
Abstract
This article is one of ten reviews selected from the Annual Update in Intensive Care and Emergency Medicine 2017. Other selected articles can be found online at http://ccforum.com/series/annualupdate2017. Further information about the Annual Update in Intensive Care and Emergency Medicine is available from http://www.springer.com/series/8901.
Originally published in the Annual Update in Intensive Care and Emergency Medicine 2017. The number of authors differs in the two versions due to constraints regarding the number of authors in the Annual Update in Intensive Care and Emergency Medicine. In the Annual Update version of the review, the three senior authors appear in the acknowledgement section. In the Critical Care version, these three senior authors appear as full authors of the manuscript. All authors helped draft and revise the manuscript for critical intellectual content.
Similar content being viewed by others

Background
Sepsis is defined as a life‐threatening organ dysfunction caused by a non‐homeostatic response of the host to an infection [1]. Sepsis associated encephalopathy (SAE) is a transient and reversible brain dysfunction, occurring when the source of sepsis is located outside of the central nervous system. SAE affects approximately a third of septic patients [2] and is a risk factor for long term disability and mortality [2, 3]. SAE is characterized by an altered mental status, which may range from delirium to coma. Electrophysiological and brain imaging anomalies may be present [4, 5]. The electroencephalogram (EEG) anomalies of SAE were described and classified by Young et al., including, from the most benign to the most severe, excessive theta rhythms, predominant delta rhythms, triphasic waves and burst suppression [6]. A recent study reported that seizures were recorded in up to 15% of patients [4]. However, the exact place of EEG monitoring in sepsis remains to be defined. Evoked potentials, when investigated in sepsis, show evidence of increased latencies and decreased amplitudes. Cerebral imaging shows various patterns of brain injury during SAE. Brain magnetic resonance imaging (MRI) can be normal or show focal injuries, such as white matter hyperintensities or ischemic stroke, in the presence of disseminated intravascular coagulation (DIC) or of cardiac arrhythmia [5]. Although there is no hallmark biomarker for SAE, elevated plasma levels of S100β or neuron‐specific enolase (NSE) have been reported. Their exact significance, however remains unclear [7].
Syndromes
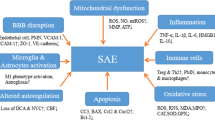
The neurological anomalies associated with sepsis include, from the least to the most severe, sickness behavior, delirium and coma (Fig. 1). The most common physiological and behavioral modifications associated with any viral or bacterial infection are called “sickness behavior”. This condition is characterized clinically by reduced attention and alertness, as well as by eating and drinking disorders, anxiety and social withdrawal [8]. Biological characteristics include activation of the adrenal axis and the adrenergic system. Sickness behavior relates to the general feeling of being unwell. These modifications result from the action of proinflammatory cytokines (interleukin [IL]‐1α and IL‐1β, tumor necrosis factor (TNF)‐α and IL-6) on centers controlling the behavioral, neuroendocrine and autonomic responses. Sickness behavior is considered to be a physiological reaction to an infectious insult, enabling subjects to better cope with the disease and to protect them from a dangerous environment. Sickness behavior can be maladaptive when its intensity or duration does not correlate with the intensity of the insult [8].

Schematic representation of the interaction between sepsis‐associated neurological syndromes, the affected structures and clinical outcomes. Modified from [15] with permission
A more severe form of neurological impairment occurring during sepsis is delirium [9]. Delirium is defined by the DSM‐5 as an acute and fluctuating disturbance in attention and awareness that is not explained by a preexisting neurological condition and does not occur in the context of a severely reduced level of arousal such as coma [10]. Standardized tools for assessing delirium in the ICU have been developed, such as the Confusion Assessment Method for the ICU (CAM‐ICU). Using such a score, delirium occurs in up to 80% of critically ill patients [9]. Two major forms of delirium are described – hypoactive and hyperactive – with a range of possible forms in between. Delirium is associated with prolonged mechanical ventilation, increased ICU length of stay and death [11]. An imbalance between neurotransmitters is implicated in the pathophysiology of delirium in the critically ill [12]. However, randomized trials assessing the neuromodulatory effects of psychotropes in the critically ill failed to show benefit and even suggested worsened outcomes [3, 13, 14]. Finally, substance withdrawal, kidney or liver failure, electrolyte anomalies as well as the toxic effect of drugs may all be implicated in the genesis of delirium [15].
The most severe form of neurological impairment during the course of sepsis is the occurrence of coma. Coma may in part be linked to the administration of sedative drugs in a critically ill patient. The depth of coma may easily be assessed using the Glasgow Coma Scale score or the FOUR score [16] and is associated with mortality [2]. The FOUR score adds an additional layer of sophistication to the assessment of coma by scoring brainstem reflexes in addition to the classical eye, motor and verbal responses elicited spontaneously or in response to a nociceptive stimuli. For example, an absent cough reflex is associated with 28‐day mortality in sedated critically ill patients [17].
Long‐term neuropsychological sequelae, including memory, attention, verbal fluency and executive function impairment, are frequent in the critically ill [18]. Sepsis is a major risk factor for long‐term cognitive impairment [3]. Hospitalization for severe sepsis increases the prevalence of moderate to severe cognitive decline by more than 10% [19]. An episode of acute brain dysfunction may lead to anxiety, depression, post‐traumatic stress disorders [20] and self‐harm [21]. Two out of five patients who have had sepsis are subsequently unable to return to full‐time employment [22].
Structures
Two distinct pathways transmit inflammatory signals to the brain in the absence of blood brain barrier disruption: (1) the vagal nerve senses local inflammation and transfers the information to the medullary autonomic nuclei [23, 24]; and (2) circumventricular organs allow the trafficking of inflammatory mediators into the brain [25]. The vagal nerve is stimulated shortly after a lipopolysaccharide (LPS) challenge [24]. The vagal nerve reaches the autonomic and neuroendocrine systems [23, 24]. Deprived of a blood brain barrier, the circumventricular organs are located in the vicinity of the third ventricle and include the area postrema, which controls nausea and transmits peripheral vagal nerve information to the rest of the central nervous system (CNS). Sickness behavior is induced by the action of pro‐inflammatory cytokines on the brain. In rodents, the systemic or central administration of IL‐1β induces all the behavioral, endocrine and autonomic features of sickness behavior [26]. The other proinflammatory cytokines that affect the brain are IL‐1α, TNF‐α and IL‐6.
Brainstem
A host of arguments point to the existence of brainstem dysfunction during sepsis, which may be the consequence of neuroinflammation. The area postrema allows the passage of circulating inflammatory mediators into the brainstem. Its surgical ablation in rats is equivalent to a central vagotomy [27]. Therefore, it is conceivable that excessive inflammatory signaling can be deleterious. Experimentally, injecting endotoxin in rodents models the apoptosis of autonomic centers that is induced by oxidative stress. We have reported multifocal necrotizing leukoencephalopathy in a septic shock patient, characterized by an intense expression of inflammatory cytokines and neuronal apoptosis within the pons [28]. Neuronal apoptosis within autonomic centers has also been described [29]. The brainstem is involved in three general functions: arousal, brainstem reflexes, and control of vital functions and modulation of the immune response [23]. Brainstem dysfunction might result in impaired alertness and impaired cardiovascular and immune controls, thereby leading to increased mortality. We found that the early abolition of the cough reflex and oculocephalogyric response was independently associated with increased ICU mortality in sedated critically ill patients [17]. Decreased heart rate variability, reflecting an impaired sympathetic activity originating from the brainstem autonomic centers, is also associated with a worse outcome in sepsis [30]. Absent EEG reactivity, indicating a dysfunction of the reticular activating ascending substance, is a predictor of death in septic patients [4].
Hypothalamus and pituitary axis
Sepsis is associated with multiple neuroendocrine disorders [23,32,33,34,, 25, 31–35]. While the initial stage of sepsis is marked by increased secretion of pituitary hormones notably in response to a decreased sensitivity in peripheral hormonal receptors, prolonged sepsis is characterized by a decrease in pituitary hormone release [36]. For example, septic shock is associated with relative adrenal insufficiency and increased mortality, responding favorably to substitutive opotherapy [33, 37]. Septic shock may also be associated with impaired production of vasopressin [31, 34, 38] due to hyperpolarization of neurons of the organum vasculosum lamina terminalis (OVLT) region [38].
Amygdala
Psychological consequences of sepsis include anxiety, depression and post‐traumatic stress disorder (PTSD) [39] all of which involve the amygdala and its network with the prefrontal cortex, hypothalamus or brainstem. The amygdala is particularly affected by microglial activation during sepsis [29, 39]. Studies in military personnel presenting with PTSD showed that the amygdala was desensitized in response to stressful stimuli. The incriminated mechanism is a dampened response to corticosteroids which could be overcome by direct corticosterone administration [40]. A recent study translated these findings in order to prevent PTSD in sepsis patients [41]. Such a beneficial effect of corticosteroids may involve non‐genetic or epigenetic effects of these drugs [42], inducing a major modulation of aversive memory formation [43].
Hippocampus
Memory impairment can be induced by insults to the hippocampus or the frontal cortex. Hippocampal atrophy is associated with loss of memory, such as in Alzheimer’s disease. In rodents, sepsis is also associated with hippocampal dysfunction and subsequent spatial memory alterations evidenced by the Morris Water Maze test [44], which are correlated with long term potentiation, late neuronal death, gliosis and increased production of reactive oxygen species (ROS). TNF‐α, caspase 1 and hypoxia seem to be implicated in the pathogenesis of this hippocampal dysfunction [45].
Frontal cortex
The prefrontal cortex and other sensorimotor areas constitute the frontal cortex. Cognitive defects occurring after frontal cortex injuries include mainly impaired memorization, attention and executive functions. Ultrastructural neuronal anomalies were shown within the sensorimotor cortex in a dog model of endotoxic shock. Pre‐frontal cortex‐related cognitive functions including memorization and verbal fluency are frequently affected after sepsis [19]. Neuroinflammation within the frontal cortex results in cognitive impairment [46].
White matter
As shown by MRI studies, sepsis can induce axonal damage; which may contribute to the cognitive decline [46].
Processes
Three major processes are involved in the pathophysiology of SAE including diffuse neuroinflammation, ischemic processes and excitotoxicity.
Neuroinflammation
Microglial cells are the mononuclear phagocytes of the brain, implicated in immune defense and synaptic plasticity. Microglia express membrane‐bound receptors that are able to detect endogenous or exogenous danger signals. Microglial activation implies a level of blood brain barrier disruption or the local release of proinflammatory mediators. Microglial activity ranges from proinflammatory to anti‐inflammatory. Microglial activation is a frequent neuropathological feature of SAE [29, 47]. The modulation of microglial activity, although still in the pre‐clinical phase, may be an interesting option for treating SAE [48].
Astrocytes are supportive glial cell components. Astrocytes are involved in neuroinflammation as they are reactive and able to secrete various mediators [49]. Astrocytes are thus involved in cerebral blood flow control through the release of mediators, such as nitric oxide (NO), prostaglandins and arachidonic acid [50]. Astrocytes regulate the concentration of neurotransmitters, such as glutamate, GABA, and glycine, in the synaptic space by taking up any excess neurotransmitter, preventing their potential for accumulation [51]. Animal studies consistently report the presence of astrogliosis during SAE. By contrast, no such finding has been reported in man, possibly due to technical issues.
Neuroinflammation affects cellular metabolism, potentially leading to oxidative stress and mitochondrial dysfunction [52]. Mitochondrial dysfunction has been found in the brain of septic animals, associated with the production of oxygen/nitrogen reactive species [53].
Ischemic processes
The gliovascular unit associates endothelial cells, astrocytes and pericytes [54]. Exchanges between the brain and components of the blood occur through endothelial cells. Astrocytes regulate water and ionic homeostasis through the transmembrane channel aquaporin 4 (AQP4). Gliovascular units are implicated in adjusting blood flow at the arteriolar and capillary levels in order to provide the energetic requirement of a given cerebral area [54].
Ischemia can result from macrocirculatory dysfunction, including hypotension, decreased cerebral flow and impaired autoregulation. Sepsis induces activation of cerebral endothelial cells which, by impairing microvascular tone and activating coagulation, promotes ischemic and/or hemorrhagic lesions of the brain [55]. Some experimental data indicate that during the early stages of sepsis, there is an increased expression of endothelial NO synthase (eNOS), which causes additional NO release and enhances the activation of cerebral endothelial cells [56]. This endothelial activation, coupled with the neuroinflammation process, can result in impaired blood brain barrier permeability, upregulated AQP4, parenchymal infiltration by neutrophils, and the activation of astrocytes [57]. MRI studies have shown blood brain barrier breakdown in patients with septic shock [58], possibly originating from the perivascular spaces of Wirchow‐Robin, which may go on to involve the whole white matter. It has to be noted that posterior reversible encephalopathy syndromes have been reported in septic patients [59]. Experimental studies confirm that blood brain barrier impairment relates to a dysfunction of tight junction proteins, such as occludin, ZO‐1, ZO‐2, claudin‐3 and claudin‐5 [60].
Neurovascular coupling is a specific brain mechanism of vasoregulation that adapts local cerebral blood flow to the neural metabolic needs. SAE is associated with neurovascular uncoupling due to microcirculatory dysfunction and low blood flow. In septic patients, a discrepancy between neuronal activation, increased metabolic needs and vascular reactivity is observed [61].
The combination of macro‐ and micro‐circulatory defects, endothelial dysfunction and neurovascular uncoupling contribute to ischemic lesions, which were constantly observed in a post‐mortem study of patients who died of septic shock. These ischemic lesions were found in brain areas sensitive to ischemia (i. e., the hippocampus, the basal ganglia, frontal cortex, dentate gyrus and the bulbar olive) but also, and specifically to sepsis, in the nuclei of the autonomic nervous system (i. e., amygdala, anterior and posterior hypothalamus, locus coeruleus). Similarly, pronounced neuronal apoptosis was observed in these areas. Furthermore, brain hemorrhages were found in 17–26% of these neuropathological cases [47].
Neurotransmitter dysfunction
A small number of brain structures, namely the amygdala, nucleus tractus solitarii and locus coeruleus, are particularly vulnerable to sepsis and liable to neuronal apoptosis [29]. Glutamate plays a role in neuronal apoptosis during sepsis, through excitoxicity [62]. For example, the recycling and glutamate‐stimulated export of ascorbate by astrocytes are inhibited during sepsis [63]. Interestingly, analysis of the cerebrospinal fluid of patients with SAE showed a decrease in the levels of ascorbate, an anti‐oxidant produced by astrocytes that plays a role in neuroprotection [64]. When activated, microglia cells also release large amounts of glutamate [65].
The dopaminergic, β‐adrenergic [66], the GABA receptor [67], and the cholinergic [52] systems are all impaired during sepsis. An imbalance between dopaminergic and cholinergic neurotransmission might be particularly involved in delirium [12]. However, cholinergic drugs, such as rivastigmine [13] as well as anti‐dopaminergic drugs, such as haloperidol [14], do not reduce the incidence or duration of delirium. The use of GABA‐agonists, such as benzodiazepines, actually increases the risk of delirium [3], which is reduced by noradrenergic drugs, such as dexmedetomidine [68]. Impaired neurotransmission might result from the production of NO [69], but also from circulating neurotoxic amino‐acids (ammonium, tyrosine, tryptophan and phenylalanine) that are released in excess by the liver and muscles during sepsis [70]. Metabolic disorders and drug toxicity (e. g., sedatives, analgesics, antibiotics …) contribute to neurotransmitter dysfunction.
Conclusion
The clinical spectrum of SAE includes sickness behavior, delirium, focal deficits and coma associated or not with brainstem reflex loss. It is associated with increased mortality and long‐term psychocognitive impairment. SAE results from inflammatory, ischemic and neurotoxic processes that affect, in particular, the frontal cortex, the hippocampus, the amygdala and the brainstem. Its diagnosis essentially relies on neurological examination and EEG findings, which may indicate the need for brain imaging. In daily clinical practice, brain infection has to be ruled out whenever suspected, as well as drug toxicity or metabolic disturbances. There is no specific treatment for SAE, apart from resolution of the underlying sepsis.
References
Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315:801–10.
Eidelman LA, Putterman D, Putterman C, Sprung CL. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA. 1996;275:470–3.
Pandharipande PP, Girard TD, Jackson JC, et al. Long-term cognitive impairment after critical illness. N Engl J Med. 2013;369:1306–16.
Azabou E, Magalhaes E, Braconnier A, et al. Early standard electroencephalogram abnormalities predict mortality in septic intensive care unit patients. PLoS One. 2015;10:e0139969.
Polito A, Eischwald F, Maho AL, et al. Pattern of brain injury in the acute setting of human septic shock. Crit Care. 2013;17:R204.
Young GB, Bolton CF, Archibald YM, Austin TW, Wells GA. The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol. 1992;9:145–52.
Grandi C, Tomasi CD, Fernandes K, et al. Brain-derived neurotrophic factor and neuron-specific enolase, but not S100β, levels are associated to the occurrence of delirium in intensive care unit patients. J Crit Care. 2011;26:133–7.
Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur J Pharmacol. 2004;500:399–411.
Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA. 2001;286:2703–10.
American Pychatric Association. Diagnostic and statistical manual of mental disorders: DSM-5. Arlington: American Psychiatric Association Publishing; 2013.
Ely EW, Shintani A, Truman B, et al. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA. 2004;291:1753–62.
van Gool WA, van de Beek D, Eikelenboom P. Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet. 2010;375:773–5.
van Eijk MM, Roes KC, Honing ML, et al. Effect of rivastigmine as an adjunct to usual care with haloperidol on duration of delirium and mortality in critically ill patients: a multicentre, double-blind, placebo-controlled randomised trial. Lancet. 2010;376:1829–37.
Page VJ, Ely EW, Gates S, et al. Effect of intravenous haloperidol on the duration of delirium and coma in critically ill patients (Hope-ICU): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2013;1:515–23.
Annane D, Sharshar T. Cognitive decline after sepsis. Lancet Respir Med. 2015;3:61–9.
Wijdicks EF, Bamlet WR, Maramattom BV, Manno EM, McClelland RL. Validation of a new coma scale: the FOUR score. Ann Neurol. 2005;58:585–93.
Sharshar T, Porcher R, Siami S, et al. Brainstem responses can predict death and delirium in sedated patients in intensive care unit. Crit Care Med. 2011;39:1960–7.
Jackson JC, Hart RP, Gordon SM, et al. Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med. 2003;31:1226–34.
Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304:1787–94.
Wintermann GB, Brunkhorst FM, Petrowski K, et al. Stress disorders following prolonged critical illness in survivors of severe sepsis. Crit Care Med. 2015;43:1213–22.
Lund-Sørensen H, Benros ME, Madsen T, et al. A nationwide cohort study of the association between hospitalization with infection and risk of death by suicide. JAMA Psychiatry. 2016;73:912–9.
Rothenhäusler HB, Ehrentraut S, Stoll C, Schelling G, Kapfhammer HP. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: results of an exploratory study. Gen Hosp Psychiatry. 2001;23:90–6.
Carlson DE, Chiu WC, Fiedler SM, Hoffman GE. Central neural distribution of immunoreactive Fos and CRH in relation to plasma ACTH and corticosterone during sepsis in the rat. Exp Neurol. 2007;205:485–500.
Reyes EP, Abarzúa S, Martin A, Rodríguez J, Cortés PP, Fernández R. LPS-induced c-Fos activation in NTS neurons and plasmatic cortisol increases in septic rats are suppressed by bilateral carotid chemodenervation. Adv Exp Med Biol. 2012;758:185–90.
Sharshar T, Hopkinson NS, Orlikowski D, Annane D. Science review: The brain in sepsis – culprit and victim. Crit Care. 2005;9:37–44.
Anforth HR, Bluthe RM, Bristow A, et al. Biological activity and brain actions of recombinant rat interleukin-1alpha and interleukin-1beta. Eur Cytokine Netw. 1998;9:279–88.
Skoog KM, Blair ML, Sladek CD, Williams WM, Mangiapane ML. Area postrema: essential for support of arterial pressure after hemorrhage in rats. Am J Physiol. 1990;258:R1472–8.
Sharshar T, Gray F, Poron F, Raphael JC, Gajdos P, Annane D. Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med. 2002;30:2371–5.
Sharshar T, Gray F, Lorin de la Grandmaison G, et al. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet. 2003;362:1799–805.
Annane D, Trabold F, Sharshar T, et al. Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am J Respir Crit Care Med. 1999;160:458–65.
Siami S, Bailly-Salin J, Polito A, et al. Osmoregulation of vasopressin secretion is altered in the postacute phase of septic shock. Crit Care Med. 2010;38:1962–9.
Sonneville R, Guidoux C, Barrett L, et al. Vasopressin synthesis by the magnocellular neurons is different in the supraoptic nucleus and in the paraventricular nucleus in human and experimental septic shock. Brain Pathol. 2010;20:613–22.
Aboab J, Polito A, Orlikowski D, Sharshar T, Castel M, Annane D. Hydrocortisone effects on cardiovascular variability in septic shock: a spectral analysis approach. Crit Care Med. 2008;36:1481–6.
Siami S, Polito A, Porcher R, et al. Thirst perception and osmoregulation of vasopressin secretion are altered during recovery from septic shock. PLoS One. 2013;8:e80190.
Escartin C, Rouach N. Astroglial networking contributes to neurometabolic coupling. Front Neuroenergetics. 2013;5:1.
Schroeder S, Wichers M, Klingmüller D, et al. The hypothalamic-pituitary-adrenal axis of patients with severe sepsis: altered response to corticotropin-releasing hormone. Crit Care Med. 2001;29:310–6.
Annane D, Sébille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–71.
Stare J, Siami S, Trudel E, Prager-Khoutorsky M, Sharshar T, Bourque CW. Effects of peritoneal sepsis on rat central osmoregulatory neurons mediating thirst and vasopressin release. J Neurosci. 2015;35:12188–97.
Muscatell KA, Dedovic K, Slavich GM, et al. Greater amygdala activity and dorsomedial prefrontal-amygdala coupling are associated with enhanced inflammatory responses to stress. Brain Behav Immun. 2015;43:46–53.
Henckens MJ, van Wingen GA, Joels M, Fernandez G. Time-dependent effects of corticosteroids on human amygdala processing. J Neurosci. 2010;30:12725–32.
Schelling G, Stoll C, Kapfhammer HP, et al. The effect of stress doses of hydrocortisone during septic shock on posttraumatic stress disorder and health-related quality of life in survivors. Crit Care Med. 1999;27:2678–83.
Hunter RG. Epigenetic effects of stress and corticosteroids in the brain. Front Cell Neurosci. 2012;6:18.
Steckert AV, Comim CM, Igna DM, et al. Effects of sodium butyrate on aversive memory in rats submitted to sepsis. Neurosci Lett. 2015;595:134–8.
Liu L, Xie K, Chen H, et al. Inhalation of hydrogen gas attenuates brain injury in mice with cecal ligation and puncture via inhibiting neuroinflammation, oxidative stress and neuronal apoptosis. Brain Res. 2014;1589:78–92.
Di Paola M, Caltagirone C, Fadda L, Sabatini U, Serra L, Carlesimo GA. Hippocampal atrophy is the critical brain change in patients with hypoxic amnesia. Hippocampus. 2008;18:719–28.
Sonneville R, Derese I, Marques MB, et al. Neuropathological correlates of hyperglycemia during prolonged polymicrobial sepsis in mice. Shock. 2015;44:245–51.
Sharshar T, Annane D, de la Grandmaison GL, Brouland JP, Hopkinson NS, Françoise G. The neuropathology of septic shock. Brain Pathol. 2004;14:21–33.
Adembri C, Selmi V, Vitali L, et al. Minocycline but not tigecycline is neuroprotective and reduces the neuroinflammatory response induced by the superimposition of sepsis upon traumatic brain injury. Crit Care Med. 2014;42:e570–82.
Retamal MA, Froger N, Palacios-Prado N, et al. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. 2007;27:13781–92.
Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–76.
Seifert G, Schilling K, Steinhäuser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206.
Semmler A, Frisch C, Debeir T, et al. Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp Neurol. 2007;204:733–40.
Comim CM, Rezin GT, Scaini G, et al. Mitochondrial respiratory chain and creatine kinase activities in rat brain after sepsis induced by cecal ligation and perforation. Mitochondrion. 2008;8:313–8.
Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53.
Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun. 2015;21:827–46.
Handa O, Stephen J, Cepinskas G. Role of endothelial nitric oxide synthase-derived nitric oxide in activation and dysfunction of cerebrovascular endothelial cells during early onsets of sepsis. Am J Physiol Hear Circ Physiol. 2008;295:H1712–1719.
Alexander JJ, Jacob A, Cunningham P, Hensley L, Quigg RJ. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int. 2008;52:447–56.
Sharshar T, Carlier R, Bernard F, et al. Brain lesions in septic shock: a magnetic resonance imaging study. Intensive Care Med. 2007;33:798–806.
Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. Am J Neuroradiol. 2008;29:1036–42.
Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9:23.
Rosengarten B, Krekel D, Kuhnert S, Schulz R. Early neurovascular uncoupling in the brain during community acquired pneumonia. Crit Care. 2012;16:R64.
Viviani B, Boraso M, Marchetti N, Marinovich M. Perspectives on neuroinflammation and excitotoxicity: A neurotoxic conspiracy? Neurotoxicology. 2014;43:10–20.
Korcok J, Wu F, Tyml K, Hammond RR, Wilson JX. Sepsis inhibits reduction of dehydroascorbic acid and accumulation of ascorbate in astroglial cultures: intracellular ascorbate depletion increases nitric oxide synthase induction and glutamate uptake inhibition. J Neurochem. 2002;81:185–93.
Voigt K, Kontush A, Stuerenburg HJ, Muench-Harrach D, Hansen HC, Kunze K. Decreased plasma and cerebrospinal fluid ascorbate levels in patients with septic encephalopathy. Free Radic Res. 2002;36:735–9.
Takeuchi H, Jin S, Wang J, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–8.
Kadoi Y, Saito S, Kunimoto F, Imai T, Fujita T. Impairment of the brain beta-adrenergic system during experimental endotoxemia. J Surg Res. 1996;61:496–502.
Kadoi Y, Saito S. An alteration in the gamma-aminobutyric acid receptor system in experimentally induced septic shock in rats. Crit Care Med. 1996;24:298–305.
Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA. 2007;298:2644–53.
Jacob A, Brorson JR, Alexander JJ. Septic encephalopathy: inflammation in man and mouse. Neurochem Int. 2011;58:472–6.
Basler T, Meier-Hellmann A, Bredle D, Reinhart K. Amino acid imbalance early in septic encephalopathy. Intensive Care Med. 2002;28:293–8.
Acknowledgments
Not applicable.
Funding
Publication of this article was funded by the Groupe d’Investigation Clinique en Réanimation (GICR).
Availability of data and materials
Not applicable.
Authors’ contributions
NH and TS were responsible for conception and design of the review. NH, AM, FV FAB, FC and TS were responsible for writing the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Heming, N., Mazeraud, A., Verdonk, F. et al. Neuroanatomy of sepsis-associated encephalopathy. Crit Care 21, 65 (2017). https://doi.org/10.1186/s13054-017-1643-z
Published:
DOI: https://doi.org/10.1186/s13054-017-1643-z