
Abstract
In this paper, we have used a thermodynamic model for the first time to investigate the steam and oxidative reforming of methane over a nickel catalyst in a wide temperature range, i.e., 400–1200 K. The available literature focus on the kinetic models and hence, thermodynamic models require attention to understand the behaviour of the thermochemistry of the species involved in the mechanism. This study presents the comparison between the species concentration produced using the thermodynamic model against the available kinetic model to validate the results. The investigation is further extended, firstly, to perform the sensitivity analysis of the reactions involved in a thermodynamic model to figure out the most influential reactions at various temperatures and pressures. This allows us to compare the most influencing reactions in reforming process for kinetic and thermodynamic model to optimize the processes. Secondly, the reaction flow analysis is carried out for the thermodynamic model to comprehend the effect of the thermochemistry of the species and the major difference in the reaction pathways for both the models are noted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The production of synthesis gas via the reforming of methane is still the most widely employed route from natural gas to commodity chemicals [1]. The most common catalytic technologies for converting natural gas to synthesis gas in various compositions involves processes, such as, steam reforming (SR), partial oxidation (POX), autothermal reforming (ATR), and dry reforming (DR) [2].
The most important industrial method to produce syngas has been the steam reforming of natural gas which is carried out since 1930 and given in equation (1) [1, 3].
High concentration of hydrogen is delivered at high fuel conversion with conventional steam reformers [2,3,4,5]. However, the reaction is highly endothermic which requires an efficient external energy supply. Also, the efficiency of the process is critically affected by the catalyst deactivation due to carbon formation [4].
The growing environmental concerns about global warming and oil depletion lead to the methane reforming with \(\mathrm {CO_2}\) (given in equation (2)) in the field of catalysis [5, 6]. The reason behind this is a desire to produce synthesis gas with lower \(\mathrm {H_2/CO}\) ratios which is better suited for stream processes, such as, oxo synthesis of aldehydes, synthesis of methanol, and acetic acid synthesis [7]. However, the main challenge in dry reforming of methane is the formation of carbon leading to catalyst deactivation, especially at industrial conditions [2].
A promising alternative to the endothermic reforming processes is the catalytic partial oxidation (see equation (3)) of methane over nickel-based catalyst [8,9,10,11]. In this process, no additional steam or heat is needed. However, different pre-treatment conditions affect the state of the catalyst surface which may change the reaction mechanism making the process complicated. The kinetics behind this reaction is studied in [8,9,10,11,12].
The catalytic oxidative conversion of methane at elevated temperatures and pressures also suffers from coke formation as observed in reforming processes. The deposition of coke on catalysts and reactor pipe walls are serious problems in many industrial reactors that involve methane as fuel. This leads to the blockage of reactor tube and can also physically disintegrate catalyst structure [13,14,15,16,17].
The problem of coke formation on the catalyst surface can be reduced by replacing the traditional nickel-based catalysts with noble metal ones, sulfur passivation, using metal sulfide catalysts, and authothermal reforming [6, 13]. However, each has its drawbacks. For instance, noble metals are expensive, sulfur passivation and sulfide catalysts require higher operating temperatures, and autothermal reforming can only be used in conjunction with an inexpensive nitrogen-free oxygen source [1]. Therefore, the optimum solution may be the development of a nickel-based catalyst with an exceptional resistance to coke formation. This has been recognized in the investigations focusing on the effect of the support and promoters on the performance of nickel catalysts, paying particular attention to coke deposition and the type of carbon deposited during reforming with carbon dioxide in the absence of steam (dry reforming) [6].
One major key to develop a more coke-resistant catalyst is a better understanding of the processes of catalytic oxidation and reforming of methane at a molecular level. The elementary steps involved in the reaction mechanism have been studied using several techniques [1, 3, 7, 18,19,20,21]. However, despite all these investigations, the detailed path for the conversion of methane to syngas and carbon remains a controversial issue [22]. Additionally, there is problem in setting up a reaction mechanism due to the difficulty to define the thermodynamic data for intermediate surface species. Hence, thermodynamic data can not be used to calculate the rate coefficient of the reverse reactions and then the forward as well as reverse reaction are defined separately with their own rate laws.
In this paper, we have developed a detailed kinetically consistent surface reaction mechanism which consists of 26 reversible reactions. Our previous work [23] forms the basis of the present investigation. In [23], a kinetically consistent surface reaction mechanism containing 21 reversible reactions was developed, however, the mechanism was limited for steam reforming processes. Whereas, in this work the mechanism from [23] is extended by adding 5 more reaction pathways and the mechanism is applicable for the modelling of partial oxidation, steam reforming, and dry reforming of methane over nickel-based catalysts along with the sub systems behind these reactions. The additional 5 reactions are taken from [24]. Note that in this paper we will only focus on steam and oxidative reforming of methane.
The mechanism is validated by comparing the simulation results with the literature [24] for various inlet gas compositions in order to develop a comprehensive understanding for partial oxidation and steam reforming of methane over a nickel-based catalyst in a wide temperature range. A detailed sensitivity analysis of reactions and reaction pathways for the developed kinetically consistent surface reaction mechanism has been presented in this paper.
The major difference of the present work from the literature is that the model is using thermochemistry of the intermediate species unlike the kinetic models [24, 25] where only the kinetic data is used to specify the forward and backward reactions explicitly. In our case, the kinetic data is needed only for the direct reactions whereas for the indirect reactions, the thermodata is used to calculate the reaction rate to establish equilibrium. The difference between this work from our previous studies is:
-
1.
In [23], 21 reactions were used to investigate the concentrations of species considered in the mechanism at the reactor outlet at various reactor conditions but only for the steam reforming of methane.
-
2.
The mechanism from [23] was upgraded by adding 5 more reactions and one more species so that the major reforming processes, for instance, steam, dry, and oxidative reforming, can be analysed by using the same mechanism. The new mechanism was tested for dry reforming and dry reforming with hydrogen and water in [26].
-
3.
In this study the above mechanism is validated for steam as well as oxidative reforming of methane over nickel catalyst.
-
4.
Also, the sensitive analysis for the reactions involved in the thermodynamic model was not discussed in any of the previous studies, so we furthermore present the reaction sensitivity analysis and reactions flow pathways for the thermodynamic model and compared it with the kinetic model from literature.
Surface reaction mechanism
The conversion of methane into a mixture of hydrogen, carbon monoxide, and carbon dioxide includes different molecular paths considered as a combination of the overall reaction given in Table 1 [24, 25]. In [25], a detailed surface reaction mechanism was developed using the global reactions in Table 1 to model reforming of methane on nickel. This established model served as basis in [24] for the development of a kinetic scheme for the extended region of operating conditions which covers all the ways from total oxidation to pyrolysis. In [24], the thermodynamic consistent surface reaction mechanism consists of 52 reactions with 6 gas-phase and 14 surface species.
The chemical source terms due to catalytic reactions are modeled by elementary-step based reaction mechanisms [27] and the homogeneous gas-phase reactions can be neglected under the considered conditions, example, temperature and pressure. In the present study, the mechanism from [24] serves as basis for developing a new kinetically consistent surface reaction mechanism. This work is also an extension of our previously developed surface reaction mechanism [23] which comprises of 21 reactions. The reaction scheme in [23] is extended here by adding new reaction paths involving carboxyl species as intermediate along with carbon formation paths.
The equilibrium of a chemical reaction is given below,
which is defined by the thermodynamic properties of the species i. To model the chemical source terms due to catalytic reactions, elementary-step based reaction mechanisms is used [27]. The forward rate constants, \(k_{f,r}\), for the reactions r are assumed to have the Arrhenius temperature dependence (for gas-phase as well as surface species) given as,
with \(A_{r}\) is the pre-exponential factor, \(\beta _r\) is the temperature exponent, \(E_{r}\) is the activation energy, T is the gas temperature and \(R_{c}\) is the gas constant in units consistent with activation energy.
The equilibrium constant is calculated by using the thermodynamic data associated with each species in a reaction and reverse rate coefficients for a reaction. In the thermal systems, the reverse rate constants, \(k_{b,r}\), are related to the forward rate constants and the equilibrium constants given as,
\(K_{c,r}\) is the equilibrium constant given in concentration units which considers the surface state as well as the gas state in case of reactions involving surface species and is given as,
here, \(P_{atm}\) denotes a pressure of 1 atm, \(R_{0}\) is the universal gas constant, \(\nu _{i,r}\) represents the stoichiometric coefficients for reaction r and species i, \(\Gamma _{n}^{0}\) is the standard-state surface site density of site type n and \(\sigma _{i}\) is the number of sites that the surface species occupies. The sum in the first exponent runs only over the gas-phase species and in the second exponent, it runs only over surface species in surface phase n.
The equilibrium constants, \(K_{p,r}\) (in pressure units) has the same form as for gas-phase reactions given as,
where \(\Delta\) as the change that occurs in passing completely from reactants to products in the \(r^{th}\) reaction. S and H are the symbols used for entropy and enthalpy, respectively with superscript \(^{0}\) denoting the standard-state which are calculated using thermodynamic data as,
So, the forward rate constants are calculated using equation (5) and the reverse rates are calculated using equation (6) which also uses the equilibrium rate constant obtained from thermodynamic data. We do not aim to cover further details in this section regarding the rate calculations and hence refer the reader to [24].
The kinetically consistent surface reaction mechanism developed in the present investigation is summarized in Table 2 and it consists of 26 reactions in total with 7 gas-phase and 14 surface species. The predictive behaviour of the overall reactor model is assessed by adaption of the thermochemistry of the species. The kinetic data is taken from [24] and the thermodynamic data for CO(s) and COOH(s) is taken from [28] whereas for all the remaining species, it is taken from [24]. Note that the thermodynamic data for CO(s) is modified by carrying out the thermochemistry sensitivity analysis presented in our previous study [23] and we have used the updated thermodynamic data for CO(s).
The interface for the calculation of thermodynamic properties and transport coefficients of gas-phase species is provided by describing the species properties based on atomic models for reactions among gas-phase and surface species based on elementary-step reaction [27]. To represent different types of catalysts, individual surface types can be defined by assigning an individual microscopic surface site density and macroscopic surface area. The microscopic surface site density influences the rate of surface reactions, and the macroscopic surface area scales the net fluxes in the interaction with the gas phase. By solving a set of reaction-diffusion equations or by applying an effectiveness-factor model, diffusion in washcoats can be accounted [27]. However, the reforming reaction are endothermic in nature and the kinetics of the reactions is very fast. All the changes happen only in the beginning of the pipe and then it reached the steady state. The diffusion is very slow and hence, is not noticeable with this fast kinetics. Therefore, we focus only on the kinetics of the reactions to consider what happens on the surface and will not discuss about the internal diffusion.
The activation for desorption of hydrogen (R1) from nickel is taken as 95.20 kJ/mol from [24]. However, a wide range for activation energy is proposed in literature, for instance, between 82-89 kJ/mol in [29, 30], 97 kJ/mol in [19], 95 kJ/mol in [31], 96 kJ/mol in [1, 32] and many more.
The activation energy for oxygen desorption (R2) is used as 468.90 kJ/mol [24]. In literature, the activation energies for desorption of oxygen for the three low index crystal planes of Ni are 520, 470, and 485 kJ/mol for the Ni(100), Ni(111), and Ni(110) planes, respectively [33].
The desorption of \(\mathrm {H_2}\)O (R3) is taken as 60.79 kJ/mol [24] which is comparable with the experimental and theoretical values reported in the literature [1, 34]. Further, the activation energy for desorption of \(\mathrm {CO_2}\) and CO (R4,R5) is 25.98 and 111.27 kJ/mol [24], respectively. The desorption of CO is studied in [1, 19, 21, 35].
In Table 2, R6-R10 show the methane dehydrogenation, R11-R14 indicate the oxygen assisted methane dehydrogenation, R15-R17 represent water activation and R18 illustrates CO dissociation. The surface reaction mechanism includes reaction paths to describe the formation of up to one monolayer of carbon from adsorbed CO species through CO(s) dissociation (R18,R19) and via Boudouard reaction (R20).
The oxidation of CO is shown by two reaction paths, (a) direct oxidation of CO(s) with O(s) (R21), and (b) through the surface intermediate carboxyl (COOH) formed from CO(s) and OH(s) (R22,R23). The formyl species (HCO) is coordinated through carbon and acts as intermediate during the reforming and oxidation of methane and is shown by R24, R25, and R26.
The kinetic data for the backward reactions given in Table 2 is calculated with the help of thermodynamic data listed in Table 3 for all the species. In [24], the heats of adsorption of adsorbed species, reaction enthalpy changes, and the original activation barriers for all relevant steps of the mechanism is determined by the unity bond index-quadratic exponential potential (UBI-QEP) approach. So, we have taken the coefficients for NASA polynomials from [24, 28] and given the entropy and enthalpy for all the surface species in Table 3.
The details about calculation of forward and backward rate constants is discussed in our previous work [23] for both the approaches, i.e., thermodynamically as well as kinetically consistent surface reaction mechanism. Hence, we refer the reader to [23] for more details.
The main pathways for methane reforming and oxidation processes on the nickel catalyst for syngas production is shown in Fig. 1 [24] which is further discussed in upcoming sections.

Reaction pathways for methane reforming and oxidation
Model description
The 1D model, LOGEcat [36] is used to perform the simulations using the mechanism provided in previous section and the results obtained are discussed in the upcoming sections. The model has been successfully tested in previous studies, for instance, [23, 37,38,39]. This model is a part of the LOFEsoft software suite [36] for chemical reaction calculations.
The diameter of the catalytic channel is small and hence, the pressure gradient along the inhomogeneity of the mixture can be neglected. The external diffusion is modeled by a thin layer indicated by a separate pore gas zone close to the wall. The model is based on the single-channel 1D catalyst model shown in Fig. 2, where the single channel is divided into a finite number of cells (25 cells for this study) with \(\Delta x\) as their length.

Schematic illustration of the 1D modelling approach
Each cell is treated as a perfectly stirred reactor (PSR) and the conservation equations (gas species mass fraction, gas enthalpy, surface enthalpy, pore layer gas species mass fraction, and surface site fractions) given in Table 4 are solved in each PSR for each time step in addition to the 1D Navier–Stokes equations for flow velocity solved by an operator splitting method over all cells.
The mathematical treatment of the model is presented in our previous publication [40]. Here, we have only summarized the important equations without considering the details. A list of abbreviations and symbols used in this section or elsewhere are given in [40]. For more details related to the model and the conservation equations, we refer the reader to our previous papers [23, 40].
Simulation set-up
In the experiments [24], a fixed bed reactor consisting of a quartz tube with an inner diameter of 10 mm filled with 20 mg of a nickel-based catalyst with a reaction zone of 27 mm surrounded by a quartz frit and glass wool is considered. The nickel-based catalyst has a particle size between 500 and 1000 µm and was synthesized by BASF.
For the simulations of methane reforming and partial oxidation processes, a Ni-coated monolithic catalyst is considered. In order to compare the results from a fixed bed reactor experiments to a monolithic reactor, we use the same open volume and all other physical and numerical parameters in the simulations are taken from experiments. The heat transfer properties with the fixed bed and monolithic reactor are not one to one similar and this is one of the limitations of the model. There is on-going work to develop a fixed bed reactor to overcome such restriction. However, our prime focus in this paper is to test the thermodynamic model for the SR and CPOX process. Note that the different pre-treatment conditions affect the state of the catalyst surface which might change the reaction mechanism. The applicability of the developed detailed surface reaction mechanism is tested by comparing the simulation results with literature.
A single layer of washcoat is used for the analysis. The reactor dimensions are taken same as experiments which consists of a circular catalyst with radius 5 mm and a reaction zone of 27 mm length. The geometric data and catalyst parameters utilized to carry out the simulations are taken from [24]. A sensitivity analysis is done to adjust the surface area per catalyst length. \(96\%\) nitrogen dilution is used and the surface site density, \(\tau\) for Ni is \(2.6\times 10^{-5}\) mol/\(\mathrm{{m}}^{2}\). The overall mass transfer efficiency factor, heat transfer efficiency factor, and efficiency factor for surface chemistry are taken as one.
The simulations are carried out at 4 slpm (standard liters per minute, T = 298.15 K and p = 1.01325 bar) as done in literature experiments. The reaction temperatures are varied in a range, [400–1200] K for different reactor conditions in terms of parameters, such as, inlet gas composition. Analysis is done for different reforming process in the considered temperature range while keeping all the other parameters constant. Only, the inlet gas composition is varied in the entire temperature range. These cases cover the steam reforming and partial oxidation of methane over nickel, and the simulation cases are summarized in Table 5. The simulation results for these cases are discussed in next section.
Methane Steam Reforming (SR)
The mechanism developed in the present study is used for validation and discussion using the simulation set-up given in previous section. The simulations are performed in a temperature range [400–1200] K at 1 bar with C/S (carbon-to-steam) ratio as 0.8 diluted in \(\mathrm {{N}_2}\) (case SR in Table 5) and the results are compared with [24] in order to validate the model. This will also check the predictability of the developed surface reaction mechanism and will allow us to use the thermodynamic data in similar investigations in future.
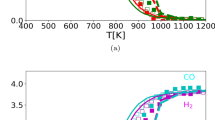
Fig. 3a shows the methane and water concentration computed with our model as a function of temperature along with the reference data from [24]. The equilibrium calculations from [24] for the given species are also plotted. According to the experimental trend, the simulated concentration of methane and water decrease with increasing temperature [24]. According to the experiment, we observe the start of the methane consumption at \(\approx\)600 K and a full consumption of methane happens at temperature \(\approx\)1000 K giving nearly 100\(\%\) conversion of the species. Most of the water consumption takes place between T\(\approx\)600-900 K. Inline with the simulation results of [24], the present simulation results of water conversion deviate from the experimental results. Note that the equilibrium calculations from [24] for both the reactants, \(\mathrm {CH_4}\) and \(\mathrm {H_2}\)O, are below the ones obtained with the simulation results at temperatures below \(\approx\)1000 K indicating that the reforming is away from the thermodynamic equilibrium at the considered simulation conditions.

The concentration of a reactants and b products are shown as a function of temperature with C/S=0.8 along with the reference data from [24]. The unfilled squares represents reference simulations, filled squares: reference experiments, solid lines: reference equilibrium calculations and dash lines: LOGEcat calculations with the new mechanism
The concentration of the products are illustrated in Fig. 3b for species \(\mathrm {H_2}\), CO, and \(\mathrm {CO_2}\). The formation of \(\mathrm {H_2}\), CO, and \(\mathrm {CO_2}\) begins with methane consumption at temperatures above \(\approx\)600 K and it increases while \(\mathrm {CH_4}\) and \(\mathrm {H_2}\)O concentration decreases, thus, leading almost to the equilibrium composition at given operating conditions. However, the concentration of the computed species for our case as well as from [24] are below the equilibrium calculations shown in Fig. 3 in the medium temperature range. The thermodynamic equilibrium is reached for our simulations as well as reference data from [24] for temperatures above 1000 K.
The consumption of \(\mathrm {CH_4}\) and \(\mathrm {H_2}\)O is utilized for the formation of \(\mathrm {H_2}\), Fig. 3b, which is maximum for the considered operating conditions. The hydrogen concentration for the present investigation as a function of temperature shows similar behaviour as shown by the reference data [24] which is zero at low temperature (T<700 K) and the formation of hydrogen increases gradually in the temperature range \(\approx\)700-1000 K and then saturates at higher temperatures. At higher temperatures, where the conversion of \(\mathrm {CH_4}\) and \(\mathrm {H_2}\)O is high, as expected, high concentration of hydrogen is delivered. The developed reaction mechanism produce the results which are in good agreement with the reference data from [24].
The concentration for CO, Fig. 3b, shows qualitatively similar behaviour as shown by the reference data [24]. However, the concentration for CO using the new detailed surface reaction mechanism as well as reference simulations is slightly under-predicted in comparison with the reference experiments and equilibrium calculations. Nevertheless, the results are acceptable in comparison with the reference data.
The \(\mathrm {CO_2}\) as a product, Fig. 3b, might be formed due to the availability of OH(s) species on the surface originating from the dissociation of water and is noted in very small amount in the temperature range 700–1000 K. The concentration of \(\mathrm {CO_2}\) is also qualitatively similar in behaviour with the reference data. Initially, the concentration is very low at low temperatures which begins to rise from 700–900 K reaching maximum at T\(\approx\)900 K and then drops from T>900 K. The \(\mathrm {CO_2}\) concentration peak for experiments is noted at T\(\approx\)750 K, for reference simulations at T\(\approx\)800 K and for the present investigation at T\(\approx\)900 K. The overall concentration for \(\mathrm {CO_2}\) is under-predicted in comparison with the reference results using the new reaction mechanism. We recommend further investigations in this direction in order to find the error in species concentrations and therefore perform the sensitivity analysis. We also look into various reaction pathways to find the differences in the kinetic model and thermodynamic model to provide a useful insight into the key rate determining steps.

Sensitivity analysis of CO gas-phase concentration for steam reforming methane at different temperatures for C/S = 0.80 and 1 bar
The normalized sensitivity coefficients on the CO yield at three different temperatures (723, 923, and 1123 K) is shown in Fig. 4. The sensitivity analysis is performed for the inlet C/S ratio 0.8 and 1 bar. This Figure indicates the sensitivity of 26 reversible reactions considered in the surface reaction mechanism developed in this investigation and comply with the reaction coefficient calculation at two different temperatures given for all the reactions in Table 2. The desorption of CO (R5), methane dehydrogenation (R7), and oxygen assisted methane dehydrogenation (R11) are highly sensitive for all the temperatures considered for simulations. At low temperatures (723 K), the methane dehydrogenation (R8, R9), carbon formation from adsorbed CO species through CO(s) dissociation (R19) and the oxidation of CO through the surface intermediate carboxyl formed from CO(s) (R22) are sensitive to the CO yield. For a better understanding, we have summarised the most sensitive reactions in SR process in Table 6 for the thermodynamic model developed in this work along with the kinetic model from [24] to make a comparison between the two models at same conditions as [24]. The common reaction found in both the models is highlighted in bold font in the table.

Reaction flow analysis for steam reforming of methane on nickel for constant C/S ratio and pressure at 723, 823, 923, and 1123 K based on C-atom
Further, the reaction flow analysis based on C-atom in Fig. 5 and based on H-atom in Fig. 6 is performed at 723 K where the formation of the products started, 823 K where the maximum \(\mathrm {CO_2}\) formation is noted for the reference case, 923 K where the maximum \(\mathrm {CO_2}\) formation is observed for our simulations and 1123 K where the formation of products is saturated.
Based on C-atom (Fig. 5), at low temperatures (723 K), the production of \(\mathrm {CO_2}\) through the direct oxidation of CO(s) with O(s) is dominated which is \(\approx 71\%\) whereas the indirect route through the surface intermediate carboxyl formed from CO(s) and OH(s) is \(\approx 27\%\). The \(\mathrm {CO_2}\) formation via direct route decreases with increasing temperature and is \(\approx 64\%\), \(\approx 62\%\), and \(\approx 44\%\) for 823, 923, and 1123 K, respectively. This leads to the increase in formation of CO as \(\approx 2\%\), \(\approx 6\%\), \(\approx 14\%\), and \(\approx 46\%\) for 723, 823, 923, and 1123 K, respectively. The formation of \(\mathrm {CO_2}\) via indirect route lies in the range \(\approx 24-29\%\) for 723-923 K where there is a sharp drop for 1123 K (\(\approx 10\%\)). It can be seen that the main path for \(\mathrm {CO_2}\) production in this model is preferred through the direct oxidation of CO(s) with O(s) in comparison with the carboxyl path.

Reaction flow analysis for steam reforming of methane on nickel for same conditions as previous Fig. based on H-atom
The pathways based on H-atom also have influence of temperature and are illustrated in Fig. 6. Some of the pathways are independent of temperature whereas some are highly dependent on temperature, for instance, formation of COOH(s) via OH(s) and H(s) via \(\mathrm {CH_4}\)(s) increases with increasing temperature. Also, formation of OH(s) from H(s) is showing similar trend, i.e., increases with increasing temperature and the OH(s) formation for 723 K is 0.0\(\%\) increasing up to 90.3\(\%\) for 1123 K. All the other pathways, for example, formation \(\mathrm {H_2}\) from H(s), OH(s) from \(\mathrm {CH_4}\)(s), \(\mathrm {H_2O}\)(s) from H(s) and \(\mathrm {H_2O}\)(s) from OH(s) decreases with increasing temperature.
The reaction mechanism from Delgado et al. [24] consist of 52 reactions (explicit forward and backward reactions), whereas our mechanism consist of 26 reversible reactions which are kinetically consistent. Nevertheless, the results produced with the developed surface reaction mechanism in this work shows reasonable agreement with the experimental data as well as the simulation results from Delgado et al. [24] for steam reforming of methane over a nickel catalyst.
Methane catalytic partial oxidation (CPOX)
In open literature, the partial oxidation of methane is suggested by two main paths. In first path, \(\mathrm {H_2}\) is directly originated from methane decomposition and this is the direct oxidation mechanism. Here, interaction of adsorbed hydrocarbon species \(\mathrm {CH_x}\), \(\textrm{x}\)=0,1,2,3, with adsorbed atomic oxygen produces carbon monoxide [41]. The second path deals with the methane totally oxidized to \(\mathrm {CO_2}\) and \(\mathrm {H_2O}\) in presence of oxygen close to the catalyst surface where the remaining \(\mathrm {CH_4}\) is reformed with \(\mathrm {H_2O}\) or \(\mathrm {CO_2}\) to \(\mathrm {H_2}\) and CO and is referred as indirect oxidation mechanism [8, 42].
The concentration of the numerically predicted inlet species, methane and oxygen, for our model are shown in comparison with the reference data in Fig. 7a as a function of temperature. The computed concentration of methane decreases with increasing temperature and the conversion starts at \(\approx\)700 K. This leads to the formation of only \(\mathrm {CO_2}\) and \(\mathrm {H_2O}\) given in Fig. 7b. No significant amount of other products at this temperature, \(\mathrm {H_2}\) and CO is detected. The formation of \(\mathrm {H_2}\) and CO increase with increase in temperature from 800 K, whereas for other products, \(\mathrm {CO_2}\) and \(\mathrm {H_2O}\), the concentrations decreases leading almost to the equilibrium composition at the considered conditions.

The concentration of a reactants and b products are shown as a function of temperature along with the reference data from [24]. The unfilled squares represents reference simulations, filled squares: reference experiments, solid lines: reference equilibrium calculations and dash lines: LOGEcat calculations with the new mechanism
In [24], the concentrations of species \(\mathrm {CO_2}\) and \(\mathrm {H_2O}\) are more than the equilibrium calculations in the temperature range \(\approx\)800-1000 K, whereas, in the same temperature range, our computed species concentrations are less than the ones obtained at equilibrium. This difference might be due the consideration of the monolithic reactor for the present simulations whereas the reference results are based on the fixed bed reactor or it may be because of the new mechanism where different paths might be dominating. It needs further investigations in order to clarify these issues. Nevertheless, the concentrations of all the species computed with our model in the entire temperature range is qualitatively in agreement with the reference experimental and simulation data.
The \(\mathrm {CO_2}\) and \(\mathrm {H_2O}\) concentrations in [24] for kinetic model are above the equilibrium calculations in the medium temperature range indicating for the indirect way to \(\mathrm {H_2}\) and CO via formation of \(\mathrm {CO_2}\) and \(\mathrm {H_2O}\). Whereas, for the thermodynamic model, the \(\mathrm {CO_2}\) and \(\mathrm {H_2O}\) concentrations are below the equilibrium calculations leading to the formation of \(\mathrm {H_2}\) and CO by direct oxidation mechanism originated from methane decomposition.
The numerically predicted CO selectivity as a function of temperature is presented in Fig. 8 for methane catalytic partial oxidation. The reference simulation and experimental data from [24] is also shown. The model predictions agree very well with the reference data at temperatures T<800 and T > 900 K. Some deviations are notes in the temperature range 800-900 K which is also seen in gas-phase species concentrations discussed above. The ignition temperature for our simulation is also slightly different in comparison with the reference data, however, once the fuel is ignited (T > 900 K), the agreement between the present simulation and reference result is good with a selectivity nearly 90\(\%\) for 1000 K and above.

Selectivity of CO concentration as a function of temperature along with the reference data from [24]. The unfilled squares represents reference simulations, filled squares: reference experiments and dash lines: LOGEcat calculations with the new mechanism
Fig. 9 illustrates the normalized sensitivity coefficients of CO gas-phase concentration for 3 different pressures (1, 3, and 10 bar) at a temperature after ignition, 973 K. The fuel composition to carry out the simulations is kept fixed as reference data. This Figure shows that the oxygen assisted methane dehydrogenation reaction, R11 is very sensitive out of all 26 reversible reactions considered in the surface reaction mechanism developed in this investigation at all considered pressures. The formation of carbon from adsorbed CO species via CO(s) dissociation is indicated by R19 and this reaction is highly dominating for low pressures, i.e., 1 and 3 bar. However, at high pressure (10 bar) R19 is not the main reaction anymore and the desorption reaction for CO, R5 becomes the main reaction for CO yield. There are some other reaction, show in the figure, which are sensitive towards the CO concentration, however, these reaction are changing with the change in pressure.

Sensitivity analysis of CO gas-phase concentration for catalytic partial oxidation reaction at 973 K for different pressures
Further, the most dominating reactions for kinetic and thermodynamic model for CPOX process are summarised in Table 7 by marking the common reactions for both the models in bold font. For these models, the differences in sensitivity analysis and influence of different reactions have been observed which explains the deviations in the computed species with the developed thermodynamic model from kinetic scheme used in [24].
Conclusions
A one-dimensional model, LOGEcat is used to develop a thermodynamic model for investigating the steam reforming (SR) and catalytic partial oxidation (CPOX) of methane over a nickel-based catalyst. The thermodynamic model is tested against literature experiments and available simulation results with kinetic model for the considered reforming processes by reproducing the reactants and products distribution in a wide temperature range. The differences in reaction pathways for kinetic and thermodynamic model explains the error in the species concentration for both the models. However, a further investigation is recommended to improve the concentration of the species under different conditions, for example, \(\mathrm {CO_2}\) concentration for SR and \(\mathrm {H_2O}\) as well as \(\mathrm {CO_2}\) concentration for CPOX.
This work is new in a sense that a detailed sensitivity analysis for reversible reactions considered in the mechanism is performed in addition with the reaction flow analysis at various conditions. Such analysis has been done for the first time for a thermodynamic model. In the thermodynamic model, the main path for \(\mathrm {CO_2}\) formation is concluded to be the direct oxidation of CO(s) with O(s) in comparison with the formation of the carboxyl group in steam reforming process. For oxidative reforming, the formation of \(\mathrm {H_2}\) and CO by direct oxidation mechanism originated from methane decomposition is preferred in contract to the kinetic model. This study illustrates major differences in reaction pathways for kinetic and thermodynamic model and therefore, we recommend a further investigation to understand the origin of the differences which might improve the mechanism and can also help to understand the influence of thermochemistry towards coke formations in various reforming processes.
Data availability
Not applicable
References
Aparicio LM (1997) Transient isotopic studies and microkinetic modeling of methane reforming over nickel catalysts. J Catal 165:262–274. https://doi.org/10.1006/jcat.1997.1468
Rostrup-Nielsen JR, Sehested J, Nørskov JK (2002) Hydrogen and synthesis gas by steam- and \(CO_2\) reforming. Adv Catal 47:65–139
Xu J, Froment GF (1989) Methane steam reforming, methanation and water-gas shift: I. Intrinsic kinetics. Am Instit Chem Eng AIChE 35:88–96. https://doi.org/10.1002/aic.690350109
Rostrup-Nielsen JR (2000) New aspects of syngas production and use. Catal Today 63:159–164
Rostrup-Nielsen JR (1984). In: Anderson JR, Boudart M (eds) Catalytic steam reforming in catalysis-science and technology. Springer-Verlag, Berlin
Rostrup-Nielsen JR, Hansen JHB (1993) CO2-reforming of methane over transition metals. J Catal 144:38–49. https://doi.org/10.1006/jcat.1993.1312
Bradford MCJ, Vannice MA (1999) CO2 reforming of CH4. Catal Rev 41:1–42
Dissanayake D, Rosynek MP, Kharas KCC et al (1991) Partial oxidation of methane to carbon monoxide and hydrogen over a Ni/Al2O3 catalyst. Catal Today 132:117–127. https://doi.org/10.1016/0021-9517(91)90252-Y
Zhu T, Flytzani-Stephanopoulos M (2001) Catalytic partial oxidation of methane to synthesis gas over Ni-Ce\(O_2\). Appl Catal A 208:403–417
Tang S, Lin J, Tan KL (1998) Partial oxidation of methane to syngas over Ni/MgO, Ni/CaO and Ni/Ce\(O_2\). Catal Lett 51:169–175
Vernon PF, Green MH, Cheetham A et al (1990) Partial oxidation of methane to synthesis gas. Catal Lett 6:181–186
Li C, Yu C, Shen S (2000) Role of the surface state of Ni/A\(l_2\)\(O_3\) in partial oxidation of \(CH_4\). Catal Lett 67:139–145
Rostrup-Nielsen JR (1984) Sulfur-passivated nickel-catalysts for carbon-free steam reforming of methane. J Catal 85:31–43
Trimm DL (1977) Formation and removal of coke from nickel-catalyst. Catal Rev- Sci Eng 16:155–189
Bartholomew CH (1982) Carbon deposition in steam reforming and methanation. Catal Rev 24:67–112
Blekkan EA, Myrstad R, Olsvik O et al (1992) Characterization of tars and coke formed during the pyrolysis of methane in a tubular reactor. Carbon 30:665–673
Wang S, Lu GQM (1998) \(CO_2\) reforming of methane on Ni catalysts: effects of the support phase and preparation technique. Appl CatalB 16:269–277
Chen D, Lodeng R, Anundskas A et al (2001) Deactivation during carbon dioxide reforming of methane over ni catalyst: microkinetic analysis. Chem Eng Sci 56:1371–1379
Chen D, Lødeng R, Svendsen H et al (2010) Hierarchical multiscale modeling of methane steam reforming reactions. Ind Eng Chem Res 50:2600–2612
Wei J, Iglesia E (2004) Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts. J Catal 224:370–383. https://doi.org/10.1016/j.jcat.2004.02.032
Blaylock DW, Ogura T, Green WH et al (2009) Computational investigation of thermochemistry and kinetics of steam methane reforming on Ni(111) under realistic conditions. J Phys Chem A 113:4898–4908
Iglesia E (1997) Design, synthesis, and use of cobalt-based fischer-tropsch synthesis catalysts. Appl Catal A 161:59–78
Rakhi Shrestha KP, Günther V et al (2022) Kinetically consistent detailed surface reaction mechanism for steam reforming of methane over nickel catalyst. React Kinet, Mech Catal. https://doi.org/10.1007/s11144-022-02314-7
Delgado KH, Maier L, Tischer S et al (2015) Surface reaction kinetics of steam- and \(CO_2\)-reforming as well as oxidation of methane over nickel-based catalysts. Catalysts 5:871–904. https://doi.org/10.3390/catal5020871
Maier L, Schädel B, Delgado KH et al (2011) Steam reforming of methane over nickel: development of a multi-step surface reaction mechanism. Top Catal 54:845–858. https://doi.org/10.1007/s11244-011-9702-1
Rakhi Günther V, Mauss F (2023) Insights into dry reforming of methane over nickel catalyst using a thermodynamic model. React Kinet, Mech Catal. https://doi.org/10.1007/s11144-023-02426-8
Tischer S, Deutschmann O (2005) Recent advances in numerical modeling of catalytic monolith reactors. Catal Today 105:407–413. https://doi.org/10.1016/j.cattod.2005.06.061
Liu M, Dana AG, Johnson MS et al (2021) Reaction mechanism generator v3.0: advances in automatic mechanism generation. J Chem Inf Model 61:2686–2696. https://doi.org/10.1021/acs.jcim.0c01480
Bartholomew C (1990) Hydrogen adsorption on supported cobalt, iron, and nickel. Catal Lett 7:27–51
Weatherbee GD, Bartholomew CH (1984) Effects of support on hydrogen adsorption/desorption kinetics of nickel. J Catal 87:55–65
Zhu XY, White JM (1988) Hydrogen interaction with nickel (100): a static secondary ion mass spectroscopy study. J Phys Chem A 92:3970–3974
Kratzer P, Hammer B, Norskov JK (1996) A theoretical study of \(CH_4\) dissociation on pure and gold-alloyed Ni(111) surfaces. J Chem Phys 105:5595–5604
Stuckless JT, Wartnaby CE, Al-Sarraf N et al (1997) Oxygen chemisorption and oxide film growth on Ni100, 110, and 111: sticking probabilities and microcalorimetric adsorption heats. J Chem Phys 106:2012–2030
Stulen RH, Thiel PA (1985) Electron-stimulated desorption and thermal desorption spectrometry of \(H_2O\) on nickel (111). Surf Sci 157:99–118
Shustorovich E, Sellers H (1998) The UBI-QEP method: a practical theoretical approach to understanding chemistry on transition metal surfaces. Surf Sci 31:1–119
LOGEsoft (2008) V1.10 www.logesoft.com
Aslanjan J, Klauer C, Perlman C, et al (2017) In: SAE International, Simulation of a Three-Way Catalyst Using Transient Single and Multi-Channel Models. ISSN: 0148-7191, 2017-01-0966
Fröjd K, Mauss F (2011) A three-parameter transient 1D catalyst model. SAE Int J Eng 4(1):1747–1763. https://doi.org/10.4271/2011-01-1306
Müller K, Rachow F, Günther V et al (2019) Methanation of coke oven gas with nickel-based catalysts. Int J Environ Sci 4:73–79
Rakhi Günther V, Richter J et al (2022) Steam reforming of methane over nickel catalyst using a one-dimensional model. Int J Environ Sci 5(1):1–32
Hickman DA, Schmidt LD (1993) Production of syngas by direct catalytic oxidation of methane. Science 259:343–346
Schwiedernoch R, Tischer S, Correa C et al (2003) Experimental and numerical study of the transient behavior of a catalytic partial oxidation monolith. Chem Eng Sci 58(3):633–642. https://doi.org/10.1016/S0009-2509(02)00589-4
Acknowledgements
Financial support by the federal ministry of education and research (Bundesministerium für Bildung und Forschung, BMBF) under the Grant Number 03SF0693A of the collaborative research project “Energie-Innovationszentrum” is gratefully acknowledged. The authors also thank Vivien Günther for discussions related to the model.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rakhi, Mauss, F. Thermodynamic model: steam and oxidative reforming of methane over nickel catalyst. Reac Kinet Mech Cat 137, 791–812 (2024). https://doi.org/10.1007/s11144-024-02571-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-024-02571-8