DCI). Polyunsaturated fatty acids need a second auxiliary enzyme, 2,4-dienoyl CoA reductase 1 (DECR1). The FAO pathway as described above is displayed in Fig. 1.
An acyl-coenzyme A dehydrogenase (ACAD) enzyme performs the first step of each FAO cycle. ACAD enzymes have several interesting aspects. First, they have tightly bound FAD as a prosthetic group. The reduced flavoproteins are re-oxidised by electron transfer flavoprotein (ETF). ETF is a heterodimer consisting of two flavoproteins, the α and β subunits (ETFA and ATFB). The reducing equivalents are subsequently transferred to ETF dehydrogenase (ETFDH), another flavoprotein that feeds the electrons into the respiratory chain via ubiquinone. Second, whereas there seems to be only one membrane-associated enzyme system and one matrix localised enzyme system for the second, third and fourth FAO steps, five different ACAD enzymes may catalyse the first step in FAO. VLCAD is specific for long chain acyl-CoAs, whereas LCAD has broad substrate specificity and is active with medium and long chain acyl-CoAs. Apart from straight-chain substrates, LCAD has been shown to catalyse the degradation of branched-chain acyl-CoAs and certain mono-unsaturated and polyunsaturated acyl-CoAs (Wanders et al. 1998; Le et al. 2000; Chegary et al. 2009). In contrast to the membrane-associated VLCAD, LCAD is present in the mitochondrial matrix. In humans, LCAD does not seem to play a prominent role in FAO, due to the extremely low expression levels (Chegary et al. 2009). In rodents, however, LCAD plays a significant role in FAO, as evidenced by the phenotype of LCAD−/− mice (Fig. 1a,b (Kurtz et al. 1998; Cox et al. 2001; Chegary et al. 2009)). ACAD9 is a second enzyme associated with the mitochondrial inner membrane that is able to dehydrogenate long chain acyl-CoAs (Zhang et al. 2002; Ensenauer et al. 2005). Its exact function in FAO remains to be elucidated, despite the recent description of three patients suggested to suffer from ACAD9-deficiency (He et al. 2007). Medium and short chain acyl-CoAs are handled by MCAD and SCAD, respectively, that reside in the matrix.
Regulation of fatty acid oxidation and substrate use in energy homoeostasis
Organs are flexible in the choice of which substrate they will use to maintain energy homoeostasis. Under normal, well-fed conditions, glucose will be the preferred substrate for oxidation. During fasting, however, fatty acids and ketone bodies will become a more important energy source as an alternative to glucose. This reciprocal relationship or competition between the oxidation of fatty acids and glucose is also known as the glucose–fatty acid or Randle cycle. A general explanation for the occurrence of this cycle is the glucose-sparing effect of FAO and ketone body utilisation. Thus, glucose can be saved for use in the brain when nutritional sources of glucose are scarce. In muscle, the situation is more complex. During exercise, the choice of substrate is determined by exercise intensity and duration. Glucose utilisation increases as exercise intensity increases. In contrast, FAO is already maximal at moderate exercise intensity (Helge et al. 2007). In the heart, the situation is also exceptional. FAO is extremely important for this organ, since it constantly relies on fatty acids for a major proportion (60–90%) of its energy needs (Schulz 1994).
There are several mechanisms for the regulation of cellular substrate use. We briefly describe these levels of regulation, but, for an in depth description of these and other mechanisms, the reader is referred to a recent excellent review (Hue and Taegtmeyer 2009). An important driver in substrate use is substrate availability. Hormones are crucial players at this level. Insulin is secreted by the pancreatic β-cells when blood glucose levels are high. One of the main effects of insulin is the increase of glucose transport by translocation of the glucose transporter Glut4 (SLC2A4) from intracellular vesicles to the plasma membrane. Insulin also inhibits lipolysis, and thus prevents the availability of its competing substrate fatty acid. On the other hand, during fasting or exercise, the hormones adrenalin, noradrenaline, glucagon and adrenocorticotropic hormone (ACTH) induce lipolysis by stimulating hormone-sensitive lipase activity in the adipose tissue. This leads to the release of free fatty acids into the circulation.
Metabolites can also affect the transcriptional regulation of genes involved in metabolism such as enzymes. For example, free fatty acids are activating ligands for peroxisome proliferator-activated receptors [PPARs (Krey et al. 1997; Forman et al. 1997; Murakami et al. 1999; Xu et al. 1999)], a subclass of the nuclear hormone receptor family. Nuclear hormone receptors typically consist of a DNA-binding domain and a ligand-binding domain. The DNA-binding domain binds to specific so-called response elements in the promoter of target genes. Upon ligand binding, nuclear receptors (NRs) undergo a conformation change, which induces dissociation of co-repressors and the recruitment of transcriptional cofactors, resulting in transcription activation. This property makes NRs on the one hand interesting pharmaceutical targets and on the other hand potential mediators in the pathology of disorders in which potential ligands accumulate. Activation of PPARα and PPARβ by (dietary) fatty acids such as oleic acid makes tissues more dependent on FAO, by stimulating cellular fatty acid utilisation pathways including transport, esterification, and oxidation (Mandard et al. 2004).
Effects on transcriptional regulation are often long-term adaptations. Several mechanisms exist for the short-term regulation of substrate use. The activity of specific enzymes such as pyruvate dehydrogenase (PDH), phosphofructokinase and CPT1, is controlled by metabolites (Hue and Taegtmeyer 2009).
CPT1 catalyses one of the rate-limiting steps in FAO (Drynan et al. 1996). CPT1 activity is regulated by its inhibitor: malonyl-CoA. The cellular energy status is one of the regulators of malonyl-CoA production. During energy shortage, adenosine triphosphate (ATP) levels fall and adenosine monophosphate levels (AMP) rise, resulting in the activation of AMP-activated protein kinase (AMPK). Activated AMPK initiates a signalling cascade aimed to restore cellular energy levels. A principal event is the phosphorylation of acetyl-CoA carboxylase (ACC), the enzyme that converts acetyl-CoA in malonyl-CoA. Thus, AMPK activation aims to increase energy production by FAO.
PDH catalyses the oxidative decarboxylation of pyruvate to acetyl-CoA and links glycolysis with oxidative metabolism. The reaction is irreversible and, therefore, well controlled. The main regulation occurs by reversible phosphorylation of PDH, which abolishes enzyme activity. The PDH complex contains associated regulatory enzymes, including pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase. The activities of these enzymes determine the proportion of PDH in its active dephosphorylated state. The activity of PDK is regulated by short- and long-term mechanisms. Short-term regulation includes activation of PDK by the reaction products of PDH, i.e. acetyl-CoA and NADH. Thus, high acetyl-CoA/CoA and NADH/NAD+ ratios activate PDK, leading to inhibitory phosphorylation of PDH. The substrate of PDH, i.e. pyruvate, inhibits PDK activity. Long-term regulation of PDK activity occurs primarily at the transcriptional level. For example, PPARs stimulate the transcription of PDK4 (Wu et al. 2001; Huang et al. 2002), an effect that can be potentiated by the activation of AMPK (Houten et al. 2009).
FAO deficiencies
For almost each enzyme involved in FAO, inherited defects have been described (Wanders et al. 1999; Rinaldo et al. 2002). These include glutaric aciduria type 2, primary carnitine deficiency and deficiencies of CPT1a, CACT, CPT2, VLCAD, MTP (including isolated LCHAD or thiolase), MCAD, M/SCHAD, SCAD and 2,4-dienoyl CoA reductase (DECR). Interestingly, CPT1b, crotonase, MCKAT and DCI deficiency have not been identified as of yet.
In general, FAO defects have three different presentations (Wanders et al. 1999; Rinaldo et al. 2002). The first is the hepatic presentation, which is a severe, often lethal, disease in infancy or the neonatal period with hypoketotic hypoglycaemia and Reye-like syndrome. This disease is triggered by a catabolic state, for example during intercurrent infections. Most importantly, this condition can be prevented, which is the main reason for the inclusion of FAO defects in neonatal screening programmes. During infancy, patients may also present with cardiac symptoms such as dilated or hypertrophic cardiomyopathy and/or arrhythmias. Alternatively, FAO defects might present as a milder, later (‘adult’) onset disease. This form is characterised by exercise-induced myopathy and rhabdomyolysis. Severely affected patients may display combinations of all three presentations. In addition, FAO defects have been associated with sudden infant death that may have been caused by hypoketotic hypoglycaemia or cardiac disease.
Cardiac presentation and muscular signs in FAO defects
The cardiac presentation and muscular signs in FAO defects have been documented in a group of 107 patients (89 families) with an FAO defect (Saudubray et al. 1999; Bonnet et al. 1999). Table 1 summarises the data from this paper. Cardiac involvement and/or muscular signs have never been described in CPT1a and MCAD deficiency. For CPT1a deficiency, this is probably explained by the specific expression of CPT1b in heart and skeletal muscle. For MCAD deficiency, there are two likely explanations. First, energy generation by the FAO pathway is not completely impaired in these patients. Second, medium chain acylcarnitines may be less toxic than long chain acylcarnitines. This explains why MCAD-deficient patients can be completely asymptomatic. The disease presents itself usually as hypoketotic hypoglycaemia triggered by prolonged fasting during intercurrent infections. If one leaves these patients and a corresponding proportion of patients with an unknown defect out of this group (87 patients), 63% of the patients have a cardiac presentation and 51% have muscular signs.
Of interest is the high frequency of heart beat disorders in patients with long chain FAO defects [Table 1, (Saudubray et al. 1999; Bonnet et al. 1999)]. Often, this is without cardiomyopathy. These include conduction abnormalities such as left bundle-branch block, atrioventricular block and sinus node dysfunction, and arrhythmias such as ventricular tachycardia, ventricular fibrillation and supraventricular tachycardia. Bonnet et al. suggested that FAO deficiencies should be considered in unexplained sudden infant death or collapse in infants, but also in infants with conduction defects or ventricular tachycardia (Bonnet et al. 1999).
Pathophysiology of FAO defects
So far, the exact pathogenetic mechanisms leading to hypoglycaemia in FAO defects have only partially been elucidated. Two different explanations are likely: increased use of glucose and/or decreased glucose production. FAO is able to stimulate gluconeogenesis (GNG) in vitro (Williamson et al. 1966) as well as in vivo (Fanelli et al. 1993). In accordance with this notion, inhibitors of FAO abolished the stimulation of GNG by FAO (Williamson et al. 1968). This effect was mediated by pyruvate carboxylase, the rate-limiting enzyme of GNG, which has an absolute requirement for acetyl-CoA as cofactor (Utter and Keech 1963; Scrutton and Utter 1967). Low acetyl-CoA levels due to a FAO defect could limit GNG.
On the other hand, patients with an FAO defect will generate less energy from fatty acids and ketones. Given the glucose–fatty acid cycle (see above), glucose use will be increased. Fuel metabolism has been assessed in patients with CPT2 and VLCAD deficiency (Orngreen et al. 2004, 2005). These patients had normal FAO at rest, but prolonged low intensity exercise revealed impaired FAO and a high respiratory exchange ratio, indicating that the defect in FAO was compensated for by increased carbohydrate metabolism, in this case enhanced muscle glycogenolysis (Orngreen et al. 2004, 2005).
To explain the pathogenesis of muscle disease in FAO defects, two hypotheses seem likely. First, energy shortage due to the FAO defect may be the primary event leading to disease. Alternatively, accumulation of metabolites could cause the myopathies. Of course, a combination of both disease mechanisms is equally possible.
Rhabdomyolysis
The pathophysiology of rhabdomyolysis is heterogeneous and, in some cases, not completely understood. In general, it seems likely that the cause lies in the breakdown of sodium/calcium homoeostasis in the muscle cell (Huerta-Alardin et al. 2005). ATP depletion due to the FAO defect could lead to insufficient function of the sodium/potassium ATPase, the calcium ATPase and the calcium/sodium exchanger (Warren et al. 2002; Huerta-Alardin et al. 2005). Alternatively, accumulation of FAO intermediates could cause increased ion leakage. The final common effector pathway is thought to be an increase in free cytosolic Ca2+, which may start a cascade of effects leading to major cell permeability and capillary leakage.
Cardiomyopathy and heart beat disorders
FAO is very important for the heart. This is illustrated by the fact that the heart constantly relies on fatty acids for a major proportion (60–90%) of its energy needs. Depending on the feeding status, glucose, lactate and ketone bodies are used as important alternative fuels. One important issue that has not been resolved is the question of whether oxidation of these substrates can fully compensate for the FAO defect. It is, however, clear that metabolic flexibility is very important for the heart. For example, type 2 diabetes patients are also at risk of developing cardiomyopathy. This coincides with increased cardiac reliance on fatty acids as the primary energy source, subsequent to the increased circulating free fatty acid levels (Huss and Kelly 2005). There are also numerous genetic mouse models in which cardiac substrate use has been modulated. In general, this leads to cardiomyopathy. It is not clear how a decrease in metabolic flexibility leads to cardiomyopathy. Moreover, it is not known if any of the accumulating metabolites in FAO defects are toxic to the heart, although acylcarnitines and fatty acids have been suggested to contribute to arrhythmias (Corr et al. 1989; Huang et al. 1992). This could provide an explanation for the heart-beat disorders in FAO defects and the defect in Ca2+ homoeostasis in the VLCAD−/− mouse (Bonnet et al. 1999; Werdich et al. 2007).
Mouse models for study of the pathogenesis of FAO defects
Several mouse models with defects in FAO have been reported. Two of these models are spontaneous mutants (jvs and SCAD); the other models have been created by genetic manipulation. Table 2 shows the mouse models and a comparison of the human disease with the mouse phenotype. With the exception of the different ACAD deficiencies, these knockout (KO) mouse models have severe or lethal phenotypes. This may be expected when one considers the severe (neonatal) disease presentation in humans, which is often caused by mutations that completely inactivate the enzyme or prevent its correct synthesis. The relatively mild phenotype of the VLCAD−/− mice is unexpected. In humans, VLCAD deficiency is a severe disease, whereas, in mice, there is only a mild phenotype. The VLCAD−/− mouse has mild hepatic steatosis and mild fatty change in the heart in response to fasting or cold (Cox et al. 2001; Exil et al. 2003). Exil et al. further characterised the VLCAD−/− hearts and demonstrated microvesicular lipid accumulation, marked mitochondrial proliferation, and facilitated induction of polymorphic ventricular tachycardia, without preceding stress (Exil et al. 2006). The LCAD−/− mouse has a more severe phenotype, more closely resembling that of human VLCAD deficiency. Characteristics include reduced fasting tolerance accompanied by tissue lipidosis, hypoglycaemia and signs of cardiomyopathy (Kurtz et al. 1998; Cox et al. 2001, 2009). In humans, LCAD deficiency has not been identified. This remarkable difference is most likely explained by LCAD expression levels. In humans, LCAD expression is very low in all organs studied, but, in mice (and rats), LCAD is expressed at levels comparable to those of VLCAD. Thus, LCAD can take over the function of VLCAD and vice versa, but the role of LCAD is somewhat more prominent, making the LCAD KO the most useful model (Chegary et al. 2009).
Conclusion
Many biochemical and genetic aspects of FAO and its deficiencies have been addressed by recent and past research. The exact pathophysiological mechanisms that cause the different disease presentations are still poorly defined, hampering the design of rational therapeutic strategies. Although feasible, studies of patients are difficult for many reasons. Detailed studies using mouse models will undoubtedly contribute to a better understanding of disease pathogenesis.
Abbreviations
- FAO:
-
mitochondrial fatty acid β-oxidation
- CPT:
-
carnitine palmitoyltransferase
- MCAD:
-
medium chain acyl-coenzyme A (CoA) dehydrogenase
- CACT:
-
carnitine acylcarnitine translocase
- (V)LCAD:
-
(very) long chain acyl-CoA dehydrogenase
- MTP:
-
mitochondrial trifunctional protein
- LCHAD:
-
long chain hydroxyacyl-CoA dehydrogenase
- SCAD:
-
short chain acyl-CoA dehydrogenase
- OCTN:
-
organic cation transporter
- DCI:
-
dodecenoyl-CoA delta isomerase
- DECR:
-
2,4-dienoyl CoA reductase
- M/SCHAD:
-
medium and short chain hydroxyacyl-CoA dehydrogenase
References
Adams SH, Hoppel CL, Lok KH et al (2009) Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 139:1073–1081
Bartlett K, Eaton S (2004) Mitochondrial beta-oxidation. Eur J Biochem 271:462–469
Binas B, Han XX, Erol E et al (2003) A null mutation in H-FABP only partially inhibits skeletal muscle fatty acid metabolism. Am J Physiol Endocrinol Metab 285:E481–E489
Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J (2004) Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med 25:495–520
Bonnet D, Martin D, de Lonlay P et al (1999) Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation 100:2248–2253
Chegary M, te Brinke H, Ruiter JP et al (2009) Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta 1791:806–815
Corr PB, Creer MH, Yamada KA, Saffitz JE, Sobel BE (1989) Prophylaxis of early ventricular fibrillation by inhibition of acylcarnitine accumulation. J Clin Invest 83:927–936
Cox KB, Hamm DA, Millington DS et al (2001) Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum Mol Genet 10:2069–2077
Cox KB, Liu J, Tian L, Barnes S, Yang Q, Wood PA (2009) Cardiac hypertrophy in mice with long-chain acyl-CoA dehydrogenase or very long-chain acyl-CoA dehydrogenase deficiency. Lab Invest 89:1348–1354
Dakin HD (1908) Comparative studies of the mode of oxidation of phenyl derivatives of fatty acids by the animal organism and by hydrogen peroxide. J Biol Chem 4:419–435
DiMauro S, DiMauro PM (1973) Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 182:929–931
Doege H, Stahl A (2006) Protein-mediated fatty acid uptake: novel insights from in vivo models. Physiology (Bethesda) 21:259–268
Doege H, Baillie RA, Ortegon AM et al (2006) Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis. Gastroenterology 130:1245–1258
Drynan L, Quant PA, Zammit VA (1996) Flux control exerted by mitochondrial outer membrane carnitine palmitoyltransferase over beta-oxidation, ketogenesis and tricarboxylic acid cycle activity in hepatocytes isolated from rats in different metabolic states. Biochem J 317(Pt 3):791–795
Ensenauer R, He M, Willard JM et al (2005) Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids. J Biol Chem 280:32309–32316
Erol E, Kumar LS, Cline GW, Shulman GI, Kelly DP, Binas B (2004) Liver fatty acid binding protein is required for high rates of hepatic fatty acid oxidation but not for the action of PPARalpha in fasting mice. FASEB J 18:347–349
Exil VJ, Roberts RL, Sims H et al (2003) Very-long-chain acyl-coenzyme a dehydrogenase deficiency in mice. Circ Res 93:448–455
Exil VJ, Gardner CD, Rottman JN et al (2006) Abnormal mitochondrial bioenergetics and heart rate dysfunction in mice lacking very-long-chain acyl-CoA dehydrogenase. Am J Physiol Heart Circ Physiol 290:H1289–H1297
Fanelli C, Calderone S, Epifano L et al (1993) Demonstration of a critical role for free fatty acids in mediating counterregulatory stimulation of gluconeogenesis and suppression of glucose utilization in humans. J Clin Invest 92:1617–1622
Forman BM, Chen J, Evans RM (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A 94:4312–4317
Fritz IB (1955) The effect of muscle extracts on the oxidation of palmitic acid by liver slices and homogenates. Acta Physiol Scand 34:367–385
Fritz IB, McEwen B (1959) Effects of carnitine on fatty-acid oxidation by muscle. Science 129:334–335
Furuta S, Miyazawa S, Hashimoto T (1981) Purification and properties of rat liver acyl-CoA dehydrogenases and electron transfer flavoprotein. J Biochem 90:1739–1750
Gimeno RE, Ortegon AM, Patel S et al (2003) Characterization of a heart-specific fatty acid transport protein. J Biol Chem 278:16039–16044
Glatz JF, Bonen A, Ouwens DM, Luiken JJ (2006) Regulation of sarcolemmal transport of substrates in the healthy and diseased heart. Cardiovasc Drugs Ther 20:471–476
Gregersen N, Lauritzen R, Rasmussen K (1976) Suberylglycine excretion in the urine from a patient with dicarboxylic aciduria. Clin Chim Acta 70:417–425
He M, Rutledge SL, Kelly DR et al (2007) A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am J Hum Genet 81:87–103
Helge JW, Stallknecht B, Richter EA, Galbo H, Kiens B (2007) Muscle metabolism during graded quadriceps exercise in man. J Physiol 581:1247–1258
Houten SM, Chegary M, te Brinke H et al (2009) Pyruvate dehydrogenase kinase 4 expression is synergistically induced by AMP-activated protein kinase and fatty acids. Cell Mol Life Sci 66:1283–1294
Huang JM, Xian H, Bacaner M (1992) Long-chain fatty acids activate calcium channels in ventricular myocytes. Proc Natl Acad Sci U S A 89:6452–6456
Huang B, Wu P, Bowker-Kinley MM, Harris RA (2002) Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes 51:276–283
Hue L, Taegtmeyer H (2009) The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297:E578–E591
Huerta-Alardin AL, Varon J, Marik PE (2005) Bench-to-bedside review: Rhabdomyolysis—an overview for clinicians. Crit Care 9:158–169
Huss JM, Kelly DP (2005) Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest 115:547–555
Ibdah JA, Paul H, Zhao Y et al (2001) Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J Clin Invest 107:1403–1409
Janssen U, Stoffel W (2002) Disruption of mitochondrial beta-oxidation of unsaturated fatty acids in the 3, 2-trans-enoyl-CoA isomerase-deficient mouse. J Biol Chem 277:19579–19584
Ji S, You Y, Kerner J et al (2008) Homozygous carnitine palmitoyltransferase 1b (muscle isoform) deficiency is lethal in the mouse. Mol Genet Metab 93:314–322
Karpati G, Carpenter S, Engel AG et al (1975) The syndrome of systemic carnitine deficiency. Clinical, morphologic, biochemical, and pathophysiologic features. Neurology 25:16–24
Kelly DP, Whelan AJ, Ogden ML et al (1990) Molecular characterization of inherited medium-chain acyl-CoA dehydrogenase deficiency. Proc Natl Acad Sci U S A 87:9236–9240
Kiens B (2006) Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev 86:205–243
Knoop F (1904) Der Abbau aromatischer Fettsäuren im Tierkörper. Beitr Chem Physiol Pathol 6:150–162
Koizumi T, Nikaido H, Hayakawa J, Nonomura A, Yoneda T (1988) Infantile disease with microvesicular fatty infiltration of viscera spontaneously occurring in the C3H-H-2(0) strain of mouse with similarities to Reye's syndrome. Lab Anim 22:83–87
Koves TR, Ussher JR, Noland RC et al (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7:45–56
Krey G, Braissant O, L'Horset F et al (1997) Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol 11:779–791
Kurtz DM, Rinaldo P, Rhead WJ et al (1998) Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci U S A 95:15592–15597
Kuwajima M, Kono N, Horiuchi M et al (1991) Animal model of systemic carnitine deficiency: analysis in C3H-H-2 degrees strain of mouse associated with juvenile visceral steatosis. Biochem Biophys Res Commun 174:1090–1094
Le W, Abbas AS, Sprecher H, Vockley J, Schulz H (2000) Long-chain acyl-CoA dehydrogenase is a key enzyme in the mitochondrial beta-oxidation of unsaturated fatty acids. Biochim Biophys Acta 1485:121–128
Mandard S, Muller M, Kersten S (2004) Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci 61:393–416
Matsubara Y, Narisawa K, Miyabayashi S et al (1990) Identification of a common mutation in patients with medium-chain acyl-CoA dehydrogenase deficiency. Biochem Biophys Res Commun 171:498–505
Utter MF, Keech DB (1963) Pyruvate carboxylase. I. Nature of the reaction. J Biol Chem 238:2603–2608
Miinalainen IJ, Schmitz W, Huotari A et al (2009) Mitochondrial 2, 4-dienoyl-CoA reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. PLoS Genet 5:e1000543
Murakami K, Ide T, Suzuki M, Mochizuki T, Kadowaki T (1999) Evidence for direct binding of fatty acids and eicosanoids to human peroxisome proliferators-activated receptor alpha. Biochem Biophys Res Commun 260:609–613
Mynatt RL (2009) Carnitine and type 2 diabetes. Diabetes Metab Res Rev 25(Suppl 1):S45–S49
Noland RC, Koves TR, Seiler SE et al (2009) Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem 284:22840–22852
Nyman LR, Cox KB, Hoppel CL et al (2005) Homozygous carnitine palmitoyltransferase 1a (liver isoform) deficiency is lethal in the mouse. Mol Genet Metab 86:179–187
Orngreen MC, Norgaard MG, Sacchetti M, van Engelen BG, Vissing J (2004) Fuel utilization in patients with very long-chain acyl-coa dehydrogenase deficiency. Ann Neurol 56:279–283
Orngreen MC, Duno M, Ejstrup R et al (2005) Fuel utilization in subjects with carnitine palmitoyltransferase 2 gene mutations. Ann Neurol 57:60–66
Price N, van der Leij FR, Jackson V et al (2002) A novel brain-expressed protein related to carnitine palmitoyltransferase I. Genomics 80:433–442
Ramsay RR, Gandour RD, van der Leij FR (2001) Molecular enzymology of carnitine transfer and transport. Biochim Biophys Acta 1546:21–43
Rinaldo P, Matern D, Bennett MJ (2002) Fatty acid oxidation disorders. Annu Rev Physiol 64:477–502
Saudubray JM, Martin D, de Lonlay P et al (1999) Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis 22:488–502
Schaap FG, Binas B, Danneberg H, van der Vusse GJ, Glatz JF (1999) Impaired long-chain fatty acid utilization by cardiac myocytes isolated from mice lacking the heart-type fatty acid binding protein gene. Circ Res 85:329–337
Schiffer SP, Prochazka M, Jezyk PF, Roderick TH, Yudkoff M, Patterson DF (1989) Organic aciduria and butyryl CoA dehydrogenase deficiency in BALB/cByJ mice. Biochem Genet 27:47–58
Schulz H (1994) Regulation of fatty acid oxidation in heart. J Nutr 124:165–171
Scrutton MC, Utter MF (1967) Pyruvate carboxylase. IX. Some properties of the activation by certain acyl derivatives of coenzyme A. J Biol Chem 242:1723–1735
Soupene E, Kuypers FA (2008) Mammalian long-chain acyl-CoA synthetases. Exp Biol Med (Maywood) 233:507–521
Tolwani RJ, Hamm DA, Tian L et al (2005) Medium-chain acyl-CoA dehydrogenase deficiency in gene-targeted mice. PLoS Genet 1:e23
van der Leij FR, Huijkman NC, Boomsma C, Kuipers JR, Bartelds B (2000) Genomics of the human carnitine acyltransferase genes. Mol Genet Metab 71:139–153
Wanders RJ, Denis S, Ruiter JP, IJlst L, Dacremont G (1998) 2, 6-Dimethylheptanoyl-CoA is a specific substrate for long-chain acyl-CoA dehydrogenase (LCAD): evidence for a major role of LCAD in branched-chain fatty acid oxidation. Biochim Biophys Acta 1393:35–40
Wanders RJ, Vreken P, den Boer ME, Wijburg FA, van Gennip AH, IJlst L (1999) Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J Inherit Metab Dis 22:442–487
Warren JD, Blumbergs PC, Thompson PD (2002) Rhabdomyolysis: a review. Muscle Nerve 25:332–347
Werdich AA, Baudenbacher F, Dzhura I et al (2007) Polymorphic ventricular tachycardia and abnormal calcium handling in very-long-chain acyl-CoA dehydrogenase null mice. Am J Physiol Heart Circ Physiol 292:H2202–H2011
Williamson JR, Kreisberg RA, Felts PW (1966) Mechanism for the stimulation of gluconeogenesis by fatty acids in perfused rat liver. Proc Natl Acad Sci U S A 56:247–254
Williamson JR, Browning ET, Scholz R, Kreisberg RA, Fritz IB (1968) Inhibition of fatty acid stimulation of gluconeogenesis by (+)-decanoylcarnitine in perfused rat liver. Diabetes 17:194–208
Wood PA, Amendt BA, Rhead WJ, Millington DS, Inoue F, Armstrong D (1989) Short-chain acyl-coenzyme A dehydrogenase deficiency in mice. Pediatr Res 25:38–43
Wu P, Peters JM, Harris RA (2001) Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem Biophys Res Commun 287:391–396
Xu HE, Lambert MH, Montana VG et al (1999) Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell 3:397–403
Yokota I, Indo Y, Coates PM, Tanaka K (1990) Molecular basis of medium chain acyl-coenzyme A dehydrogenase deficiency. An A to G transition at position 985 that causes a lysine-304 to glutamate substitution in the mature protein is the single prevalent mutation. J Clin Invest 86:1000–1003
Zhang J, Zhang W, Zou D et al (2002) Cloning and functional characterization of ACAD-9, a novel member of human acyl-CoA dehydrogenase family. Biochem Biophys Res Commun 297:1033–1042
Acknowledgements
This work was supported by the Netherlands Organisation for Scientific Research (VIDI, grant no. 016.086.336 to S.M.H.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Ertan Mayatepek
Competing interest: None declared.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Houten, S.M., Wanders, R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis 33, 469–477 (2010). https://doi.org/10.1007/s10545-010-9061-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9061-2