
Abstract
Cellular pathophysiology of sepsis associated encephalopathy (SAE) remains poorly characterised. Brain pathology in SAE, which is manifested by impaired perception, consciousness and cognition, results from multifactorial events, including high levels of systemic cytokines, microbial components and endotoxins, which all damage the brain barriers, instigate neuroinflammation and cause homeostatic failure. Astrocytes, being the principal homeostatic cells of the central nervous system contribute to the brain defence against infection. Forming multifunctional anatomical barriers, astroglial cells maintain brain-systemic interfaces and restrict the damage to the nervous tissue. Astrocytes detect, produce and integrate inflammatory signals between immune cells and cells of brain parenchyma, thus regulating brain immune response. In SAE astrocytes are present in both reactive and astrogliopathic states; balance between these states define evolution of pathology and neurological outcomes. In humans pathophysiology of SAE is complicated by frequent presence of comorbidities, as well as age-related remodelling of the brain tissue with senescence of astroglia; these confounding factors further impact upon SAE progression and neurological deficits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: The Concept of Sepsis-Associated Encephalopathy
Systemic inflammation often develops as a component of diverse somatic diseases of infectious and non-infectious nature, as well as degenerative and metabolic pathologies [1,2,3]. In sepsis the combination of systemic inflammatory factors is of paramount importance for the development of multiple organ dysfunction syndrome (MODS). The pathogenesis of MODS reflects the uncontrolled systemic inflammatory response associated with pathological changes in systemic circulation manifested by vasodilatation and hypotension. These in turn result in the microcirculatory abnormalities, activation of the coagulation cascade with predominance of procoagulant factors and development of systemic ischemia, hypoxia, cell death of both necrotic and apoptotic nature ultimately leading to tissue and organ insufficiency, the classic functio laesa [4]. The brain damage in a form of sepsis-associated encephalopathy (SAE) is one of the most frequent and early components of MODS in sepsis [5]. The SAE is generally recognised as a fundamental factor that determines the clinical evolution, prognosis and outcome of sepsis [6].
Although possessing the immune privilege, the brain is actively involved in the regulation of immune system homeostasis; the immune system reciprocally affects the brain, which develops defined responses to systemic inflammation [7]. Being the omnipresent controller of the immune-neuroendocrine system, the brain contains the highly complex and interconnected signalling system based on specific interfaces that support a wide range of homeostatic and defensive functions. On a cellular level the homeostatic control of the central nervous system (CNS) belongs to neuroglia, the heterogeneous cell population responsible for CNS homeostasis [8]. Systemic inflammation violates the functions of the brain impacting upon both neurones and neuroglia with glial activation in particular, this latter providing a critical contribution to immune reactivity of the nervous tissue upon inflammatory damage and in SAE [9]. In systemic inflammation pathogens have haematogenous and cerebrospinal origin and hence damage to the blood–brain barrier (BBB) and to the blood–cerebrospinal-fluid-barrier (BCSFB) are critical for pathology. Astrocytes, in a form of glia limitans, maintain the integrity of these barriers and are the first parenchymal neural cells facing invading pathogens.
Sepsis, SAE and Models of Sepsis
Acute CNS dysfunction frequently accompanies sepsis and, in particular, the abdominal sepsis (AbS) [10]. Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection [6]. Therefore, sepsis has only two degrees of severity: sepsis itself and septic shock, which complicates the diagnosis of sepsis at the earlier stages (preceding organ failure) and ultimately may increase the severity of the disease [11]. Sometimes sepsis proceeds in a hidden state and remain undiagnosed, despite the presence of systemic inflammatory response syndrome (SIRS) [12]. The SIRS represents a non-specific complex reaction of the organism to severe damage whether it is an infection, mechanical or thermal injury, pancreatitis etc. [13]. For SIRS diagnosis more than 2 criteria should be met: body temperature > 38 °C or < 36 °C; heart rate > 90/min; respiratory rate > 20/min or PaCO2 < 32 mm Hg (4.3 kPa); white blood cell count > 12 000/mm3 or < 4000/mm3 or > 10% immature bands [6].
Brain dysfunction, assessed according to the Glasgow coma scale in septic patients, increases the risk of death by 10%, that determines the management strategy in such patients [6, 14]. The emergence of SAE in septic patients is a marker of the severity of septic state, although an increase in mortality is not always interpreted as a direct outcome of cerebral dysfunction [15]. Nevertheless appearance of brain symptoms impacts upon clinical strategies and may necessitate distinct therapeutic approaches. Cerebral dysfunction developing in sepsis has a wide range of potentially reversible cognitive manifestations ranging from reduced attention, disrupted sleep–wakefulness balance, impaired memory, speech, orientation, as well as focal neurological deficits and seizure activity, perception disorders (delusion-hallucinatory complex) terminating with a decreased consciousness and coma [16]. This complex of symptoms, which complicates sepsis in up to 70% of cases [17, 18] is defined as SAE [19], or sepsis-associated delirium or sepsis-associated brain dysfunction [20]. Of note, the term “septic encephalopathy” is not considered appropriate for this condition, since such term reflects direct infection of the cerebral parenchyma that is not consistent with modern views on this pathology [21].
Conceptually the SAE can be defined as a clinical syndrome indicating general brain dysfunction developing in sepsis without direct infectious lesion to the nervous tissue. At the same time diagnostic and clinical usage of this term remains somewhat blurred due to a polymorphism of pathological processes, which cause brain damage. Histological appearance of the SAE similarly remains poorly defined with rather non-specific pathomorphological hallmarks. With increased duration and severity of septic conditions the integrity of brain barriers is compromised and the brain parenchyma becomes infected with frequent development of abscesses.
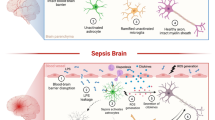
The main histopathologic changes in the brain during sepsis include infarctions, petechial and small focal hemorrhages, septic-embolic abscesses and septicopyemic microabscesses, signs of disseminated intravascular coagulation (DIC) syndrome with fibrinous microthrombi, multifocal necrotizing leukoencephalopathy, selective necrosis and apoptosis of neurones in the regions most sensitive to ischemia and neurotoxicity with more prominent hippocampal and brainstem damage, proliferation of astrocytes and microglia in the cerebral cortex, diffuse perivascular and cytotoxic oedema, BBB damage and reactive neuroinflammation [22,23,24,25,26]. In this regard, the signal example can be septic endocarditis characterized by microbial emboli entering the brain parenchyma and causing ischemic infarction foci in conjunction with formation of abscesses. However, at present, this form of brain lesion is classified as septic-embolic encephalitis [27]. Also there are other indications for direct brain microbe invasion with formation of microabscesses in human SAE [28, 29]. Emergence of abscesses and microabscesses seems to be directly associated with sepsis (Fig. 1); although abscess formation can represent a distinct nosological entity.

Astrocytes in the formation of brain microabscess during SAE. Infiltration of brain parenchyma by infectious agents in SAE leads to the formation of microabscesses. The abscess core contains germs and purulent exudate, surrounded by hematogenous polymorphonuclear leukocytes (PMNc). a Infiltration of germs and endotoxins triggers astroglial activation; reactive astrocytes form a dense protective border composed of their tightly coupled and overlapping processes and the extracellular matrix, which prevents the spread of microbes and inflammatory cells toward the surrounding tissue. A lesser degree of tissue damage is manifested in the form of less pronounced tissue oedema and less changes in neurones. b Astroglial asthenia and compromised astroglial reactivity facilitate abscess enlargement. Polymorphonuclear leukocytes spread freely to the adjacent parenchyma and convey damage, aggravating leakage of the BBB. Astrocytes become oedematous, undergo clazmatodendrosis, loss of functions thus leading to acute perifocal parenchymal oedema of cytotoxic or combined type. Secondary acute swelling of neurones develops, followed by their karyocytolysis. c Astroglial scar formation establishes protective wall localising the infectious lesion and thus protecting the brain parenchyma
At the early stages of SAE the complex of behavioural symptoms referred to as “sickness behaviour” (which accompanies virtually every infectious disease or febrile inflammatory process) is often observed. Clinically, this syndrome is manifested by anorexia, anxiety, irritability, depression, anhedonia, decreased social communication and environmental interest, cognitive changes, including decreased concentration, learning ability and memory [30]. Research into mechanisms of this syndrome began in 1950s [31], and remains active since [32,33,34]. The biological background for sickness behaviour was formalised in 1988 as an adaptive mechanism of a macroorganism mounting an immune response to the invading infection aimed at preservation of energy potential for an adequate defence against the pathogen [35]. Alternatively, sickness behaviour is considered as a motivational behavioural mechanism [36, 37] or as a component of the body’s anti-pain system [38]. Development of sickness behaviour is associated with pro-inflammatory factors, such as interleukin (IL)-1α, IL-1β, tumour necrosis factor-α (TNF)-α and IL-6 with particular role attributed to IL-1 [39]. In rodents, the systemic or central IL-1β administration induces typical behavioural and neuro-endocrine symptoms of sickness behaviour [40] due to an activation of hypothalamic–pituitary–adrenal axis and adrenergic system [22]. Sickness behaviour acquires maladaptive features with increased severity and duration [30].
In sepsis, impairment of brain function develops upon the background of multiorgan failure (cardio-vascular, respiratory, hepato-renal) in conjunction with systemic overload with microbes and their related toxins (septicaemia and endotoxinaemia). High blood concentrations of endogenous metabolites (which are not properly eliminated or neutralised) produce detrimental effects on all physiological barriers and interfaces. Therefore, brain dysfunction during sepsis results from multicomponent, complex pathophysiology and could be defined as “mixed encephalopathy”. This reflects pathological remodelling of the vascular system and blood components in the uncontrolled immune response, DIC syndrome with violation of systemic microcirculation as well as global circulatory, respiratory and tissue hypoxia, and in addition—accumulation of endogenous toxins (such as ammonia and urea), electrolyte imbalance, altered osmolarity, altered blood proteins etc. [4]. Thus encephalopathy resulting from this complex pathogenesis may include acute circulatory encephalopathy (mostly ischemic variants in the form of mosaic foci of ischemic cellular damage and small infarcts due to the insufficient blood supply to the brain), acute hepatic or uremic encephalopathies manifested by cytotoxic oedema and swelling of the brain, and others, with great dependence on the concomitant pathology and a primary source of the infection [19]. There is also a direct relationship between the age of the septic patients and the severity of the disease and its complications, as well as the frequency of the SAE appearance [41]. Increased severity of sepsis in old patients can be explained by deficient immune responses and frequent concomitant pathologies such as atherosclerosis, arterial hypertension, diabetes mellitus etc. Consequently, in sepsis systemic inflammatory agents often face the already compromised brain and its barriers.
The most common animal models used to reproduce polymicrobial sepsis and endotoxemia are cecal ligation and puncture (CLP) and systemic injection of lipopolysaccharide (LPS). These models however are far from ideal. Significant differences exist between human AbS and CLP model. The latter mostly results in pathomorphological changes observed in “septic focus” into abdominal cavity, specificity of spreading the infection upon peritoneum, the time span of septicaemia appearance and staging of the disease course [42, 43]. The faecal peritonitis in humans, arising from perforated intestinal tract damage and characterised by a significant purulent–fibrinous reaction of the peritoneum, diffuse spread of faecal masses over the abdominal cavity, is absent in CPL subjected rats [43]; the intestinal contents rapidly becomes restricted, leading to formation of intestinal abscess or even infiltration depending on the size of the perforation hole (own observations). Microscopic examination of peritoneal samples from these infiltrates does not reveal purulent-fibrinous inflammation characteristic for human disease. Alternative models of AbS, such as colon ascendens stent peritonitis show more similarity to human pathology [43]. Despite almost complete replication of the conditions of human peritonitis, or at least of intra-abdominal abscess, and similar blood cytokine profile as well as presence of other common pathological features, full reproduction of the human pathology seems unattainable due to different immune and peritoneal reactivity. Another factor affecting model validity is associated with concomitant human pathologies, especially in older patients; modelling these in animals is of course impossible. The same applies to the LPS model of systemic Gram-negative infections; this model only partially mimics the composition of harmful toxins observed in human pathology and does not reproduce concomitant morbidities [42].
Principles of Astrogliopathology
Astrocytes are homeostatic cells of the CNS with wide array of functions including ionostasis and neurotransmitters metabolism, fluid balance, regulation of functional hyperaemia, neuronogenesis, maintenance of synaptic connectivity and plasticity etc. [44]. In the healthy brain astrocytes are characterised by significant region-dependent morphological and functional heterogeneity [45,46,47], although astroglial cells share common features such as absence of electrical excitability, highly hyperpolarised resting membrane potential and abundant expression of Na+-dependent solute carrier (SLC) transporters fundamental for CNS homoeostasis [44, 48]. In the CNS astroglial perisynaptic processes which closely enwrap ~ 50% to 60% of synapses [49] form the synaptic cradle that oversees synaptogenesis, synaptic maturation, maintenance, synaptic isolation and extinction [50,51,52]. Astrocytes contribute to the experience-dependent synaptic plasticity as well as to learning and memory, assisting morphological and functional synaptic plasticity [53,54,55,56]. Through dedicated transporters and enzymes astrocytes control homeostasis and catabolism of major neurotransmitters such as glutamate, GABA, noradrenaline and adenosine and supply neurones with neurotransmitter precursors [44, 57, 58]. Increase in computing power of the brain with its increased homeostatic demands is reflected in increase in astroglial complexity in the primate and human brains, which also possess several types of specialised astroglial cells (such as interlaminar and polarised astrocytes) that are absent in other species [59]. In the grey matter protoplasmic astrocytes occupy independent territorial domains [60, 61] and form glio-neurovascular units which integrate neural cells with blood vessels. Astrocytes are integrated into anatomically segregated syncytia through gap junctional connexons [62]. Astrocytes also form (with other neuroglia) the endocranial secretory system, which releases in excess of 200 molecules from neuromodulators and trophic factors to hormones [63]; these gliogenic molecules spread through the extracellular space by volume transmission and convective flow.
Astrocytes are key players in neuropathology; contemporary views regard astroglial changes as an obligatory component of neurological diseases [64,65,66,67,68,69]. Pathological changes in astrocytes are multifaceted and are disease and disease-stage specific. Conceptually, astrogliopathology is sub-classified into (i) reactive astrogliosis and (ii) astrocytopathies represented by astodegeneration with astroglial atrophy and loss of function and astroglial pathological remodelling [65, 66, 70].
Reactive astrogliosis is the evolutionary conserved programme triggered by nervous tissue damage which leads to genetic, morphological and functional remodelling of astrocytes; astroglial cells become hypertrophic, increase expression of intermediate filament proteins (glial fibrillary acidic protein GFAP and vimentin), modify receptors expression and the secretome [65, 71,72,73,74]. Astrogliotic programme produces multiple reactive phenotypes which are context and pathology specific; intrinsically astrogliosis is a defensive response aimed at neuroprotection and limiting the damage to the nervous tissue; in certain conditions, however, astrogliosis may acquire neurotoxic and deleterious potential [73, 75]. Astrogliotic changes vary depending on the nature and severity of the damage and are regulated in a context-specific manner by specific and complex inter- and intracellular signalling. Reactive phenotype is characterised by remodelling of multiple astrocytic properties through amplification or loss of functions [72, 76]. Reactive astrogliosis is classified according to morphological features (into isomorphic with preserved territorial domain organisation and anisomorphic, with violation of territorial domains, formation of astroglial palisades and ultimate scar formation) and according to the severity. The latter classification divides astrogliosis into (i) mild to moderate astrogliosis; (ii) severe diffuse astrogliosis; (iii) severe astrogliosis with compact scar formation [65, 72,73,74, 76].
Astrocytopathies represent widespread class of pathology-associated changes in astrocytes which affect their function and may lead to neuropathology. Pathological remodelling of astroglia results from genetic (Alexander disease), environmental (some variants of schizophrenia) or systemic (hepatic encephalopathy) factors which render astrocytes to acquire pathological phenotype that promote pathological evolution (disappearance of white matter in Alexander disease, neurotransmission imbalance leading to reactive psychosis in hepatic encephalopathy or aberrant synthesis of kynurenic acid in T. gondii infected brains leading to schizophrenic developments; see [77,78,79]). Astroglial degeneration with astroglial atrophy and loss of function has been detected in various diseases including toxic encephalopathies, neurodegenerative disorders, major psychiatric disorders (schizophrenia and major depression), addictive disorders and some forms of familial migraine [80,81,82,83,84,85,86,87].
Neurobiology of Septic Encephalopathy
Aberrant neurotransmission represents one of the leading pathological alterations in SAE, directly associated with cognitive, psychotic and consciousness-related symptomatology. In particular, changes in expression of receptors for γ-aminobutyric acid (GABA), serotonin, dopamine and noradrenaline have been detected [88, 89]. The neurotransmitter imbalance in the brain is thought to be related to changes in amino acids levels in the blood, in particular a decrease in branched-chain amino acids together with relative increase in aromatic amino acids with consequent increase the presence of the latter in the brain tissue [90]. Excess of the aromatic amino acids in plasma probably results from increased muscle proteolysis [91] and reduced hepatic clearance caused by liver insufficiency in sepsis [92]. Loss of BBB integrity may also contribute to an increased ratio of aromatic/branched chain amino acids in the brain of patients with SAE. Subsequently, large amounts of aromatic amino acids (tyrosine, phenylalanine, and tryptophan) can act as false neurotransmitters and affect the synthesis of bona fide neurotransmitters such as dopamine, noradrenaline, and serotonin [93].
Similar pathological developments are observed in acute hepatic encephalopathy (AHE) resulting from liver failure, which frequently complicates sepsis [94, 95]. In AHE excessive plasma-borne ammonium crosses often compromised BBB; ammonium is subsequently converted to glutamine by astrocytic glutamine synthetase, which is the key component of glutamate/GABA–glutamine cycle. Overload with ammonia leads to accumulation of glutamine in astrocytes resulting in an osmotic astrocyte swelling and subsequent cytotoxic brain oedema as well as aberrant neurotransmission with relative predominance of GABAergic tone [96,97,98]. The shift in aromatic/branched chain amino acids balance is present in the brain tissue in hepatic encephalopathy [96] similarly to that observed in SAE, indicating common mechanisms underlying aberrant neurotransmission and acute brain dysfunction. Hyperammonemia affects expression of several key astrocytic proteins including aquaporin-4 (AQP-4), inward rectifier Kir4.1 channels, GFAP, glutamine transporter SNAT5, glutamate transporters (EAAT-1/2), glycine transporters (GlyT-1), glucose transporters (GLUT-1) and peripheral type benzodiazepine receptors [96, 99,100,101]. Increased expression of AQP-4 in the astrocytic vascular end-feet and marked decrease in GFAP expression emerges at the background of the brain cytotoxic oedema [99], although aquaporin changes in the AHE remain controversial with indications for both increased and decreased expression of AQP-4 [99, 102]. In conclusion, astroglial response plays a critical role in pathogenesis of both AHE and SAE, with indications for both astroglial atrophy with loss of function and astroglial pathological remodelling.
Astroglia Protect the CNS Against Invasive and Systemic Infection
The instant and stereotyped immune reaction against injury is an activation of innate immunity, the humoural arm of which includes complement system and factors of coagulation system, whereas cellular arm is represented by mast cells, polymorphonuclear leukocytes (PMNc) and other myeloid cells with phagocytic properties [103]. Innate immune response, in turn, promotes adaptive immunity scenario, which usually has a delayed onset and develops during first few weeks after injury [104]. The adaptive immune mechanisms are initiated by pro-inflammatory cells antigen presentation to effector T- and B-lymphocytes, which enter the CNS (even in the presence of the intact BBB) from the periphery and destroy invasive pathogens [105, 106]. Leukocyte migration into the cerebrospinal fluid (CSF) is considered to occur through the choroid plexus (CP), whereas their trafficking in the blood-to-brain route occurs mainly through perivascular space (PVS) [107]. The immune surveillance of the brain has been claimed to be associated with CD4+ T-lymphocytes [108]. Activated T-lymphocytes penetrate the barriers using their surface enzymes and adhesion molecules either without altering vascular integrity or inducing mild and transient breakdown of the BBB. Activated T-cells exit the CNS back to the circulation or are eliminated by apoptosis [109]. The T-cells, which are capable to react with presented CNS antigens and recognise them in the appropriate major histocompatibility complex (MHC) context, remain in the parenchyma to initiate neuroinflammation [105]. Autoimmune and infectious neuroinflammation as well as blood-derived cytokine signalling during septic state lead to an increased expression of adhesion molecules by BBB endothelium and epithelial cells of choroid plexus, including P- and E-selectins, intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1 (VCAM1), platelet/endothelial cell adhesion molecule 1 (PECAM1), and members of the integrin family that mediate the adherence of activated leukocytes and facilitate their entry into the brain [107, 110, 111]. The T-cells activated by pathological CNS signals adhere and bind to brain endothelial cells (BECs) and subsequently cross the barrier [112]. In the CLP model of sepsis the BBB permeability is compromised within first 24 h after sepsis induction, which correlates with increase in brain cytokines and chemokines and is associated with increased adhesion of leucocytes to the brain microvasculature [113].
Strategic position of astroglia, which form the parenchymal part of brain barriers, as well as their territorial organisation and intimate contacts with neurones and other types of glia, makes astrocytes the principal guardians of the CNS; they control permeability of BBB and BCSFB and regulate microglial activation. Astrocytes therefore control the relationship between intracerebral and systemic physiology and pathology, including neuroimmune communication. Regional heterogeneity of neuroglia defines specificity of the innate immune response, thus underlying differences in pathogen dissemination and immunopathology in different CNS regions [114].
Reactive astrocytes contribute to neuroprotection and limit the propagation of pathological agents through the nervous tissue. In particular, astrocytes establish “physical” and “molecular” barriers for the spread of pathological agents [115]. The physical barrier is provided by astrocytes adjacent to the lesion site; these reactive astroglial cells undergo hypertrophy, polarisation and elongation, they aggregate into bundles (or palisades) perpendicular to the lesion border and organised in parallel distantly to the injured foci [116, 117]. These reactive astrocytes produce and secrete components of extracellular matrix (ECM) (for example chondroitin sulfate proteoglycans [118,119,120]), brevican, aggrecan [121], tenascin C [122], fibronectin and laminin [123] and increase the density of gap junctions [124, 125], thus reinforcing barriers. In case of diverse brain infections these barriers isolate tissue areas of pathogen replication and limit immune cells invasion [126, 127].
Experimental suppression of astrogliosis affects the barrier function and exacerbates the brain infection. Transgenic GFAP−/− mice with Staphylococcus aureus-induced brain abscess and Toxoplasma encephalitis (TE) demonstrated large and poorly demarcated inflammatory lesions with highly increased bacterial penetration into the brain, vasculitis, purulent ventriculitis and severe brain oedema as well as increased intracerebral Toxoplasma load and parasite-induced tissue necrosis [128]. In the GFAP-Cre gp130(fl/fl) mice lacking astroglial receptor for IL-6 family cytokines the inflammatory response was exacerbated, the restriction of inflammatory lesions was ineffective, parasite control was impaired, and necrotising variant of the TE developed [129]. Similarly, inhibition of astrogliotic response (manifested by reduced GFAP expression, aberrant astrocyte morphology and decreased expression of the ECM components) by activation of NF-κB signalling cascade in retinal ischemia or in spinal cord injury, is associated with an increased neuronal damage [130, 131]. Likewise ablation of scar-forming reactive astrocytes in experimental autoimmune encephalomyelitis (EAE) compromised both acute innate inflammatory response and chronic adaptive immune reaction leading to increased immune cell (monocytes, macrophages, neutrophils and lymphocytes) infiltration, BBB failure and neurodegeneration [132, 133]. Astrocytic perivascular barrier therefore restricts the influx of leukocytes into parenchyma and protects nervous tissue during peripherally triggered immune response of the CNS [132]. Astroglial reactivity defines the formation and progression of infectious abscesses in the brain parenchyma; inability of astrocytes to acquire reactive phenotype results in swelling, cytotoxic oedema and spread of damage (Fig. 1).
In addition to physical barrier, astrocytes also mould the molecular or functional barrier that restricts dissemination of infectious and inflammatory agents. Astroglial cells express a wide range of receptors to damage-associated and pathogen-associated molecular patters (DAMPs and PAMPs); these receptors include Toll-like receptors (TLRs), nucleotide-binding oligomerisation domains (NOD)-like receptors (NLRs), double-stranded RNA-dependent protein kinase, scavenger receptors, mannose receptor, complement components and mediators, such as CXCL10, CCL2, interleukin-6, B-cell-activating factor of the TNF family (BAFF) [134].
Infectious neuroinflammation is supported by the rapid recruitment of peripheral and activation of resident immunocompetent cells. Astrocytes play significant role in engaging, instructing and retaining leukocytes at distinct sites of CNS damage, thus controlling inflammatory response magnitude and outcome. Reactive astrocytes release molecules that attract immune cells specifically to the injured region and facilitate their extravasation and tissue infiltration. In models of ischemia, bacterial infection and EAE, astrocytes express leukocyte adhesion molecules, such as intercellular adhesion molecules and vascular cell adhesion molecules, which are linked to astrocytic NF-κB signalling [71, 130, 135]. Following CNS injury activation of BECs and astrocytes can lead to impairment of tight junction (TJ) integrity and formation of transendothelial cell channels [136] thus facilitating passage of leukocytes, pathogens and other inflammatory agents through the BBB. In case of infections or chronic neuroinflammatory diseases such as multiply sclerosis (MS) neutrophils, eosinophils, T-lymphocytes, monocytes, and other immune cells are routinely found in the CNS parenchyma [107]. Glia limitans, formed by astroglial endfeet is associated with its own basement membrane (also known as parenchymal basement membrane) consisting of laminin, type IV collagen and fibronectin, which serves as an additional barrier restricting access of immune cells to the CNS [107]. Crossing of this glial basement membrane by immune cells is facilitated by matrix metalloproteinases (MMPs) [137, 138].
Astrocytes secrete both anti-inflammatory and pro-inflammatory factors that can induce and/or regulate neuroinflammation. Among anti-inflammatory agents astroglial cells release (i) cytokines and growth factors: TGF-β, IL-6, IL-10, IL-11, IL-19, IL-27, SHH; (ii) intracellular signalling factors: STAT3, A20, Gal9, CRYAB; (iii) receptors: oestrogen receptor-α (ERα), dopamine D2 receptors (DRD2), glycoprotein 130 (GP130); (iv) small intercellular effector molecules: retinoic acid (RA); microRNAs: miR-181, Dicer1, miR-17-5p. Astrocyte-derived factors with pro-inflammatory activity are represented by: (i) chemokines: monocyte chemoattractant protein-1 (MCP-1/CCL2), CCL5 (RANTES), CCL7, CCL8, CCL12, CXCL1, CXCL8 (IL-8), CXCL9, IFN-γ-inducible protein-10 (IP-10/CXCL10), CXCL12, CXCL16; (ii) cytokines and growth factors: IL1-β, IL-6, IL-11, IL-15, IL-17, TNF-α, BAFF, VEGF; (iii) intracellular signalling factors: NF-κB, SOCS3, Act1; (iv) small intercellular effector molecules: PGE and NO [116, 139]. In systemic inflammation astrocytes tend to up-regulate production of pro-inflammatory cytokines and chemokines and develop more delayed pro-inflammatory phenotype comparing with microglia [140, 141]. The IL-1β, one of the main cytokines involved in sepsis pathophysiology, induces astrocyte production of vascular endothelial growth factor A (VEGF-A) and thymidine phosphorylase (TYMP/endothelial cell growth factor 1, ECGF1), which contribute to down-regulation of TJ proteins expression in BECs, thus promoting BBB breakdown [142]. At the same time astrocytes can attenuate microglial activation via TGF-β secretion, thus containing the inflammatory process [143]. The IL-1β is also recognised as potent instigator of astrocyte reactivity and scar formation. Stimulation of astrocytes with IL-1β up-regulates mRNA and protein expression of both TNF-α and IL-6 [144]. Constitutive astroglial over-expression of IL-6 (in a transgenic model) is associated with neurodegeneration, astrogliosis, breakdown of the BBB, angiogenesis, and increased expression of complement proteins [145, 146].
Chemokines secreted by reactive astrocytes activate specific receptors on both invading macrophages and microglia to attract them to the lesion focus. Astrocytes are the major source of monocyte chemoattractant protein 1 (MCP1 or CCL2) within the diseased CNS [139], for example in Toxoplasma encephalitis [147]. In rodent prion model of chronic neurodegeneration and inflammation astrocytes upregulate immunoreactivity of microglial chemoattractant chemokine CX3CL1 as well as its receptor CX3CR1 [148]. Reactive astrocytes regulate the course of immune response by releasing IFN-γ, IL-12, TNF-α, IL-10 and TGF-β that control pro- or anti-inflammatory macrophage phenotype. In rodent models of meningoencephalitis caused by Borna disease virus or HIV-1 reactive astrocytes serve as a major source of T-cell chemoattractants CCL5, and CXCL10, initiating adaptive immune response [149, 150]. In infectious and non-infectious neuroinflammation reactive astrocytes via pro-inflammatory (IFN-γ, TNF-α, IL-17) and anti-inflammatory (IL-10, TGF-β) cytokines can induce incoming naive precursor T-cells to develop their pro-inflammatory (Th1) or anti-inflammatory (Th2) phenotypes [131]. In particular, Th1 cells contribute to activation of macrophages and aggravate inflammation by producing IL-2, IFN-γ, TNF, while Th2 cells inhibit a range of macrophagial inflammatory functions, producing IL-4, IL-5, IL-6, IL-10, IL-13 mediated humoural immunity [151]. Astrocytes control endothelial cells (by secreting VEGF, TGFα, bFGF, TNF-α, IL-1β, IL-3, IL-6, Ang-1, BAFF), and glial-derived neurotrophic factor (GDNF) thus supporting the integrity of BBB [103]. Astroglial function is known to be regulated by glutamate; at the same time astrocytes are the major glutamate sink in the CNS [44]. Treatment of cultured astrocytes with glutamate decreased synthesis of CCL5, which potentially may reduce neuroinflammation [152]. At the same time exposure to the inflammatory media diminished astroglial uptake of glutamate, thus enhancing excitotoxicity [153]; whereas and microglia-derived TNF-α may stimulate astroglial release of glutamate with the same effect [154].
Astrocytes express several pattern recognition receptors (PRRs), including TLR 2, 3, 4, 9 [155], NLRs, retinoic acid-inducible gene (RIG)-like receptors (RLRs) and cytokine receptors [156]. Stimulation of astrocytes with ATP (classical DAMP) induces formation of inflammasomes comprised of the NLR protein-1 or -2 (NLRP1, NALP2), the adaptor protein apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC) and caspase-1; activation of this inflammasome leads to the processing of inflammatory caspase-1 and IL-1β [157]. The NOD2, expressed by astrocytes, acts as a receptor for a minimal motif present in all bacterial peptidoglycans and it is required for initiation of astrogliosis in response to Neisseria meningitidis (meningococcus) and Borrelia burgdorferi infections [158]. Astrocytes arguably express class II MHC molecules thus acting as antigen presenting cells (APCs) linked to CD4+ T-cells [159, 160], however, this expression is lower than in microglia [161]. All in all, the ability of astrocytes to present antigen through class II MHC remains controversial [139, 162, 163]. It seems that astroglial cells behave as non-professional APCs because of their supposed inability to express co-stimulatory molecules CD40 and B7-1, B7-2 that are critical for complete antigen presentation and further T-cell clonal expansion [139]. Of note, astrocytes form immunological synapses with CD8+ T-cells, which corroborates their ability to antigen presentation through class I MHC receptors [164]. Astroglial expression of class II MHC remains to be clarified [139].
Astrocytes and Cerebral Infection
Only a limited number of neutrotropic viruses, fungi and parasites can invade the brain parenchyma with relative ease; the majority of pathogens are effectively fenced by the brain barriers [114]. Pathogens are able to cross the BBB using the paracellular route, via transcytotic mechanism, inside entering monocytes (the Trojan horse hypothesis) as well as by other mechanisms such as hijacking of β-adrenergic receptors as shown for N. meningitides [165].
Activation of astroglial PRRs (for example by LPS) triggers astrogliotic response, which is a common feature of brain infections, with well appreciated defensive role [128, 134, 166]. Alternatively, astrocytes may undergo pathological remodelling and form a reservoir for infection; in addition astrocytes can trigger apoptotic death of their uninfected neighbours through gap junction route [167, 168]. Several bacteria including Streptococcus pneumonia, B. burgdorferi and N. meningitides interact with astrocytes, inducing reactive response and increasing production of pro-inflammatory cytokines and chemokines such as IL-6, TNF-α, IL-8, CXCL-1 and CXCL-10 [168].
Several protozoa with a tropism for the brain infect astroglia, including Toxoplasma gondii and Plasmodium falciparum. The T. gondii, a wide-spread neurotropic obligate intracellular protozoan parasite, naturally infects astrocytes causing their prominent activation. During T. gondii infection astrocytic cytokines can directly target invading pathogens. For instance, IL-6 and IFN-γ reduce parasite burden, whereas animals with deleted gp130 (IL-6 receptor) in astrocytes show rapidly progressive lethal necrotising encephalitis [129, 166, 169, 170]. The IFN-γ similarly inhibits parasite replication in astrocytes. This inhibition is mediated by signal transducer and activator of transcription 1 (STAT1); deletion of STAT1 facilitates parasite replication and dissemination [171]. Astrocytes infected by T. gondii alter neurotransmission (arguably linked to an increased risk of schizophrenia) because of aberrant production of kinyrenic acid [79]. In cerebral malaria, caused by P. falciparum, astrocytes show early reactive changes and increase production of TNF-α with loss of astrocytes associated with BBB failure and spread of infection [168].
Cats with feline infectious peritonitis, caused by Coronavirus, characterised by leptomeningitis, focal encephalitis and necrosis of subventricular regions, showed pronounced astrocytic reactivity throughout the brain, including periventricular areas, molecular layer of the cerebellum and cerebral cortex. Astrocytic reactivity is mainly associated with areas of granulomatous vasculitis; astrocytes demonstrated strong vimentin expression in parenchyma with severe inflammatory and necrotic changes, while GFAP expression was mild or absent [172], probably indicating combination of reactivity and astrodegeneration.
Violation of Astroglial Gap Junction Communication During Infection
Exposure to pro-inflammatory stimuli reduces astroglial gap junction communication [173,174,175,176]. The peptidoglycan of the bacterial wall of Staphylococcus aureus (one of the main pathogens causing abscesses in the human brain) induces time-dependent reduction in expression of connexin-43 and 30 (Cx43/Cx30) and decrease of gap junctional communications between astrocytes surrounding the infection foci; this may reflect a defensive mechanism limiting the propagation of infectious agents and toxins [177, 178]. In the animal model of staphylococcal brain abscess decrease in gap junctional connectivity between reactive astrocytes developed in parallel with an increase in activity of Cx43, Cx30 and pannexin-1 (Panx1) hemichannels, which may contribute to aberrant astroglial secretion [179]. In the LPS model of systemic inflammation expression of Cx43 was inhibited by IL-1β and TNF-α released from activated microglia [180]. Similarly, treatment of cultured rat astrocytes with LPS reduced expression of Cx43 through TLR4-mediated signalling pathway involving activation of stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and the ubiquitin–proteasome proteolytic pathway [173]. At the same time partial deletion of astrocytic Cx43 in mice model of neonatal sepsis suppressed LPS-induced inflammatory response and attenuated microglial activation [181].
Astroglial Signalling Associated with Sepsis Induced Lesion
Receptors of TLR family are closely involved in regulation of the neuroimmune response and astroglial pathology. The subtypes of TLRs are sensitive to distinct PAMPs: the LPS signals through TLR4, double stranded RNA activates TLR3; peptidoglycans interacts with TLR2, while CpG DNA acts through TLR9 [182]. In polymicrobial sepsis TLR2 and TLR4 are considered to play the major role [183]. After stimulation, astrocytic TLRs bind adaptor proteins myeloid differentiation factor 88 (MyD88) and/or a TIR-containing adaptor molecule, Toll/interferon-1 receptor domain-containing adaptor inducing interferon-β (TRIF), which act as part of signalling cascade [182]. Both the MyD88- and TRIF-dependent pathways induce activation of p38, JNK/SAPK, extracellular signal-regulated kinase and NF-κB signalling [173]. Astrocytes in simulated polymicrobial sepsis express the cyclin-dependent kinase inhibitor p21, a cell cycle regulator that important for NF-κB activation, as well as for activation of an inducible nitric oxide synthase (iNOS) [140]. In turn, NF-κB is a key regulator of the inflammatory response that triggers production of pro-inflammatory cytokines and chemokines (TNF-α, IL-1β, and IL-6) and promotes generation of reactive oxygen/nitrogen species (ROS/RNS), including iNOS and subsequently increases NO levels [184]. In turn, NO forms oxidant species, which cause cellular toxicity and neuronal death [185].
Endotoxemia (another model of sepsis) results in up-regulation of astrocytic AQP4 with associated development of oedema (a common sign of human SAE), mediated by TNF-α signalling through TNFR1 [186]. Hippocampus appears to be the most vulnerable to experimental sepsis with the peak of astrocyte activation attained 24 h after sepsis induction [187]. The pro-inflammatory astroglial profile (increased levels of TNF-α, IL-1β, IL-6, IL-18, cyclooxygenase-2 with decreased levels of IL-10) was accompanied with an increase in mRNA expression of TLR2, the latter playing a crucial role in Gram-positive sepsis. In contrast, activated microglia was characterised by an increased expression of TLR4 [140].
Intraperitoneal injection of LPS rapidly increased expression of cytokines CCL2, CXCL1, CXCL2 and IL-6 in cellular components of the brain-immune interfaces (CP epithelial and stromal cells, leptomeningeal stromal cells, and hippocampal vascular BECs), while receptors for these cytokines were simultaneously expressed in astrocytic endfeet. Up-regulation of cytokines CCL11, CXCL10, and granulocyte-colony stimulating factor (G-CSF) in hippocampal astrocytes increased with 24 h delay. At the same time astroglial reactivity developed much earlier—as early as 4 h after systemic LPS injection [21]. In response to endotoxemia astrocytes also secrete CCL11, which impairs learning and memory in adult brain, and instigates microglia migration and ROS production, thereby causing hippocampal neuronal damage, behavioural changes and memory impairment [21]. It seems that cells of the brain–immune interface respond to endotoxemia by releasing cytokines, and astrocytes respond to these signals using endfeet cytokine receptors [21]. Astrocytes also regulate neuroinflammatory environment in sepsis context by controlling microglial activation. Activated microglia in inflamed brain is characterised by pronounced neurotoxic phenotype reflected by the release of multiple cytokines and ROS/RNS, which contribute to cell death within specific vulnerable brain areas [19]. It has to be noted however, that data on reactive features of astrocytes and microglia in the presence of systemic LPS are controversial; there are for example findings indicating that pro-inflammatory cytokine expression in microglia peaked much earlier then cytokine and chemokine induction of astrocytes [141].
Role of Astroglia in BBB Abnormalities in Systemic Inflammation
The abnormalities of BBB are classified as disruptive and non-disruptive with both variants present in systemic inflammation (Figs. 2, 3). The non-disruptive BBB pathology (Fig. 2) is represented by alterations at the molecular level when BBB permeability is compromised following up- or down-regulation of receptors and transporters expressed in BECs and astrocytes, cytokine production, astrocytic dysfunction and increased pathogen neuroinvasion [188]. Disruptive BBB alterations (Fig. 3) are manifested by anatomical changes, which include degradation of glycocalyx, loss of tight junctions integrity, mitochondrial damage, re-induction of fenestrae, increased vesicular traffic and apoptosis of BECs, breakdown of glia limitans and astrocytopathy. Disruption of BBB in systemic inflammation is mediated by blood-derived MMPs, prostanoids, NO and ROS [189, 190]. Systemic LPS challenge also affects astrocytes by modulating transcriptional profile and promoting pro-inflammatory phenotype [71] associated with morphological changes, including early reactivity [191, 192], structural remodelling of astrocytic end-feet and even astrocytic loss [192], all these alterations facilitate disruption of BBB. Astroglia-produced cytokines, including IL-1β, IL-6, TNF-α, as well as prostaglandins mediate both disruptive and non-disruptive alterations of BBB [188].

Tissue reactivity in non-disruptive BBB changes during mild systemic inflammation. Non-disruptive BBB changes are represented by functional changes of cellular components of BBB. Thus, activation of BEC by systemic agents leads to aberrant expression of transporters, receptors and adhesion molecules and production of various cytokines that stimulate cytokine receptors of other cellular constituents, in particular in astrocytes. Such stimulation causes reactive changes primarily in glia limitans. Sufficient level of astrocytic reactivity, accompanied by upregulation of cytoskeletal interfilaments GFAP and vimentin, prevents penetration of infectious and toxic agents into the brain parenchyma. BBB blood brain barrier, BEC brain endothelial cell, PVS perivascular space, rMcG ramified microglial cell, Olg oligodendrocyte

Tissue reactivity in disruptive BBB changes during severe systemic inflammation. Disruptive BBB changes are represented by functional and anatomical changes of all its components. Activation of BEC by systemic agents leads both to upreglation of transporters, receptors and adhesion molecules and production of various cytokines that stimulate cytokine receptors of other cellular constituents, in particular astrocytic ones. BECs cytokines initiate reactive changes primarily in glia limitans. The list of systemic aggressive factors in septic condition, in particular hematogenious MMPs, causes alteration and destructive changes of endothelial TJ. Together with increased pinocytosis, transcytosis and apoptotic changes in BECs all these lead to BBB become leaky and highly permeable for LPS, pathogens, various immune cells and other aggressive factors observed in sepsis. Components of glia limitans also appeared damaged and show disruptive changes. Reactive changes of astrocytes in this case are supposed to be diverse. Pathological remodeling is seems to be predominant and display various phenotypes. Astrogliotic changes can be combined with oedematous ones accompanied by loss of functions. Besides loss of homeostatic functions reactive astrocytes release a number of chemokines, cytokines, enzymes, reactive species, neurotoxins that all together lead to aggravation of neuroinflammation, tissue oedema, parenchymal cell death and further increasing of the BBB leakage. BBB blood brain barrier, PVS perivascular space, aMcG activated microglial cell, Olg oligodendrocyte
The type of BBB impairment varies with the severity of systemic inflammation. Thus, in mild systemic infections, manifested by sickness behaviour or at the very early stages of septic process non-disruptive changes prevail, while both non-disruptive and disruptive changes probably occur in sepsis [188]. In the human brain disruptive changes are frequent and are enhanced by comorbide pathologies especially in older patients. Atherosclerosis and hypertension related vessel damage impair autoregulation of brain blood circulation, which may rapidly decompensate in systemic inflammation and MODS. This is particularly evident in ischemic-reperfusional and hemorrhagic damage over the most vulnerable regions of the brain [193, 194].
Conclusion
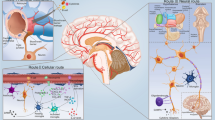
Astrocytes are responsible for essential homeostatic and defensive functions in the CNS. In particular astroglial cells act as surveillant and integrative hubs overseeing CNS immune responses by supporting the balance between beneficial and detrimental effects of immunity in the nervous tissue. In sepsis, astrocytes define the depth and severity of encephalopathy and cognitive impairment (Fig. 4). Being an essential component of brain-immune interfaces in SAE astrocytes can gain abnormal functions that facilitate the unfavourable course of neuroinflammation and brain dysfunction. Pathophysiology of sepsis in humans is complicated by frequent presence of comorbide or background pathologies, especially the vascular deficits, as well as by specificity of immune response and tissue reactions to injury. In older patients age-related remodelling of the brain tissue define a distinct impact on SAE outcome.

Astroglial reactivity as a defining factor in pathological evolution of septic encephalopathy
Systemic infection and peripheral immune activation instigate rapid and concerted response of CNS neuroglia regulated through fine-tuned communications between astrocytes, microglia and cells of brain barriers, which propagate inflammatory signals over the brain and induce physiological and behavioural changes that correspond to sickness behaviour or more severe cognitive disruptions characteristic for sepsis associated encephalopathy.
References
Kaperonis EA, Liapis CD, Kakisis JD, Dimitroulis D, Papavassiliou VG (2006) Inflammation and atherosclerosis. Eur J Vasc Endovasc Surg 31:386–393. https://doi.org/10.1016/j.ejvs.2005.11.001
Badawi A, Klip A, Haddad P, Cole DE, Bailo BG, El-Sohemy A, Karmali M (2010) Type 2 diabetes mellitus and inflammation: prospects for biomarkers of risk and nutritional intervention. Diabetes Metab Syndr Obes 3:173–186
De Miguel C, Rudemiller NP, Abais JM, Mattson DL (2015) Inflammation and hypertension: new understandings and potential therapeutic targets. Curr Hypertens Rep 17:507. https://doi.org/10.1007/s11906-014-0507-z
Remick DG (2007) Pathophysiology of sepsis. Am J Pathol 170:1435–1444. https://doi.org/10.2353/ajpath.2007.060872
Dal-Pizzol F, Tomasi CD, Ritter C (2014) Septic encephalopathy: does inflammation drive the brain crazy? Rev Bras Psiquiatr 36:251–258
Singer M et al (2016) The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315:801–810. https://doi.org/10.1001/jama.2016.0287
Schwartz M, Deczkowska A (2016) Neurological disease as a failure of brain-immune crosstalk: the multiple faces of neuroinflammation. Trends Immunol 37:668–679. https://doi.org/10.1016/j.it.2016.08.001
Verkhratsky A, Butt AM (2013) Glial physiology and pathophysiology. Wiley, Chichester
Michels M, Sonai B, Dal-Pizzol F (2017) Polarization of microglia and its role in bacterial sepsis. J Neuroimmunol 303:90–98. https://doi.org/10.1016/j.jneuroim.2016.12.015
Shuliatnikova TV, Shavrin VO (2018) Sepsis associated encephalopathy and abdominal sepsis: current state of problem. Art Med 3:158–165. https://doi.org/10.14739/2310-1237.2017.3.118773
Sartelli M, Coccolini F, Catena F, Ansaloni L (2016) Sepsis is a dynamic syndrome. J Peritoneum 1:5–6. https://doi.org/10.4081/joper.2016.23
Mosevoll KA et al (2018) Inflammatory mediator profiles differ in sepsis patients with and without bacteremia. Front Immunol 9:691. https://doi.org/10.3389/fimmu.2018.00691
Balk RA (2014) Systemic inflammatory response syndrome (SIRS): where did it come from and is it still relevant today? Virulence 5:20–26. https://doi.org/10.4161/viru.27135
Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, Hinshaw LB (1990) Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med 18:801–806
Piva S, McCreadie VA, Latronico N (2015) Neuroinflammation in sepsis: sepsis associated delirium. Cardiovasc Hematol Disord Drug Targets 1:10–18. https://doi.org/10.2174/1871529X15666150108112452
Sonneville R et al (2013) Understanding brain dysfunction in sepsis. Ann Intensive Care 3:15. https://doi.org/10.1186/2110-5820-3-15
Lamar CD, Hurley RA, Taber KH (2011) Sepsis-associated encephalopathy: review of the neuropsychiatric manifestations and cognitive outcome. J Neuropsychiatry Clin Neurosci 23:237–241. https://doi.org/10.1176/appi.neuropsych.23.3.237
Bleck TP, Smith MC, Pierre-Louis SJ, Jares JJ, Murray J, Hansen CA (1993) Neurologic complications of critical medical illnesses. Crit Care Med 21:98–103
Chaudhry N, Duggal AK (2014) Sepsis associated encephalopathy. Adv Med 2014:762320. https://doi.org/10.1155/2014/762320
Tsuruta R, Oda Y (2016) A clinical perspective of sepsis-associated delirium. J Intensive Care 4:18. https://doi.org/10.1186/s40560-016-0145-4
Hasegawa-Ishii S, Inaba M, Umegaki H, Unno K, Wakabayashi K, Shimada A (2016) Endotoxemia-induced cytokine-mediated responses of hippocampal astrocytes transmitted by cells of the brain-immune interface. Sci Rep 6:25457. https://doi.org/10.1038/srep25457
Heming N, Mazeraud A, Verdonk F, Bozza FA, Chretien F, Sharshar T (2017) Neuroanatomy of sepsis-associated encephalopathy. Crit Care 21:65. https://doi.org/10.1186/s13054-017-1643-z
Mazeraud A, Pascal Q, Verdonk F, Heming N, Chretien F, Sharshar T (2016) Neuroanatomy and physiology of brain dysfunction in sepsis. Clin Chest Med 37:333–345. https://doi.org/10.1016/j.ccm.2016.01.013
Sharshar T, Annane D, de la Grandmaison GL, Brouland JP, Hopkinson NS, Francoise G (2004) The neuropathology of septic shock. Brain Pathol 14:21–33
Jackson AC, Gilbert JJ, Young GB, Bolton CF (1985) The encephalopathy of sepsis. Can J Neurol Sci 12:303–307
Pendlebury WW, Perl DP, Munoz DG (1989) Multiple microabscesses in the central nervous system: a clinicopathologic study. J Neuropathol Exp Neurol 48:290–300
Tonges L, Pilgram-Pastor S, Puls M, Schmidt H (2009) Septic embolic encephalitis after Staphylococcus aureus endocarditis of a prosthetic valve in a 57-year-old woman: a case report. Cases J 2:6653. https://doi.org/10.4076/1757-1626-2-6653
Young GB, Bolton CF, Austin TW, Archibald YM, Gonder J, Wells GA (1990) The encephalopathy associated with septic illness. Clin Investig Med 13:297–304
Singer BH et al (2018) Bacterial dissemination to the brain in sepsis. Am J Respir Crit Care Med 197:747–756. https://doi.org/10.1164/rccm.201708-1559OC
Dantzer R, Kelley KW (2007) Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun 21:153–160. https://doi.org/10.1016/j.bbi.2006.09.006
Garcia J, Kimeldorf DJ, Koelling RA (1955) Conditioned aversion to saccharin resulting from exposure to gamma radiation. Science 122:157–158
Mitchell D, Kirschbaum EH, Perry RL (1975) Effects of neophobia and habituation on the poison-induced avoidance of exteroceptive stimuli in the rat. J Exp Psychol Anim Behav Process 1:47–55
Bluthe RM, Dantzer R, Le Moal M (1985) Peripheral injections of vasopressin control behavior by way of interoceptive signals for hypertension. Behav Brain Res 18:31–39
Dantzer R, Bluthe RM, LeMoal M (1988) Experimental assessment of drug-induced changes in cognitive function: vasopressin as a case study. Neurotoxicology 9:471–477
Hart BL (1988) Biological basis of the behavior of sick animals. Neurosci Biobehav Rev 12:123–137
Miller NE (1964) Some psychophysiological studies of motivation and of the behavioral effects of illness. Bull Br Psychol Soc 17:1–20
Dantzer R (2001) Cytokine-induced sickness behavior: mechanisms and implications. Ann N Y Acad Sci 933:222–234
Watkins LR, Maier SF (2000) The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol 51:29–57. https://doi.org/10.1146/annurev.psych.51.1.29
Larson SJ, Dunn AJ (2001) Behavioral effects of cytokines. Brain Behav Immun 15:371–387. https://doi.org/10.1006/brbi.2001.0643
Anforth HR et al (1998) Biological activity and brain actions of recombinant rat interleukin-1α and interleukin-1β. Eur Cytokine Netw 9:279–288
Kempker JA, Martin GS (2016) The changing epidemiology and definitions of sepsis. Clin Chest Med 37:165–179. https://doi.org/10.1016/j.ccm.2016.01.002
Lewis AJ, Seymour CW, Rosengart MR (2016) Current murine models of sepsis. Surg Infect (Larchmt) 17:385–393. https://doi.org/10.1089/sur.2016.021
Schabbauer G (2012) Polymicrobial sepsis models: CLP versus CASP. Drug Discov Today 9:e17–e21
Verkhratsky A, Nedergaard M (2018) Physiology of astroglia. Physiol Rev 98:239–389. https://doi.org/10.1152/physrev.00042.2016
Oberheim NA, Goldman SA, Nedergaard M (2012) Heterogeneity of astrocytic form and function. Methods Mol Biol 814:23–45. https://doi.org/10.1007/978-1-61779-452-0_3
Morel L et al (2017) Molecular and functional properties of regional astrocytes in the adult brain. J Neurosci 37:8706–8717. https://doi.org/10.1523/JNEUROSCI.3956-16.2017
Hewett JA (2009) Determinants of regional and local diversity within the astroglial lineage of the normal central nervous system. J Neurochem 110:1717–1736. https://doi.org/10.1111/j.1471-4159.2009.06288.x
Kirischuk S, Parpura V, Verkhratsky A (2012) Sodium dynamics: another key to astroglial excitability? Trends Neurosci 35:497–506. https://doi.org/10.1016/j.tins.2012.04.003
Ventura R, Harris KM (1999) Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci 19:6897–6906
Eroglu C, Barres BA (2010) Regulation of synaptic connectivity by glia. Nature 468:223–231. https://doi.org/10.1038/nature09612
Verkhratsky A, Nedergaard M (2014) Astroglial cradle in the life of the synapse. Philos Trans R Soc Lond B 369:20130595. https://doi.org/10.1098/rstb.2013.0595
Nedergaard M, Verkhratsky A (2012) Artifact versus reality—how astrocytes contribute to synaptic events. Glia 60:1013–1023. https://doi.org/10.1002/glia.22288
Hirrlinger J, Hulsmann S, Kirchhoff F (2004) Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur J Neurosci 20:2235–2239. https://doi.org/10.1111/j.1460-9568.2004.03689.x
Benediktsson AM, Schachtele SJ, Green SH, Dailey ME (2005) Ballistic labeling and dynamic imaging of astrocytes in organotypic hippocampal slice cultures. J Neurosci Methods 141:41–53. https://doi.org/10.1016/j.jneumeth.2004.05.013
Nishida H, Okabe S (2007) Direct astrocytic contacts regulate local maturation of dendritic spines. J Neurosci 27:331–340. https://doi.org/10.1523/JNEUROSCI.4466-06.2007
Zorec R, Horvat A, Vardjan N, Verkhratsky A (2015) Memory formation shaped by astroglia. Front Integr Neurosci 9:56. https://doi.org/10.3389/fnint.2015.00056
Oliet SH, Piet R, Poulain DA (2001) Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292:923–926. https://doi.org/10.1126/science.1059162
Verkhratsky A, Nedergaard M, Hertz L (2015) Why are astrocytes important? Neurochem Res 40:389–401. https://doi.org/10.1007/s11064-014-1403-2
Oberheim NA et al (2009) Uniquely hominid features of adult human astrocytes. J Neurosci 29:3276–3287. https://doi.org/10.1523/JNEUROSCI.4707-08.2009
Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG (2007) Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27:6473–6477. https://doi.org/10.1523/JNEUROSCI.1419-07.2007
Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002) Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22:183–192
Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N (2010) Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11:87–99
Verkhratsky A, Matteoli M, Parpura V, Mothet JP, Zorec R (2016) Astrocytes as secretory cells of the central nervous system: idiosyncrasies of vesicular secretion. EMBO J 35:239–257. https://doi.org/10.15252/embj.201592705
Parpura V et al (2012) Glial cells in (patho)physiology. J Neurochem 121:4–27. https://doi.org/10.1111/j.1471-4159.2012.07664.x
Pekny M et al (2016) Astrocytes: a central element in neurological diseases. Acta Neuropathol 131:323–345. https://doi.org/10.1007/s00401-015-1513-1
Verkhratsky A, Steardo L, Parpura V, Montana V (2016) Translational potential of astrocytes in brain disorders. Prog Neurobiol 144:188–205. https://doi.org/10.1016/j.pneurobio.2015.09.003
Sofroniew MV (2014) Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 20:160–172. https://doi.org/10.1177/1073858413504466
Giaume C, Kirchhoff F, Matute C, Reichenbach A, Verkhratsky A (2007) Glia: the fulcrum of brain diseases. Cell Death Differ 14:1324–1335. https://doi.org/10.1038/sj.cdd.4402144
Verkhratsky A, Parpura V (2016) Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiol Dis 85:254–261. https://doi.org/10.1016/j.nbd.2015.03.025
Verkhratsky A, Zorec R, Parpura V (2017) Stratification of astrocytes in healthy and diseased brain. Brain Pathol 27:629–644. https://doi.org/10.1111/bpa.12537
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410. https://doi.org/10.1523/JNEUROSCI.6221-11.2012
Sofroniew MV (2014) Astrogliosis. Cold Spring Harb Perspect Biol 7:a020420. https://doi.org/10.1101/cshperspect.a020420
Pekny M, Pekna M (2014) Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev 94:1077–1098. https://doi.org/10.1152/physrev.00041.2013
Liddelow SA, Barres BA (2017) Reactive astrocytes: production, function, and therapeutic potential. Immunity 46:957–967. https://doi.org/10.1016/j.immuni.2017.06.006
Liddelow SA et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647. https://doi.org/10.1016/j.tins.2009.08.002
Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE (2012) Alexander disease. J Neurosci 32:5017–5023. https://doi.org/10.1523/JNEUROSCI.5384-11.2012
Norenberg MD (1987) The role of astrocytes in hepatic encephalopathy. Neurochem Pathol 6:13–33
Schwarcz R, Hunter CA (2007) Toxoplasma gondii and schizophrenia: linkage through astrocyte-derived kynurenic acid? Schizophr Bull 33:652–653. https://doi.org/10.1093/schbul/sbm030
Cotter D, Mackay D, Landau S, Kerwin R, Everall I (2001) Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry 58:545–553
Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, Meltzer H, Overholser J, Stockmeier C (2002) Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophr Res 57:127–138
Verkhratsky A, Rodriguez JJ, Steardo L (2014) Astrogliopathology: a central element of neuropsychiatric diseases? Neuroscientist 20:576–588. https://doi.org/10.1177/1073858413510208
Rajkowska G, Legutko B, Moulana M, Syed M, Romero DG, Stockmeier CA, Miguel-Hidalgo JJ (2018) Astrocyte pathology in the ventral prefrontal white matter in depression. J Psychiatr Res 102:150–158. https://doi.org/10.1016/j.jpsychires.2018.04.005
Scofield MD et al (2016) Cocaine self-administration and extinction leads to reduced glial fibrillary acidic protein expression and morphometric features of astrocytes in the nucleus accumbens core. Biol Psychiatry 80:207–215. https://doi.org/10.1016/j.biopsych.2015.12.022
Plata A et al (2018) Astrocytic atrophy following status epilepticus parallels reduced Ca(2+) activity and impaired synaptic plasticity in the rat hippocampus. Front Mol Neurosci 11:215. https://doi.org/10.3389/fnmol.2018.00215
Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58:831–838. https://doi.org/10.1002/glia.20967
Capuani C et al (2016) Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol Med 8:967–986. https://doi.org/10.15252/emmm.201505944
Kadoi Y, Saito S (1996) An alteration in the gamma-aminobutyric acid receptor system in experimentally induced septic shock in rats. Crit Care Med 24:298–305
van Gool WA, van de Beek D, Eikelenboom P (2010) Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet 375:773–775. https://doi.org/10.1016/S0140-6736(09)61158-2
Basler T, Meier-Hellmann A, Bredle D, Reinhart K (2002) Amino acid imbalance early in septic encephalopathy. Intensive Care Med 28:293–298. https://doi.org/10.1007/s00134-002-1217-6
Murton A et al (2018) Sepsis increases muscle proteolysis in severely burned adults, but does not impact whole-body lipid or carbohydrate kinetics. Shock. https://doi.org/10.1097/SHK.0000000000001263
Yan J, Li S, Li S (2014) The role of the liver in sepsis. Int Rev Immunol 33:498–510. https://doi.org/10.3109/08830185.2014.889129
Skowronska M, Albrecht J (2012) Alterations of blood brain barrier function in hyperammonemia: an overview. Neurotox Res 21:236–244. https://doi.org/10.1007/s12640-011-9269-4
Wang D, Yin Y, Yao Y (2014) Advances in sepsis-associated liver dysfunction. Burns Trauma 2:97–105. https://doi.org/10.4103/2321-3868.132689
Aldridge DR, Tranah EJ, Shawcross DL (2015) Pathogenesis of hepatic encephalopathy: role of ammonia and systemic inflammation. J Clin Exp Hepatol 5:S7–S20. https://doi.org/10.1016/j.jceh.2014.06.004
Butterworth RF (2015) Pathogenesis of hepatic encephalopathy and brain edema in acute liver failure. J Clin Exp Hepatol 5:S96–S103. https://doi.org/10.1016/j.jceh.2014.02.004
Montana V, Verkhratsky A, Parpura V (2014) Pathological role for exocytotic glutamate release from astrocytes in hepatic encephalopathy. Curr Neuropharmacol 12:324–333. https://doi.org/10.2174/1570159X12666140903094700
Shulyatnikova TV, Shavrin VA (2017) Modern view on hepatic encephalopathy: basic terms and concepts of pathogenesis. Pathologia 14:371–380. https://doi.org/10.14739/2310-1237.2017.3.118773
Thumburu KK et al (2014) Expression of astrocytic genes coding for proteins implicated in neural excitation and brain edema is altered after acute liver failure. J Neurochem 128:617–627. https://doi.org/10.1111/jnc.12511
Obara-Michlewska M, Ding F, Popek M, Verkhratsky A, Nedergaard M, Zielinska M, Albrecht J (2018) Interstitial ion homeostasis and acid-base balance are maintained in oedematous brain of mice with acute toxic liver failure. Neurochem Int 118:286–291. https://doi.org/10.1016/j.neuint.2018.05.007
Obara-Michlewska M, Ruszkiewicz J, Zielinska M, Verkhratsky A, Albrecht J (2015) Astroglial NMDA receptors inhibit expression of Kir4.1 channels in glutamate-overexposed astrocytes in vitro and in the brain of rats with acute liver failure. Neurochem Int 88:20–25. https://doi.org/10.1016/j.neuint.2014.10.006
Bodega G et al (2012) Ammonia induces aquaporin-4 rearrangement in the plasma membrane of cultured astrocytes. Neurochem Int 61:1314–1324. https://doi.org/10.1016/j.neuint.2012.09.008
Lampron A, Elali A, Rivest S (2013) Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron 78:214–232. https://doi.org/10.1016/j.neuron.2013.04.005
Wraith DC, Nicholson LB (2012) The adaptive immune system in diseases of the central nervous system. J Clin Invest 122:1172–1179. https://doi.org/10.1172/JCI58648
Hickey WF, Hsu BL, Kimura H (1991) T-lymphocyte entry into the central nervous system. J Neurosci Res 28:254–260. https://doi.org/10.1002/jnr.490280213
Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC (2006) CNS immune privilege: hiding in plain sight. Immunol Rev 213:48–65. https://doi.org/10.1111/j.1600-065X.2006.00441.x
Wilson EH, Weninger W, Hunter CA (2010) Trafficking of immune cells in the central nervous system. J Clin Investig 120:1368–1379. https://doi.org/10.1172/JCI41911
Kivisakk P et al (2003) Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci USA 100:8389–8394. https://doi.org/10.1073/pnas.1433000100
Hu P, Pollard JD, Chan-Ling T (2000) Breakdown of the blood-retinal barrier induced by activated T cells of nonneural specificity. Am J Pathol 156:1139–1149. https://doi.org/10.1016/S0002-9440(10)64982-6
Hofer S et al (2008) Injury of the blood brain barrier and up-regulation of icam-1 in polymicrobial sepsis. J Surg Res 146:276–281. https://doi.org/10.1016/j.jss.2007.07.021
Coisne C, Faveeuw C, Delplace Y, Dehouck L, Miller F, Cecchelli R, Dehouck B (2006) Differential expression of selectins by mouse brain capillary endothelial cells in vitro in response to distinct inflammatory stimuli. Neurosci Lett 392:216–220. https://doi.org/10.1016/j.neulet.2005.09.028
Takeshita Y, Ransohoff RM (2012) Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immunol Rev 248:228–239. https://doi.org/10.1111/j.1600-065X.2012.01127.x
Comim CM et al (2011) Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Intensive Care Med 37:711–718. https://doi.org/10.1007/s00134-011-2151-2
Klein RS, Hunter CA (2017) Protective and pathological immunity during central nervous system infections. Immunity 46:891–909. https://doi.org/10.1016/j.immuni.2017.06.012
Cekanaviciute E, Buckwalter MS (2016) Astrocytes: integrative regulators of neuroinflammation in stroke and other neurological diseases. Neurotherapeutics 13:685–701. https://doi.org/10.1007/s13311-016-0477-8
Sofroniew MV (2015) Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci 16:249–263. https://doi.org/10.1038/nrn3898
Wanner IB et al (2013) Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33:12870–12886. https://doi.org/10.1523/JNEUROSCI.2121-13.2013
Hsu JY et al (2008) Matrix metalloproteinase-9 facilitates glial scar formation in the injured spinal cord. J Neurosci 28:13467–13477. https://doi.org/10.1523/JNEUROSCI.2287-08.2008
Jones LL, Margolis RU, Tuszynski MH (2003) The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 182:399–411
McKeon RJ, Jurynec MJ, Buck CR (1999) The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci 19:10778–10788
McKillop WM, Dragan M, Schedl A, Brown A (2013) Conditional Sox9 ablation reduces chondroitin sulfate proteoglycan levels and improves motor function following spinal cord injury. Glia 61:164–177. https://doi.org/10.1002/glia.22424
Smith GM, Hale JH (1997) Macrophage/Microglia regulation of astrocytic tenascin: synergistic action of transforming growth factor-beta and basic fibroblast growth factor. J Neurosci 17:9624–9633
Wang Y, Moges H, Bharucha Y, Symes A (2007) Smad3 null mice display more rapid wound closure and reduced scar formation after a stab wound to the cerebral cortex. Exp Neurol 203:168–184. https://doi.org/10.1016/j.expneurol.2006.08.006
Haupt C, Witte OW, Frahm C (2007) Up-regulation of Connexin43 in the glial scar following photothrombotic ischemic injury. Mol Cell Neurosci 35:89–99. https://doi.org/10.1016/j.mcn.2007.02.005
Kielian T, Esen N (2004) Effects of neuroinflammation on glia-glia gap junctional intercellular communication: a perspective. Neurochem Int 45:429–436. https://doi.org/10.1016/j.neuint.2003.09.010
Sikasunge CS, Johansen MV, Phiri IK, Willingham AL 3rd, Leifsson PS (2009) The immune response in Taenia solium neurocysticercosis in pigs is associated with astrogliosis, axonal degeneration and altered blood-brain barrier permeability. Vet Parasitol 160:242–250. https://doi.org/10.1016/j.vetpar.2008.11.015
Schittone SA, Dionne KR, Tyler KL, Clarke P (2012) Activation of innate immune responses in the central nervous system during reovirus myelitis. J Virol 86:8107–8118. https://doi.org/10.1128/JVI.00171-12
Stenzel W, Soltek S, Schluter D, Deckert M (2004) The intermediate filament GFAP is important for the control of experimental murine Staphylococcus aureus-induced brain abscess and Toxoplasma encephalitis. J Neuropathol Exp Neurol 63:631–640
Drogemuller K et al (2008) Astrocyte gp130 expression is critical for the control of Toxoplasma encephalitis. J Immunol 181:2683–2693
Dvoriantchikova G et al (2009) Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. Eur J Neurosci 30:175–185. https://doi.org/10.1111/j.1460-9568.2009.06814.x
Brambilla R et al (2005) Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med 202:145–156. https://doi.org/10.1084/jem.20041918
Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci 29:11511–11522. https://doi.org/10.1523/JNEUROSCI.1514-09.2009
Bush TG et al (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145. https://doi.org/10.1016/j.it.2007.01.005
Gimenez MA, Sim JE, Russell JH (2004) TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol 151:116–125. https://doi.org/10.1016/j.jneuroim.2004.02.012
Lossinsky AS, Shivers RR (2004) Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Rev Histol Histopathol 19:535–564. https://doi.org/10.14670/HH-19.535
Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM (2006) Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med 203:1007–1019. https://doi.org/10.1084/jem.20051342
Geurts N, Opdenakker G, Van den Steen PE (2012) Matrix metalloproteinases as therapeutic targets in protozoan parasitic infections. Pharmacol Ther 133:257–279. https://doi.org/10.1016/j.pharmthera.2011.11.008
Dong Y, Benveniste EN (2001) Immune function of astrocytes. Glia 36:180–190
Bellaver B et al (2018) Systemic inflammation as a driver of brain injury: the astrocyte as an emerging player. Mol Neurobiol 55:2685–2695. https://doi.org/10.1007/s12035-017-0526-2
Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP (2016) Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 64:300–316. https://doi.org/10.1002/glia.22930
Chapouly C et al (2015) Astrocytic TYMP and VEGFA drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain 138:1548–1567. https://doi.org/10.1093/brain/awv077
Vincent VA, Tilders FJ, Van Dam AM (1997) Inhibition of endotoxin-induced nitric oxide synthase production in microglial cells by the presence of astroglial cells: a role for transforming growth factor beta. Glia 19:190–198
Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW (1993) Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J Immunol 150:2659–2667
Barnum SR, Jones JL, Muller-Ladner U, Samimi A, Campbell IL (1996) Chronic complement C3 gene expression in the CNS of transgenic mice with astrocyte-targeted interleukin-6 expression. Glia 18:107–117. https://doi.org/10.1002/(SICI)1098-1136(199610)18
Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, Mucke L (1993) Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci USA 90:10061–10065
Strack A, Asensio VC, Campbell IL, Schluter D, Deckert M (2002) Chemokines are differentially expressed by astrocytes, microglia and inflammatory leukocytes in Toxoplasma encephalitis and critically regulated by interferon-gamma. Acta Neuropathol 103:458–468. https://doi.org/10.1007/s00401-001-0491-7
Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH (2002) Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia 37:314–327
Sauder C et al (2000) Chemokine gene expression in astrocytes of Borna disease virus-infected rats and mice in the absence of inflammation. J Virol 74:9267–9280
Kim BO, Liu Y, Zhou BY, He JJ (2004) Induction of C chemokine XCL1 (lymphotactin/single C motif-1 alpha/activation-induced, T cell-derived and chemokine-related cytokine) expression by HIV-1 Tat protein. J Immunol 172:1888–1895
Rengarajan J, Szabo SJ, Glimcher LH (2000) Transcriptional regulation of Th1/Th2 polarization. Immunol Today 21:479–483
Besong G et al (2002) Activation of group III metabotropic glutamate receptors inhibits the production of RANTES in glial cell cultures. J Neurosci 22:5403–5411 doi:20026585
Prow NA, Irani DN (2008) The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J Neurochem 105:1276–1286. https://doi.org/10.1111/j.1471-4159.2008.05230.x
Bezzi P et al (2001) CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci 4:702–710. https://doi.org/10.1038/89490
El-Hage N, Podhaizer EM, Sturgill J, Hauser KF (2011) Toll-like receptor expression and activation in astroglia: differential regulation by HIV-1 Tat, gp120, and morphine. Immunol Investig 40:498–522. https://doi.org/10.3109/08820139.2011.561904
Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW (2014) Pattern recognition receptors and central nervous system repair. Exp Neurol 258:5–16. https://doi.org/10.1016/j.expneurol.2014.01.001
Minkiewicz J, de Rivero Vaccari JP, Keane RW (2013) Human astrocytes express a novel NLRP2 inflammasome. Glia 61:1113–1121. https://doi.org/10.1002/glia.22499
Chauhan VS, Sterka DG Jr, Furr SR, Young AB, Marriott I (2009) NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia 57:414–423. https://doi.org/10.1002/glia.20770
Wong GH, Bartlett PF, Clark-Lewis I, Battye F, Schrader JW (1984) Inducible expression of H-2 and Ia antigens on brain cells. Nature 310:688–691
Vardjan N et al (2012) IFN-γ-induced increase in the mobility of MHC class II compartments in astrocytes depends on intermediate filaments. J Neuroinflamm 9:144. https://doi.org/10.1186/1742-2094-9-144
Vass K, Lassmann H (1990) Intrathecal application of interferon gamma. Progressive appearance of MHC antigens within the rat nervous system. Am J Pathol 137:789–800
Horwitz MS, Evans CF, Klier FG, Oldstone MB (1999) Detailed in vivo analysis of interferon-γ induced major histocompatibility complex expression in the the central nervous system: astrocytes fail to express major histocompatibility complex class I and II molecules. Lab Investig 79:235–242
Bo L, Mork S, Kong PA, Nyland H, Pardo CA, Trapp BD (1994) Detection of MHC class II-antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis lesions. J Neuroimmunol 51:135–146
Barcia C et al (2008) T cells’ immunological synapses induce polarization of brain astrocytes in vivo and in vitro: a novel astrocyte response mechanism to cellular injury. PLoS ONE 3:e2977. https://doi.org/10.1371/journal.pone.0002977
Combes V, Guillemin GJ, Chan-Ling T, Hunt NH, Grau GE (2012) The crossroads of neuroinflammation in infectious diseases: endothelial cells and astrocytes. Trends Parasitol 28:311–319. https://doi.org/10.1016/j.pt.2012.05.008
Wilson EH, Hunter CA (2004) The role of astrocytes in the immunopathogenesis of toxoplasmic encephalitis. Int J Parasitol 34:543–548. https://doi.org/10.1016/j.ijpara.2003.12.010
Eugenin EA, Berman JW (2007) Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J Neurosci 27:12844–12850. https://doi.org/10.1523/JNEUROSCI.4154-07.2007
Zorec R, Zupanc TA, Verkhratsky A (2018) Astrogliopathology in the infectious insults of the brain. Neurosci Lett. https://doi.org/10.1016/j.neulet.2018.08.003
Suzuki Y et al (1997) Impaired resistance to the development of toxoplasmic encephalitis in interleukin-6-deficient mice. Infect Immun 65:2339–2345
Suzuki Y, Sa Q, Gehman M, Ochiai E (2011) Interferon-γ and perforin-mediated immune responses for resistance against Toxoplasma gondii in the brain. Expert Rev Mol Med 13:e31. https://doi.org/10.1017/S1462399411002018
Hidano S et al (2016) STAT1 signaling in astrocytes is essential for control of infection in the central nervous system. MBio. https://doi.org/10.1128/mBio.01881-16
Mesquita LP, Hora AS, de Siqueira A, Salvagni FA, Brandao PE, Maiorka PC (2016) Glial response in the central nervous system of cats with feline infectious peritonitis. J Feline Med Surg 18:1023–1030. https://doi.org/10.1177/1098612X15615906
Liao CK, Jeng CJ, Wang HS, Wang SH, Wu JC (2013) Lipopolysaccharide induces degradation of connexin43 in rat astrocytes via the ubiquitin-proteasome proteolytic pathway. PLoS ONE 8:e79350. https://doi.org/10.1371/journal.pone.0079350
Liao CK, Wang SM, Chen YL, Wang HS, Wu JC (2010) Lipopolysaccharide-induced inhibition of connexin43 gap junction communication in astrocytes is mediated by downregulation of caveolin-3. Int J Biochem Cell Biol 42:762–770. https://doi.org/10.1016/j.biocel.2010.01.016
Meme W, Calvo CF, Froger N, Ezan P, Amigou E, Koulakoff A, Giaume C (2006) Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: potentiation by β-amyloid. FASEB J 20:494–496. https://doi.org/10.1096/fj.05-4297fje
Zhang FF, Morioka N, Kitamura T, Hisaoka-Nakashima K, Nakata Y (2015) Proinflammatory cytokines downregulate connexin 43-gap junctions via the ubiquitin-proteasome system in rat spinal astrocytes. Biochem Biophys Res Commun 464:1202–1208. https://doi.org/10.1016/j.bbrc.2015.07.105
Esen N, Shuffield D, Syed MM, Kielian T (2007) Modulation of connexin expression and gap junction communication in astrocytes by the gram-positive bacterium S. aureus. Glia 55:104–117. https://doi.org/10.1002/glia.20438
Kielian T (2008) Glial connexins and gap junctions in CNS inflammation and disease. J Neurochem 106:1000–1016. https://doi.org/10.1111/j.1471-4159.2008.05405.x
Karpuk N, Burkovetskaya M, Fritz T, Angle A, Kielian T (2011) Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J Neurosci 31:414–425. https://doi.org/10.1523/JNEUROSCI.5247-10.2011
Meme W, Calvo CF, Froger N, Ezan P, Amigou E, Koulakoff A, Giaume C (2006) Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: potentiation by beta-amyloid. FASEB J 20:494–496. https://doi.org/10.1096/fj.05-4297fje
Zhou JJ, Cheng C, Qiu Z, Zhou WH, Cheng GQ (2015) Decreased connexin 43 in astrocytes inhibits the neuroinflammatory reaction in an acute mouse model of neonatal sepsis. Neurosci Bull 31:763–768. https://doi.org/10.1007/s12264-015-1561-5
Carpentier PA, Duncan DS, Miller SD (2008) Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain Behav Immun 22:140–147. https://doi.org/10.1016/j.bbi.2007.08.011
Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H (2008) Role of Toll-like receptors in the development of sepsis. Shock 29:315–321. https://doi.org/10.1097/SHK.0b013e318157ee55
Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM (2011) Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59:242–255. https://doi.org/10.1002/glia.21094
Brown GC, Neher JJ (2010) Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol 41:242–247. https://doi.org/10.1007/s12035-010-8105-9
Alexander JJ, Jacob A, Cunningham P, Hensley L, Quigg RJ (2008) TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int 52:447–456. https://doi.org/10.1016/j.neuint.2007.08.006
Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT (2005) Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat 30:144–157. https://doi.org/10.1016/j.jchemneu.2005.07.003
Varatharaj A, Galea I (2017) The blood-brain barrier in systemic inflammation. Brain Behav Immun 60:1–12. https://doi.org/10.1016/j.bbi.2016.03.010
Candelario-Jalil E et al (2007) Cyclooxygenase inhibition limits blood-brain barrier disruption following intracerebral injection of tumor necrosis factor-alpha in the rat. J Pharmacol Exp Ther 323:488–498. https://doi.org/10.1124/jpet.107.127035
Minami T, Okazaki J, Kawabata A, Kawaki H, Okazaki Y, Tohno Y (1998) Roles of nitric oxide and prostaglandins in the increased permeability of the blood-brain barrier caused by lipopolysaccharide. Environ Toxicol Pharmacol 5:35–41
Biesmans S et al (2013) Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediat Inflamm 2013:271359. https://doi.org/10.1155/2013/271359
Cardoso FL et al (2015) Systemic inflammation in early neonatal mice induces transient and lasting neurodegenerative effects. J Neuroinflamm 12:82. https://doi.org/10.1186/s12974-015-0299-3
Correa DG, Cruz Junior LC, Bahia PR, Gasparetto EL (2012) Intracerebral microbleeds in sepsis: susceptibility-weighted MR imaging findings. Arq Neuropsiquiatr 70:903–904
Sumbria RK et al (2016) A murine model of inflammation-induced cerebral microbleeds. J Neuroinflamm 13:218. https://doi.org/10.1186/s12974-016-0693-5
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Special Issue: In Honor of Prof Prof. Eva Sykova.
Rights and permissions
About this article

Cite this article
Shulyatnikova, T., Verkhratsky, A. Astroglia in Sepsis Associated Encephalopathy. Neurochem Res 45, 83–99 (2020). https://doi.org/10.1007/s11064-019-02743-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02743-2