
Abstract
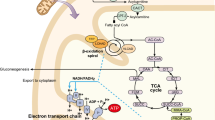
Assessing the outcome of fatty acid oxidation disorders is difficult, as most are rare. For diagnosis by newborn screening, the situation is compounded: far more cases are diagnosed by screening than by clinical presentation, representing a somewhat different cohort. The literature on outcome was reviewed. For disorders other than medium-chain acyl-coenzyme A (CoA) dehydrogenase (MCAD) deficiency there was insufficient evidence to make many firm statements. In MCAD deficiency, risk of death in the first 72 h is around 4%, with a further approximately 5–7% fatality rate in the first 6 years but very low subsequent risk in previously undiagnosed patients. The risk of death after diagnosis is very low at any age, with good management. The long-term outcome is good nowadays. Very-long-chain acyl-CoA dehydrogenase deficiency poses a risk of death in early infancy, but the condition is generally treatable, with a good outcome after diagnosis. Approximately 10–20% of patients diagnosed by newborn screening and treated nevertheless suffer episodic rhabdomyolysis. Some patients never become symptomatic. Isolated long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency is treatable, but most patients suffer episodic hypoketotic hypoglycaemia and rhabdomyolysis. Generalised mitochondrial tri-functional protein deficiency has high early mortality rate. A more insidious presentation also occurs, with symptoms sometimes confined to progressive axonal neuropathy. Among carnitine cycle disorders, carnitine transporter deficiency, potentially lethal, is uniformly successfully treated orally with carnitine. Carnitine-acylcarnitine translocase and early-onset carnitine palmitoyl transferase type II (CPT II) deficiencies have an extremely high neonatal mortality rate. Late-onset CPT II is characterised only by episodic rhabdomyolysis on severe exercise. CPT type IA deficiency may often be benign, although early presentation with hypoketotic hypoglycaemia certainly occurs.
Similar content being viewed by others


Abbreviations
- CPT IA:
-
carnitine palmitoyl transferase type IA
- CPT II:
-
carnitine palmitoyl transferase type II
- CTD:
-
carnitine transporter defect
- LCHAD:
-
long-chain 3-hydroxyacyl-coenzyme A dehydrogenase
- MCAD:
-
medium-chain acyl-coenzyme A dehydrogenase
- MTP:
-
mitochondrial tri-functional protein
- VLCAD:
-
very long-chain acyl-coenzyme A dehydrogenase deficiency
References
Andresen BS, Olpin S, Poorthuis BJ et al (1999) Clear correlation of genotype with disease phenotype In very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet 64:479–494
Andresen BS, Dobrowolski SF, O’Reilly L et al (2001) Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet 68:1408–1418
den Boer ME, Wanders RJ, Morris AA et al (2002) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics 109:99–104
den Boer ME, Dionisi-Vici C, Chakrapani A et al (2003) Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J Pediatr 142:684–689
Derks TG, Reijngoud DJ, Waterham HR et al (2006) The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. J Pediatr 148:665–670
Gillingham MB, Koeller DM (2008) Metabolic consequences of CPT-1 deficiency. Clinical trial: http://clinicaltrials.gov/ct2/show/NCT00653666. Accessed 19 August 2009
Grosse SD, Khoury MJ, Greene CL, Crider KS, Pollitt RJ (2006) The epidemiology of medium chain acyl-CoA dehydrogenase deficiency: an update. Genet Med 8:205–212
Iacobazzi V, Invernizzi F, Baratta S, Pons R, Chung W, Garavaglia B, Dionisi-Vici C, Ribes A, Parini R, Huertas MD, Roldan S, Lauria G, Palmieri F, Taroni F (2004) Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum Mutat 24:312–320
Iafolla AK, Thompson RJ Jr, Roe CR (1994) Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr 124:409–415
Ibdah JA, Yang Z, Bennett MJ (2000) Liver disease in pregnancy and fetal fatty acid oxidation defects. Mol Genet Metab 71:182–189
IJlst L, Wanders RJ, Ushikubo S, Kamijo T, Hashimoto T (1994) Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim Biophys Acta 1215:347–350
Labarthe F, Benoist JF, Brivet M, Vianey-Saban C, Despert F, de Baulny HO (2006) Partial hypoparathyroidism associated with mitochondrial trifunctional protein deficiency. Eur J Pediatr 165:389–391
Lund AM, Joensen F, Hougaard DM et al (2007) Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands. J Inherit Metab Dis 30:341–349
Maier EM, Liebl B, Roschinger W et al (2005) Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-CoA dehydrogenase deficiency. Hum Mutat 25:443–452
Pollitt RJ, Leonard JV (1998) Prospective surveillance study of medium chain acyl-CoA dehydrogenase deficiency in the UK. Arch Dis Child 79:116–119
Pourfarzam M, Morris A, Appleton M, Craft A, Bartlett K (2001) Neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency. Lancet 358:1063–1064
Roe CR, Ding JH (2005) Mitochondrial fatty acid oxidation disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B (eds) The metabolic and molecular bases of inherited disease (vol 8). McGraw-Hill, New York, pp 2297–2326
Sander J, Sander S, Steuerwald U et al (2005) Neonatal screening for defects of the mitochondrial trifunctional protein. Mol Genet Metab 85:108–114
Spiekerkoetter U, Sun B, Khuchua Z, Bennett MJ, Strauss AW (2003) Molecular and phenotypic heterogeneity in mitochondrial trifunctional protein deficiency due to beta-subunit mutations. Hum Mutat 21:598–607
Spiekerkoetter U, Lindner M, Santer R et al (2009) Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis 32:488–497
Touma EH, Charpentier C (1992) Medium chain acyl-CoA dehydrogenase deficiency. Arch Dis Child 67:142–145
Touma EH, Rashed MS, Vianey-Saban C (2001) A severe genotype with favourable outcome in very long chain acyl-CoA dehydrogenase deficiency. Arch Dis Child 84:58–60
Vijay S, Patterson A, Olpin S (2006) Carnitine transporter defect: diagnosis in asymptomatic adult women following analysis of acylcarnitines in their newborn infants. J Inherit Metab Dis 29:627–630
Wilcken B (2008a) More on medium-chain acyl-coenzyme a dehydrogenase deficiency in a neonate. N Engl J Med 358:647
Wilcken B (2008b) Disorders of the carnitine cycle. Ann Acad Med Singapore 37(Suppl):71–73
Wilcken B, Hammond J, Silink M (1994) Morbidity and mortality in medium chain acyl coenzyme A dehydrogenase deficiency. Arch Dis Child 70:410–412
Wilcken B, Haas M, Joy P et al (2007) Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet 369:37–42
Wilcken B, Haas M, Joy P et al (2009) Expanded newborn screening: outcome in screened and unscreened patients at age 6 years. Pediatrics 124:e241–e248
Wilhelm GW (2006) Sudden death in a young woman from medium chain acyl-coenzyme A dehydrogenase (MCAD) deficiency. J Emerg Med 30:291–294
Ziadeh R, Hoffman EP, Finegold DN et al (1995) Medium chain acyl-CoA dehydrogenase deficiency in Pennsylvania: neonatal screening shows high incidence and unexpected mutation frequencies. Pediatr Res 37:675–678
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Ertan Mayatepek
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Wilcken, B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis 33, 501–506 (2010). https://doi.org/10.1007/s10545-009-9001-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-009-9001-1