
Abstract
The functionalization of silica-coated Fe3O4 magnetic nanoparticles (Fe3O4@SiO2) using chlorosulfonic acid were afforded sulfonic acid-functionalized magnetic Fe3O4 nanoparticles (Fe3O4@SiO2–SO3H) that can be applied as an organic–inorganic hybrid heterogeneous catalyst. The used Fe3O4 magnetic nanoparticles are 18–30 nm sized that was rapidly functionalized and can be used as catalyst in organic synthesis. The prepared nanoparticles were characterized by X-ray diffraction analysis, magnetization curve, scanning electron microscope, dynamic laser scattering, and FT-IR measurements. The resulting immobilized catalysts have been successfully used in the synthesis of 1,8‐dioxo-octahydroxanthene derivatives under solvent free condition. This procedure has many advantages such as; a much milder method, a shorter reaction time, a wide range of functional group tolerance, and absence of any tedious workup or purification. Other remarkable features include the catalyst can be reused at least five times without any obvious change in its catalytic activity. This procedure also avoids hazardous reagents/solvents, and thus can be an eco-friendly alternative to the existing methods.
Graphical Abstract
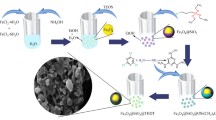
A highly efficient nano-Fe3O4 encapsulated-silica particles bearing sulfonic acid groups as a solid acid catalyst for synthesis of 1,8-dioxo-octahydroxanthene derivatives

Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Recently, Fe3O4 magnetic nanoparticles (MNPs) have been intensively investigated because of their superparamagnety, high coercively, and low Curie temperature (Kim and Kim 2003; Raj and Moskowitz 2002; Beydoun et al. 2000; McMichael et al. 1992). In addition to these characters, MNPs are also non-toxic and biocompatible.
MNPs have been used in various fields such as sealing, oscillation damping, information storage, and electronic devices (Lee et al. 2002; Yang et al. 2005; Caruntu et al. 2004; Zhang et al. 2004). One of the rapidly developing applications of MNPs in recent years is in biomedical areas, including rapid biologic separation and drug delivery (Tan et al. 2005; Garcia et al. 2007; Wang et al. 2005; Liao and Chen 2002). However, unmodified MNPs tend to aggregate because of their high specific area and strong inter particle interaction, which limit their utilization. Therefore, it is necessary to develop strategies for the chemical stabilization of the naked MNPs against aggregation over a long period. Although the development of more efficient and versatile approaches to functionalized MNPs is very important. Therefore, the outer shell of silica not only protects the inner magnetite core from oxidation but also provides sites for surface functionalization with chlorosulfonic acid. Despite the formation of Fe3O4@SiO2–SO3H materials not only stabilized the MNPs, but also endowed the MNPs with functionality (Iaoying Yang et al. 2010).
Xanthenes and their derivatives are an important class of heterocyclic compounds, which are widely used in biological applications, such as antibacterial activities (Karthikeyana and Pandurangan 2009), anti-inflammatory (Poupelin et al. 1978), and antiviral properties (Hajipour et al. 2010). Furthermore, some of the heterocycles based on xanthenes have found application as photodynamic therapy for destroying the tumor cells (Ion et al. 1998). The other useful applications of these heterocycles can be used as dyes (Imani Shakibaei et al. 2007), laser technology (Ahmad et al. 2002), and in fluorescent materials for visualization of biomolecules (Knight and Stephens 1989).
In this context, we were prepared Fe3O4 nanoparticles by chemical co-precipitation method and subsequently coated with tetraethoxysilane (TEOS) via silanization reaction. Grafting of chlorosulfonic acid on the Fe3O4@SiO2 nanoparticles was afforded sulfamic acid-functionalized MNPs (Fe3O4@SiO2–SO3H). They were found to be a mild and efficient solid acid nano catalyst for the one-pot synthesis of 1,8‐dioxo-octahydroxanthenes under solvent-free conditions.
In this method, the corresponding 1,8‐dioxo-octahydroxanthenes were afforded in shorter reaction times and excellent yields with high purity. This green procedure has many obvious advantages compared to those reported in the previous literatures, including avoiding the use of harmful catalysts, easy workup of the reaction, excellent yields, short routine, and simplicity of the methodology.
Experimental section
Materials
All commercially available reagents were used without further purification and purchased from the Merck Chemical Company in high purity. The used solvents were purified by standard procedure.
Apparatus
FT-IR spectra were obtained as KBr pellets on a Perkin-Elmer 781 spectrophotometer and on an Impact 400 Nicolet FT-IR spectrophotometer. 1H NMR and 13C NMR were recorded in CDCl3 solvents on a Bruker DRX-400 spectrometer with tetramethylsilane as internal reference. Nanostructures were characterized using a Holland Philips Xpert X-ray powder diffraction (XRD) diffractometer (CuK, radiation, k = 0.154056 nm), at a scanning speed of 2°/min from 10° to 100° (2θ). Thermo gravimetric analyses (TGA) were conducted on a Rheometric Scientific Inc. 1998 thermal analysis apparatus under a N2 atmosphere at a heating rate of 10 °C/min. Scanning electron microscope (SEM) of nanoparticles was performed on a FESEM Hitachi S4160. Dynamic laser scattering (DLS) was performed on Malvern ZEN 3600. The Bandelin ultrasonic HD 3200 with probe model KE 76.6 mm diameter, was used to produce ultrasonic irradiation and homogenizing the reaction mixture. Melting points obtained with a Yanagimoto micro melting point apparatus are uncorrected. The purity determination of the substrates and reaction monitoring were accomplished by TLC on silica-gel polygram SILG/UV 254 plates (from Merck Company).
Preparation of catalyst
Preparation of nano-Fe3O4
Fe3O4 MNPs were prepared according to a previously reported procedure by the chemical co-precipitation method (Hu et al. 2012). Typically, FeCl3·6H2O (2.7 g) and FeCl2·4H2O (1 g) were dissolved in 100 ml of 1.2 mmol l−1 aqueous HCl by ultrasonic bath for 30 min. Then, 1.25 mol l−1 aqueous NaOH (150 ml) was added under vigorous stirring and a black precipitate was immediately formed. The resulting transparent solution was heated at 80 °C with rapid mechanical stirring under N2 atmosphere. After vigorous stirring for 2 h, the precipitate was magnetically separated and washed thoroughly with water until the supernatant liquor reached neutrality (pH ~7).
Preparation of nano-Fe3O4@SiO2
This precursor was prepared according to the reported method (Yang et al. 2009). MNPs (1 g) were initially diluted via the sequential addition of water (20 ml), ethanol (60 ml), and concentrated aqueous ammonia (2 ml, 25 wt%). The resulting dispersion was then homogenized by ultrasonic. A solution of TEOS (0.5 ml) in ethanol (10 ml) was then added to the dispersion in a drop-wise manner under continuous mechanical stirring. After vigorous stirring for 16 h, the product were collected by an external magnetic and washed three times with ethanol. Finally, the products were dried under vacuum at 70 °C for 5 h.
Preparation of Fe3O4@SiO2–SO3H
Fe3O4@SiO2–SO3H microspheres were synthesized as following; firstly, 1 g of Fe3O4@SiO2 microspheres was dispersed in dry CH2Cl2 (10 ml) by ultrasonic bath for 30 min. Subsequently, chlorosulfonic acid (1 ml) was added drop-wise to a cooled (ice-bath) solution of Fe3O4@SiO2 (1 g) over a period of 30 min at room temperature. After completion of the addition, the mixture was stirred for a further 6 h until to allow for the complete dissipation of HCl from the reaction vessel. The resulted MNPs were separated using an external magnet and washed with ethanol and water before being dried in an oven at 70 °C to give Fe3O4@SiO2–SO3H as a brown powder.
General procedure for the synthesis of 1,8-dioxo-octahydroxanthenes
A mixture of an aromatic aldehydes (1 mmol), dimedone (2 mmol) and nano Fe3O4@SiO2–SO3H (0.05 g) was heated at 110 °C under solvent-free conditions. The progress of the reactions was monitored by TLC (ethyl acetate/petroleum ether 3/7). After completion of the reaction, the reaction mixture was cooled and CH2Cl2 (5 ml) was added and the catalyst was separated by an external magnet and reused for the next experiment. The reaction mixture was concentrated on a rotary evaporator under reduced pressure. The residue was purified by recrystallization from ethanol. They were characterized by comparison of their physical and spectral data with those of authentic samples (Girijesh et al. 2011; Mahdavinia et al. 2009; Swapna et al. 2011; Kantevari et al. 2007).
9-Phenyl-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8-octahydroxanthene-1,8-dione (3a): white solid, m.p. = 202–203 °C, (m.p. = 203–204 °C Girijesh et al. 2011), IR (KBr)/υ(cm−1): 2956, 1666, 1460, 1363, 1198, 698; 1H NMR (CDCl3, 400 MHz)/δ ppm: 7.27–7.30 (t, 3H, J = 7.6 Hz, ArH), 7.20–7.23 (t, 3H, J = 7.2 Hz, ArH), 7.10–7.12 (t, 1H, J = 7.2 Hz, ArH), 4.75 (s, 1H, CH); 2.47 (s, 4H, 2CH2), 2.15–2.26 (q, 4H, 2CH2), 1.10 (s, 6H, 2CH3), 0.99 (s, 6H, CH3).
9-(4-Nitrophenyl)-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8-octahydroxanthene-1,8-dione (3g): pale yellow solid, m.p. = 224–226 °C, (m.p. = 226–227 °C, Mahdavinia et al. 2009), IR (KBr)/υ(cm−1): 2959, 1664, 1517, 1359, 1199, 865; 1H NMR (CDCl3, 400 MHz)/δ ppm: 8.08–8.10 (d, 2H, J = 8.0 Hz, ArH), 7.46–7.48 (d, 2H, J = 8.0 Hz, ArH), 4.48 (s, 1H, CH), 2.50 (s, 4H, 2CH2), 2.14–2.28 (q, 4H, J = 16.4 Hz, 2CH2), 1.12 (s, 6H, 2CH3), 0.99 (s, 6H, CH3); 13C NMR (CDCl3, 100 MHz)/δ ppm: 196.32, 169.32, 163.02, 151.58, 146.45, 129.38, 123.42, 114.49, 50.60, 40.82, 32.38, 32.24, 29.24, 27.27.
9-(3-Nitrophenyl)-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8-octahydroxanthene-1,8-dione (3f): white solid, m.p. = 171–172 °C, (m.p. = 168–170 °C, Mahdavinia et al. 2009), IR (KBr)/υ(cm−1): 2960, 1664, 1527, 1357, 1198, 1138, 814; 1H NMR (CDCl3, 400 MHz)/δ ppm: 7.39–8.01 (m, 4H, ArH), 4.84 (s, 1H, CH), 2.51 (s, 4H, 2CH2), 2.15–2.28 (q, 4H, J = 16.4 Hz, 2CH2), 1.12 (s, 6H, 2CH3), 0.99 (s, 6H, CH3); 13C NMR/(CDCl3, 100 MHz)/δ ppm: 196.36, 169.03, 148.33, 146.31, 135.37, 128.80, 122.55, 121.66, 114.55, 50.64, 40.82, 32.25, 32.10, 29.21, 27.31.
9-(4-Chlorophenyl)-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8-octahydroxanthene-1,8-dione (3c): white solid, m.p. = 231–233 °C, (m.p. = 230–232 °C, Swapna et al. 2011), IR (KBr)/υ(cm−1): 2956, 1663, 1469, 1362, 1197, 1139, 846; 1H NMR (CDCl3, 400 MHz)/δ ppm: 7.27–7.52 (d, 2H ArH), 4.84 (s, 1H, CH), 2.51 (s, 4H, 2CH2), 2.15–2.28 (q, 4H, J = 16.4 Hz, 2CH2), 1.12 (s, 6H, 2CH3), 0.99 (s, 6H, CH3).
9-(4-Chloro-3-nitrophenyl)-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8-octahydroxanthene-1,8-dione (3h): white solid, m.p. = 251–253 °C, IR (KBr)/υ(cm−1): IR (KBr)/υ(cm−1): 2961, 1665, 1534, 1361, 1198, 828; 1H NMR (CDCl3, 400 MHz)/δ ppm: 7.67 (s, 1H, ArH), 7.63–7.66 (d, 1H, J = 8.4 Hz, ArH); 7.40–7.42 (d, 1H, J = 8.4 Hz, ArH), 4.77 (s, 1H, CH); 2.50 (s, 4H, 2CH2), 2.17–2.28 (q, 4H, J = 16.4 Hz, 2CH2), 1.12 (s, 6H, 2CH3), 1.02 (s, 6H, CH3).
9-(2,4-Dichlorophenyl)-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8-octahydroxanthene-1,8-dione (3d): white solid, m.p. = 250–252 °C, (m.p. = 248–250 °C, Mahdavinia et al. 2009), IR (KBr)/υ(cm−1): 2943, 2930, 1717, 1657, 1587, 1383, 1169; 1H NMR (CDCl3, 400 MHz)/δ ppm: 7.37 (s, 1H, ArH), 7.25–7.27 (d, 1H, J = 8.0 Hz, ArH), 7.14–7.16 (d, 1H, J = 8.4 Hz, ArH), 4.95 (s, 1H, CH), 2.45 (s, 4H, 2CH2), 2.14–2.25 (q, 4H, J = 16.0 Hz, 2CH2), 1.11 (s, 6H, 2CH3), 1.02 (s, 6H, CH3).
9-(3-Methoxy)-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8-octahydroxanthene-1,8-dione (3l): white solid, m.p. = 181–182 °C, (m.p. = 179–181 °C, Mahdavinia et al. 2009), IR (KBr)/υ(cm−1): 2959, 1662, 1485, 1363, 1274, 1201, 1048, 763; 1H NMR (CDCl3, 400 MHz)/δ ppm: 7.15–7.18 (t, 1H, J = 8.0 Hz, ArH), 6.86–6.89 (d, 1H, ArH), 6.64–6.67 (d, 1H, ArH), 4.74 (s, 1H, CH), 2.46 (s, 4H, 2CH2), 2.16–2.26 (q, 4H, J = 16.4 Hz, 2CH2), 1.10 (s, 6H, 2CH3), 1.01 (s, 6H, CH3).
9-(4-Methoxy)-3,3,6,6-tetramethyl-1,2,3,4,5,6,7,8-octahydroxanthene-1,8-dione (3m): white solid, mp: 245–247 °C, (m.p. = 243–245 °C, Mahdavinia et al. 2009), IR (KBr)/υ(cm−1): 2957, 1667, 1510, 1461, 1359, 1260, 1194, 1137, 1032, 568; 1H NMR (CDCl3, 400 MHz)/δ ppm: 7.19–7.21 (d, 2H, J = 8.4 Hz, ArH), 6.74–6.79 (d, 2H, ArH), 6.64–6.67 (d, 1H, ArH), 4.70 (s, 1H, CH), 3.73 (s, 3H, OCH3), 2.46 (s, 4H, 2CH2), 2.14–2.25 (q, 4H, J = 16.4 Hz, 2CH2), 1.10 (s, 6H, 2CH3), 0.99 (s, 6H, CH3); 13C NMR/(CDCl3, 100 MHz)/δ ppm: 196.47, 162.08, 157.96, 136.51, 131.99, 129.31, 115.79, 114.33, 113.46, 55.10, 50.78, 40.86, 32.19, 30.97, 29.28, 27.34, 23.43.
Results and discussion
Characterization of Fe3O4@SiO2–SO3H as solid acid catalyst
The MNPs of 18–22 nm were prepared by co-precipitation via iron(II) and iron(III) ions. For the surface modification, the MNPs coated with a layer of silica using the (TEOS) by co-precipitation method (Yang et al. 2009) to provide reaction sites for further functionalization and thermal stability. Ultimately, the reaction of Fe3O4@SiO2 with chlorosulfonic acid led to sulfamic acid-functionalized magnetic Fe3O4 nanoparticles (Fe3O4@SiO2–SO3H) (Scheme 1). The pH measurement of Fe3O4@SiO2–SO3H (10 % w/v) was obtained about 1.36.

Preparation steps for fabricating sulfamic acid-functionalized magnetic Fe3O4 nanoparticles
The X-ray diffraction patterns of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2–SO3H are shown in Fig. 1. The position and relative intensities of all peaks confirm well with standard XRD pattern of Fe3O4 indicating retention of the crystalline cubic spinel structure during functionalization of MNPs. Characteristic peak of SiO2 in core shell structure has been hidden under weak peak of Fe3O4 at 2θ = 30. The average MNPs core diameter was calculated to be 22 nm from the XRD results by Scherrer’s equation, D = kλ/βcosθ where k is a constant (generally considered as 0.94), λ is the wavelength of Cu Ka (1.54 Å), β is the corrected diffraction line full-width at half-maximum (FWHM), and θ is Bragg’s angle (Massart 1981).

XRD patterns of a nano Fe3O4 b Fe3O4@SiO2 c Fe3O4@SiO2–SO3H
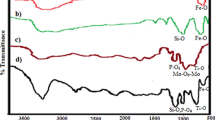
The FT-IR spectra of catalyst are presented the band in the region of 572 cm−1 is attributed to the stretching vibrations of the (Fe–O) bond and the band at about 1,100 cm−1 belongs to (Si–O) stretching vibrations. FT-IR analysis was used to characterize the presence of the –SO3H groups on the surface of the MNPs (Fig. 2). As shown in Fig. 2 c, the FT-IR spectra of Fe3O4@SiO2–SO3H was clearly different from those of Fe3O4 (Fig. 2a) and Fe3O4@SiO2 (Fig. 2b). For sulfonic acid functional group, the appeared peaks in 1,042 and 1,134 cm−1 are related to the stretching of the S–O bonds. A peak appeared at about 3,409 cm−1 due to the stretching of OH groups in the SO3H (Fig. 2c).

The comparative FT-IR spectra of a nano Fe3O4 b Fe3O4@SiO2 c Fe3O4@SiO2–SO3H
A thermogravimetric analysis (TGA) was used to study the thermal stability of the acid catalyst (Fig. 3). The TGA curve was divided into several regions corresponding to different mass lose ranges. The first region, which occurred below 150 °C, displayed a mass loss that was attributable to the loss of adsorbed solvent or trapped water from the catalyst. A weight loss of approximately 10 % weight occurred between 150 and 500 °C that was likely a consequence of the loss of SO3H groups. The occurrence of further mass losses at higher temperature was resulted from the decomposition of silica shell (Nemati et al. 2012). Thus; the catalyst was stable up to 250 °C, confirming that it could be safely used in organic reactions at temperatures between the ranges of 80–150 °C.

TGA curve of a Fe3O4@SiO2 b Fe3O4@SiO2–SO3H
The SEM image was shown that MNPs have a mean diameter of about 20 nm and a nearly spherical shape in Fig. 4a, b shows that Fe3O4@SiO2 nanoparticles still keep the morphological properties of Fe3O4 except for a slightly larger particle size and smoother surface, which silica are uniform coated on the Fe3O4 particles to form silica shell in compared to the Fe3O4@SiO2. The SEM image shown in Fig. 4 c demonstrates that Fe3O4@SiO2–SO3H nanoparticles are nearly spherical with more than 20 nm in size.

The SEM image of a Fe3O4, b Fe3O4@SiO2, c Fe3O4@SiO2–SO3H
The magnetization curve for Fe3O4@SiO2 nanoparticles and Fe3O4@SiO2–SO3H are shown in Fig. 5. Room temperature specific magnetization (M) versus applied magnetic field (H) curve measurements of the sample indicate a saturation magnetization value (Ms) of 15 emu g−1, lower than that of bare MNPs (50.86 emu g−1) due to the coated shell.

Magnetization curves for the prepared a Fe3O4@SiO2 and b Fe3O4@SiO2–SO3H at 40 °C
The dynamic laser scattering (DLS) measurement of Fe3O4@SiO2–SO3H nanoparticles was shown in Fig. 6. In order to determine the fraction of the particle population that aggregates, comparisons between the intensity averaged DLS data and number averaged DLS data were made. From this slurry, an aqueous stock dispersion (100 ml acetone at 5 g Fe3O4@SiO2–SO3H) was prepared using an ultrasonic bath for 30 min.

The dynamic laser scattering (DLS) measurement of nano Fe3O4@SiO2–SO3H
Figure 7a shows the photograph of Fe3O4@SiO2–SO3H microspheres that dispersed in water. After a magnet was placed aside, the black microspheres can be magnetized in 5 min, leaving a clear solution (Fig. 7b). That is to say, the Fe3O4@SiO2–SO3H nanoparticles were shown good magnetic responsibility even if the SiO2–SO3H layer was increased to 20 nm.

Digital camera images of the aqueous solution a with dispersed magnetic Fe3O4@SiO2–SO3H composite particles and b after applied magnetic field
Evaluation of the catalytic activity of Fe3O4@SiO2–SO3H in the synthesis 1,8-dioxo-octahydro-xanthenes
In this research, a simple energy, eco-friendly and convenient method for the synthesis of 1,8‐dioxo-octahydroxanthenes using Fe3O4@SiO2–SO3H as new catalyst are described. Initially, in order to optimize the reaction conditions, it is considered to represent the reaction of dimedone and benzaldehyde in a 2:1 ratio to afford the xanthene 3a under various reaction conditions was performed for an appropriate time (Table 1).
The obtained results from the reaction to determine the optimum amount of catalyst are presented in Table 1. As can be seen from this Table, the best results were obtained using 0.05 g of catalyst in the reaction of benzaldehyde (1 mol) with dimedone (2 mol) (Table 1, entry 5).
After optimization of the reaction conditions, the reaction of dimedone with various aldehydes was carried out in according to the general experimental procedure (Scheme 2). In all the cases, the corresponding xanthenediones were obtained in high to excellent yields and short reaction times. The obtained similar products are summarized in Table 2.

Synthesis of 1,8-dioxo-octahydroxanthene under solvent-free conditions

The presence of electron-donating (alkoxy or hydroxyl group) or electron-withdrawing groups (nitro or halide group) on the aromatic ring of the aldehydes did not have much effect on the reaction such that to afford respective products with high yields. While para-substituted aldehydes were given good results in compared to the ortho-substituents. There is more steric hindrance for the ortho substituted aldehydes (o-OCH3, –OH, –Cl, –NO2) on the product formation than the para-substituted (p-OCH3, –OH, –Cl, –NO2) aldehydes.
The possibility of recycling the catalyst was examined through the reaction of dimedone and 4-nitrobenzaldehyde catalyzed by Fe3O4@SiO2–SO3H nanoparticles under optimized conditions. Upon completion of the reaction, the catalyst was separated by an external magnet, washed with acetone, and the recycled catalyst was saved for the next reaction. The recycled catalyst could be reused five times without any decrease in catalytic activity so that the yields were ranged from 93 to 97 % (Fig. 8).

Reusability of Fe3O4@SiO2–SO3H in the reaction of dimedone, 4-nitrobenzaldehyde under solvent-free conditions
The structure of the obtained products was confirmed by IR, 1H NMR, and 13C NMR spectra. The infrared spectra of the 3g exhibit a medium band at 2,959 cm−1 represents the presence of alkane protons (4CH3 groups). In addition, a band at 1,664 cm−1 represents the presence of general carbonyl groups (C=O stretching) and a strong band at 1,199 cm−1 confirms the presence of C–O bond stretching. In 1H NMR spectra of compound 3g, the four methyl groups were appeared as two series of axial and equatorial methyl groups. Therefore, these protons were differently indicated as two singlet bands with six hydrogens at around 0.99 and 1.12 ppm, respectively. Due to the possibility of free rotation and conformational considerations, the four methyl groups are not becoming equivalent and appear as two singlet at different chemical shifts in all the compounds that is indicated the axial and equatorial positions. The proton at the bridge between two dimedone rings appears usually in the region 4.83 ppm and the signal around δ = 7.46–8.10 ppm is assigned to the protons of the aromatic rings (CH=CH). In 13C NMR, two carbon atoms of dimedone (CH3 groups) were symmetrical and give rise to one signal for each set of carbons. The carbon at the bridge between two dimedone rings is shown at 50.6 ppm. The carbonyl carbon atom appears in the expected region around 196.32 ppm.
The proposed reaction mechanism
The formation of 1,8‐dioxo-octahydroxanthene from dimedone and aldehyde in the presence of Fe3O4@SiO2–SO3H as catalyst can be explained by a tentative mechanism is presented in Scheme 3. One molecule of dimedone (I) was firstly condensed with an activated aromatic aldehyde to provide intermediate II, which can be regarded as a fast Knoevenagel addition. Then the active methylene of the second molecule of dimedone reacted with intermediate II via conjugate Michael addition to produce the intermediate III, which undergoes intramolecular cyclodehydration to give the octahydroxanthene-1,8-dione (IV).

Proposed reaction mechanism
Conclusion
In summary, we were described using Fe3O4@SiO2–SO3H as a reusable, readily available, inexpensive and efficient catalyst for the one-pot synthesis of 1,8‐dioxo-octahydroxanthenes. These compounds were prepared through treatment of dimedone with various aromatic aldehydes under solvent-free conditions at 110 °C. Our main strategy in this work is to develop a facile protocol, low cost, easily available catalyst, reduce reaction time, easy workup, and environmental friendliness.
References
Ahmad M, King TA, Ko D, Cha BH, Lee J (2002) Performance and photostability of xanthene and pyrromethene laser dyes in sol–gel phases. J Phys D Appl Phys 35:1473–1476
Beydoun D, Amal R, Low GKC, McEvoy S (2000) Novel photocatalyst: titania-coated magnetite activity and photodissolution. J Phys Chem B 104:4387–4396
Caruntu D, Caruntu G, Chen Y, O’Connor CJ, Goloverda G, Kolesnichenko VL (2004) Synthesis of variable-sized nanocrystals of Fe3O4 with high surface reactivity. Chem Mater 16:5527–5534
Garcia I, Zafeiropoulos NE, Janke A, Tercjak A, Eceiza A, Stamm M, Mondragon I (2007) Functionalization of iron oxide magnetic nanoparticles with poly(methyl methacrylate) brushes via grafting-from atom transfer radical polymerization. J Polym Sci Part A 45:925–932
Girijesh KV, Keshav R, RajivPratibha KVD, Singh MS (2011) An efficient one-pot solvent-free synthesis and photophysical properties of 9-aryl/alkyl-octahydroxanthene-1,8-diones. Tetrahedron 67:3698–3704
Hajipour AR, Ghayeb Y, Sheikhan N, Ruoho AR (2010) Bronsted acidic ionic liquid as an efficient and reusable catalyst for synthesis of 14-Aryl-14-H-dibenzo[a,j]xanthenes under solvent-free conditions. Synlett 741–744
Hu Y, Zhang Z, Zhang H, Luo L, Yao S (2012) Selective and sensitive molecularly imprinted sol–gel film-based electrochemical sensor combining mercaptoacetic acid-modified PbS nanoparticles with Fe3O4@Au–multi-walled carbon nanotubes–chitosan. J Solid State Electro Chem 16:857–867
Iaoying Yang X, Chen L, Han B, Yang X, Duan H (2010) Preparation of magnetite and tumor dual-targeting hollow polymer microspheres with pH-sensitivity for anticancer drug-carriers. Polymer 51:2533–2539
Imani Shakibaei G, Mirzaei P, Bazgir A (2007) Dowex-50 W promoted synthesis of 14-aryl-14H-dibenzo[a, j]xanthenes and 1,8-dioxo-octahydroxanthene derivatives under solvent-free conditions. Appl Catal A 325:188–192
Ion RM, Frackowiak D, Planner A, Wiktorowicz K (1998) The incorporation of various porphyrins into blood cells measured via flow cytometry, absorption and emission spectroscopy. Acta Biochim Polonica 45:833–845
Kantevari S, Bantu R, Nagarapu L (2007) HClO4-SiO4 and PPA catalyzed efficient one-pot Knovenagel condensation, Micheal addition and cyclo dihydration of dimedone and aldehydes, aqueous and solvent-free conditions scope and limitations. J Mol Catal A 53(269):53–57
Karthikeyana G, Pandurangan A (2009) Heteropolyacid (H3PW12O40) supported MCM-41: an efficient solid acid catalyst for the green synthesis of xanthenedione derivatives. J Mol Catal A 311:36–45
Kim YS, Kim YH (2003) Application of ferro-cobalt magnetic fluid for oil sealing. J Magn Magn Mater 267:105–110
Knight CG, Stephens T (1989) Xanthene-dye-labelled phosphatidylethanolamines as probes of interfacial pH. Studies in phospholipid vesicles. Bio Chem J 258:683–687
Lee DK, Kang YS, Lee CS, Stroeve P (2002) Structure and characterization of nanocomposite Langmuir-Blodgett Films of poly(maleic monoester)/Fe3O4 nanoparticle complexes. J Phys Chem B 106:7267–7271
Liao MH, Chen DH (2002) Preparation and characterization of a novel magnetic nano-adsorbent. J Mater Chem 12:3654–3659
Mahdavinia GH, Bigdeli MA, Saeidi Hayeniaz Y (2009) Covalently anchored sulfonic acid on silica gel (SiO2-R-SO3H) as an efficient and reusable heterogeneous catalyst for the one-pot synthesis of 1,8-dioxo-octahydroxanthenes under solvent-free conditions. Chin Chem Lett 20:539–541
Massart R (1981) Preparation of aqueous magnetic liquids in alkaline and acidic media. IEEE Trans Magn 17:1247–1248
McMichael RD, Shull RD, Swartzendruber LJ, Bennett LH, Watson RE (1992) Magnetocaloric effect in superparamagnets. J Magn Magn Mater 111:29–33
Nemati F, Heravi MM, Saeedirad R (2012) Nano-Fe3O4 encapsulated-silica particles bearing Sulfonic acid groups as a magnetically separable catalyst for highly efficient knoevenagel condensation and michael addition reactions of aromatic aldehydes with 1,3-cyclic diketones. Chin J Catal 33:1825–1831
Poupelin JP, Saint-Ruf G, Foussard-Blanpin O, Narcisse G, Uchida-Ernouf G, Lacroix R (1978) Synthesis and anti-inflammatory properties of bis (2-hydroxy-1-naphthyl)methane, II. Polysubstituted and polycyclic derivatives. Eur J Med Chem 13:67–71
Raj K, Moskowitz R (2002) A review of damping applications of ferrofluids. Trans Magn 16:358–363
Swapna K, Narayana MS, Venkata DNY (2011) β-Cyclodextrin mediated synthesis of 1,8-dioxooctahydroxanthenes in water. Eur J Chem 2:272–275
Tan ST, Wendorff JH, Pietzonka C, Jia ZH, Wang GQ (2005) Biocompatible and biodegradable polymer nanofibers displaying superparamagnetic properties. Chem Phys Chem 6:1461–1465
Wang PC, Lee CF, Young TH, Lin DT, Chiu WY (2005) Preparation and clinical application of immunomagnetic latex. J Polym Sci Part A 43:1342–1356
Yang TZ, Shen CM, Li Z, Zhang HR, Xiao CW, Chen ST (2005) Highly ordered self-assembly with large area of Fe3O4 nanoparticles and the magnetic properties. J Phys Chem B 109:23233–23236
Yang D, Hu J, Fu S (2009) Controlled synthesis of magnetite–silica nanocomposites via a seeded sol–gel approach. J Phys Chem C 113:7646–7651
Zhang DH, Liu ZQ, Han S, Han S, Li C, Lei B, Stewart MP, Tour JM, Zhou CW (2004) Magnetite (Fe3O4) core-shell nanowires: synthesis and magnetoresistance. Nano Lett 4:2151–2155
Acknowledgments
The authors are grateful to University of Kashan for supporting this work by Grant No. 159148/22.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Naeimi, H., Nazifi, Z.S. A highly efficient nano-Fe3O4 encapsulated-silica particles bearing sulfonic acid groups as a solid acid catalyst for synthesis of 1,8-dioxo-octahydroxanthene derivatives. J Nanopart Res 15, 2026 (2013). https://doi.org/10.1007/s11051-013-2026-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-013-2026-2