
Abstract
Vascular endothelial cells (ECs) are monolayers of cells arranged in the inner walls of blood vessels. Under normal physiological conditions, ECs play an essential role in angiogenesis, homeostasis and immune response. Emerging evidence suggests that abnormalities in EC metabolism, especially aerobic glycolysis, are associated with the initiation and progression of various diseases, including multiple cancers. In this review, we discuss the differences in aerobic glycolysis of vascular ECs under normal and pathological conditions, focusing on the recent research progress of aerobic glycolysis in tumor vascular ECs and potential strategies for cancer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Endothelial cells (ECs) form a monolayer of cells arranged along the inner lining of blood vessels. Under normal physiological conditions, ECs play a pivotal role in maintaining oxygen and nutrient supply to all bodily tissues [1]. ECs are critical for upholding the internal environment, its homeostasis, and immune function. In recent years, research on EC metabolism has gained significant momentum. A growing body of evidence has shown that EC metabolism undergoes significant alterations under various pathological conditions, profoundly impacting disease onset and progression. As an crucial link in cellular energy metabolism, aerobic glycolysis plays a remarkable role in EC activities and could offer novel targets for disease treatment. This paper aims to explore recent advances in glycolysis in ECs under normal and pathological conditions, with a particular emphasis on human cancers.
The function of ECs in physiological state
Angiogenesis
ECs are typically quiescent in healthy state but can be rapidly activated in response to pathological changes, facilitating the delivery of oxygen and nutrients to hypoxic tissues through the formation of new blood vessels, a process known as angiogenesis.
Angiogenesis is accomplished through the interaction among three distinct EC subtypes: tip cells, stalk cells, and phalanx cells, each fulfilling a specific role in the process. Neovascularization commences with the migration of tip cells, succeeded by the proliferation of stalk cells, which generate new vascular sprouts. Subsequently, phalanx cells, characterized by a nonproliferating phenotype, continue to align established vessels, facilitating the formation of mature vessels with the functions of regulating vascular homeostasis and establishing endothelial barrier [2].
The key cytokine that dynamically regulates the differentiation of ECs into one of these subtypes is vascular endothelial growth factor (VEGF), which binds to VEGF receptor 2 (VEGFR2) on ECs and induces their differentiation into tip cells. Tip cells express delta-like ligand 4 (DLL4), which binds to Notch receptors on neighboring ECs, initiating their differentiation into stalk cells. In stalk cells, DLL4 signaling induces cleavage of the Notch intracellular structural domain (NICD), which then induces the cell to stimulating the expression of produce VEGF receptor 1 (VEGFR1). VEGFR1 is substantially less sensitive to VEGF compared to VEGFR2, further ensuring that ECs adjacent to tip cells become stalk cells [3].
This seemingly strict interaction between tip and stalk cells is actually dynamic, and this dynamic competition ensures that cells with the highest VEGFR2/VEGFR1 ratio become tip cells [4].
Maintaining internal environmental and its homeostasis
ECs serve as crucial intermediaries between human blood circulation and organ tissues, playing an indispensable role in the regulation of body functions. These functions include maintaining coagulation, regulating blood pressure, and facilitating the exchange of substances both within and outside of blood vessels, with particular significance in the regulation of coagulation. For instance, ECs regulate blood pressure and maintain the balance between coagulation and anticoagulation in blood flow by releasing vasodilatory substances such as prostaglandin I2 (PGI2) and nitric oxide (NO), as well as vasoconstrictive substances like endothelin 1 (ET-1) and angiotensin II (AngII) [5]. Moreover, ECs can produce nitric oxide synthase (NOS) in response to hormonal and chemical signal stimulation, and NO synthesized by endothelial NOS (eNOS) regulates peripheral vascular tone by stimulating NO-sensitive guanylate cyclase, which plays a vital role in maintaining vascular homeostasis and controlling blood pressure [6].
Immune response
As the type of cells that are in direct contact with the circulation, ECs are pivotal participants and regulators of the inflammatory response. Their recognition and response to substances like invading circulating microorganisms are crucial for activating the body’s early immune system [7]. For instance, ECs express Nod1 to trigger the release of the inflammatory factor IL-8 [8]. There is also an inextricable link between the inflammatory response and the angiogenic process. In chronic inflammation, ECs can respond to angiogenic factors like VEGFA to sustain inflammatory angiogenesis [7].
EC Metabolism under physiological conditions
Glucose, fatty acids (FA) and amino acids (AA) are the three primary substrates for adenosine triphosphate (ATP) and biomass production in ECs, and their roles have been extensively studied and summarized. The following section will summarize the role of EC metabolism in maintaining endothelial function and disease pathogenesis, with a specific focus on glycolysis in tumor vascular ECs.
The glycolysis and bypasses of ECs
Under physiological conditions, glycolysis serves as the primary mode of energy production in ECs, exhibiting a higher proportion compared to other cells in the body. Similar to many cancer cells, up to 85% of ATP is produced by glycolysis in ECs. This phenomenon can be attributed to two possible reasons. Firstly, ECs protect themselves from oxidative stress by keeping reactive oxygen species (ROS) at a low level. Secondly, ECs would like to increase the oxygen supply to their surrounding tissues by means of anaerobic metabolism [9]. It has been demonstrated that when exposed to a low glucose environment or when glucose is competed by 2-deoxy-d-glucose (2-DG), a structural analogue of glucose, for the glycolytic process, ECs trigger ROS-induced autophagy, suggesting that ECs reduce oxygen dependence by preferentially utilizing glycolytic capacity, and thereby decrease ROS production [9, 10]. Additionally, the angiogenesis process in hypoxic tissues demands a faster rate of ATP production through glycolysis to expedite the re-establishment of blood circulation at hypoxic sites. This necessity also contributes to the high glycolytic rate observed in ECs.
In the process of angiogenesis, the initiation of the glycolytic pathway first involves the uptake of glucose into ECs via the membrane-bound glucose transporter protein (GLUT) [11]. The glucose-based glycolytic pathway is regulated by three key enzymes: phosphofructokinase-1 (PFK1), hexokinase 2 (HK2) and pyruvate kinase (PK).
PFK1 is an important rate-limiting enzyme in the glycolytic pathway, converting fructose-6-phosphate (F6P) into fructose-1,6-bisphosphate (F1,6P2). 6-phosphofructo-2-kinase/fructose-2,6bisphosphatase3 (PFKFB3), an efficient glycolytic activator, can variably activate PFK1 by producing fructose-2,6-bisphosphate (F2,6P2). PFKFB3 not only regulates the proliferation of ECs but also controls the formation and directional migration of filopodium and lamellipodium. Therefore, the deletion of PFKFB3 impairs the angiogenic function of ECs [12]. In contrast, overexpression of PFKFB3 leads to elevatedglycolysis levels in ECs exhibiting a pro-tip cell phenotype. It even inhibits Notch1 signaling in pro-stalk cells during retinal vascular development, further underscoring the importance of cellular glycolysis levels in ECs in determining the tip cell phenotype [12]. As previously previously, VEGF operates through the Notch signaling pathway to ensure that cells with the highest VEGFR2/VEGFR1 ratio become the tip cells. Similarly, it is reported that stalk cells can display higher glycolytic activity when expressing higher levels of PFKFB3, effectively taking the place of tip cells [12].
HK2 is another glycolytic rate-limiting enzyme responsible for catalyzing the initial step of glycolysis in ECs, which involves the phosphorylation of glucose in ECs to glucose-6-phosphate (G6P). Increasing evidence suggests that HK2 is upregulated in various types of tumors, correlating with enhanced aerobic glycolysis.The upregulation of HK2 expression can be attributed to fibroblast growth factor (FGF), which enhances the expression of MYC, one of the most frequently dysregulated driver genes in human cancers. Although MYC was previously deemed “undruggable” due to the lack of a suitable pocket for high-affinity binding of low-molecular-weight inhibitors, recent preclinical trials have shown significant anticancer effects of some MYC-targeted inhibitors. This suggests that such anticancer effects may also arise from the inhibition of aerobic glycolysis in tumor vascular ECs [13]. For example, it has recently been found that OTUB1 blocks MYC protein degradation through deubiquitination, thereby promoting HK2-mediated glycolysis and the development of breast cancer [14].
PK is also a glycolytic rate-limiting enzyme that converts phosphoenolpyruvate (PEP) to pyruvate. PKM2, one of the four tissue-specific isoforms of PK, exists as a dimer or tetramer in normal, malignant, and embryonic cells. The affinity of dimeric PKM2 for PEP is lower than that of tetrameric PKM2, resulting in less conversion to pyruvate, which inhibits glycolysis and leads glucose more into the glycolytic bypass for biomass synthesis. In highly proliferative endothelial cells, PKM2 tends to shift towards its dimeric form to promote biomass synthesis, aligning with their heightened proliferative properties [15,16,17]. As a glycolytic rate-limiting enzyme, silencing PKM2 in ECs inhibits neovascularization. Furthermore, Protein JMJD8 has been identified as associated with PKM2 to regulate the angiogenic process. Knocking down JMJD8 inhibits EC metabolism and angiogenesis, although the exact mechanism by which JMJD8 regulates PKM2 remains uncertain [18].
Several environmental factors and signaling proteins have been identified that can influence the glycolytic level and angiogenic capacity of ECs by regulating the three key enzymes mentioned above and their regulators. For example, mechanical signaling in the circulation can also affect angiogenesis in a metabolism-dependent manner, as evidenced by the activation of Krüppel-like factor 2 (KLF2) in response to laminar shear stress, which inhibits PFKFB3, HK2, and several other glycolytic genes [19]. The FOXO family is highly enriched in ECs. The transcription factor FOXO1 was found to inhibit glycolysis in ECs by suppressing MYC and PFKFB3 levels [20, 21]. Additionally, FOXO1 exhibits environment-dependent activity, promoting the germination and migration of lymphatic ECs by upregulating the purinergic receptor P2RY1 upon exposure to ATP [20]. Meanwhile, impairment of vascular growth and germination function was also observed after silencing PFKFB3 and HK2 in animal models, further suggesting the essential role of glycolysisl for ECs and angiogenesis in vivo [12, 22]. Furthermore, the availability of sufficient glucose in the microenvironment for uptake plays a decisive role in the level of glycolysis of ECs. Glycogen synthesis in ECs increases with sufficient glucose supply, leading to accumulation of intracellular glycogen, while deprivation of glucose causes the inhibition of glycogen phosphorylase (GP), which catalyzes glycogenolysis in ECs, impairing the ability of ECs to migrate and survive [23]. This evidence suggests that ECs utilize glycogen as reserve energy when carrying out angiogenic pro-cesses in a glucose-deficient environment.
The pentose phosphate pathway (PPP) is a bypass of glycolysis that facilitates the synthesis of raw nucleotide material and the homeostasis of redox balance by diverting G6P generated from glucose to produce nicotinamide adenine dinucleotide phosphate (NADPH) and ribose 5-phosphate (R5P). NADPH, in turn, can regenerate a common cellular antioxidant, reduced glutathione [24,25,26]. Additionally, NADPH also participates in fatty acid and NO production. The NADPH generated by the pentose phosphate pathway is directly involved in fatty acid synthesis, thus linking the PPP of ECs to fatty acid metabolism. Moreover, NO promotes the angiogenic activity of ECs [27]. Glucose 6-phosphate dehydrogenase (G6PD) is one of the rate-limiting enzymes of PPP. Overexpression of G6PD stimulates EC proliferation and migration by boosting NO and NADPH production. Another rate-limiting enzyme of PPP in the reversible non-oxidative pathway is transketolase. Inhibition of either of these two rate-limiting enzymes has been shown to significantly suppress EC survival [23, 28].
Another bypass of glycolysis is known as the hexosamine biosynthetic pathway (HBP). Although it contributes to a relatively small proportion of glucose metabolism, HBP is closely linked to post-translational protein modifications via glycosylation, including N-glycosylation and O-glycosylation. .N-glycosylation enhances the stability, membrane expression, and signaling activity of VEGFR2, while O-glycosylation influences interactions related to NOTCH signaling ligands. Consequently, HBP is implicated in determining the apical cell phenotype of ECs, underscoring its unique role in EC functions [29,30,31,32]. Further investigations are warranted to elucidate the mechanisms by which HBP impacts angiogenesis.
Other metabolic pathways of ECs
FA metabolism of ECs
Generally, mitochondria are the primary site of ATP production, and acetyl coenzyme A derived from fatty acids FA is utilized in the tricarboxylic acid (TCA) cycle to main-tain ATP production. While in proliferating endothelial cells (PECs), the main function of mitochondria is biosynthesis. The dependence of dNTP synthesis in PECs on the carbon source in fatty acids is now well established. FA is metabolized to acetyl coenzyme A to maintain the TCA cycle. In addition to the production of ATP, the TCA cycle provides precursors for the synthesis of dNTP necessary for proliferation, thereby promoting DNA synthesis and thus aiding PECs proliferation [33]. Furthermore, surprisingly, quiescent endothelial cells (QECs) regenerate NADPH for redox homeostasis by enhancing FAO levels [34].
Carnitine palmitoyltransferase 1a (CPT1a) is a rate-limiting enzyme in the FA metabolic pathway that transfers FA into mitochondria. It has been discovered that CPT1a deficiency not only leads to attenuated proliferation of PECs and defective neovascularization in vitro and in vivo, but also promotes QECs dysfunction by increasing endothelial oxidative stress. Supplementation with acetyl coenzyme A precursors, which promote the tricarboxylic acid (TCA) cycle, restored cellular dNTP levels and alleviated oxidative stress-induced EC dysfunction in CPT1a-deficient ECs. Neither glucose nor glutamine metabolism could compensate for this metabolic defect of Fatty acid oxidation (FAO) [33, 34]. These findings indicates that ECs primarily utilize fatty acid-derived carbon for biosynthesis rather than energy generation through FA metabolism. This metabolic pathway supports cell proliferation during angiogenesis and facilitates the regeneration of NADPH to maintain cellular redox balance.
Glutamine metabolism of ECs
Glutamine, the most abundant non-essential amino acid (NEAA) in circulation, plays a crucial role in maintaining proliferation and vasodilation in ECs through its metabolism. In ECs, approximately 30% of the carbon source for the TCA cycle is derived from glutamine, a proportion comparable to glycolysis and FAO-derived carbon. Glutaminase 1 (GLS1) replenishes glutamine as a carbon source for the TCA cycle, supporting protein and nucleotide synthesis. Additionally, maintaining the dynamic redox balance requires glutathione (GSH), a reducing agent produced from glutamine. Thus, glutamine depletion not only impairs biomass synthesis in ECs but also renders them more susceptible to damage induced by ROS [35]. Silencing GLS1 leads to a reduced tendency for ECs to differentiate into a tip cell phenotype [36]. However, EC-specific deletion of glutamine synthetase (GS), the enzyme that converts glutamate and ammonia to glutamine, markedly impairs EC migration but not proliferation [37]. These findings suggest that glutamine metabolism plays a vital role in regulating the phenotype of ECs during vascular sprouting.
In summary, various metabolic pathways may have significant impacts on the for-mation of different subtypes of ECs. Multiple studies have shown that The level of glycolysis mainly affects the ability of ECs to differentiate towards tip cells, while FA metabolism is closely related to the proliferation of stalk cells. Additionally, glutamine metabolism differentially affects the phenotype of ECs under different conditions.
Glucose metabolism characteristics of tumor endothelial cells
Hypoxia of the tumor microenvironment
Hypoxia is one of the characteristics of the tumor microenvironment where neovascularization is usually exposed to relatively low oxygen, and tumor endothelial cells (TECs) need to adjust their metabolism to adapt to the hypoxic tumor environment [38, 39]. In general, cancer cells are characterized by their high metabolism, thus stimulating angiogenesis to supply oxygen and nutrients; however, unlike normal vascular endothelium, blood vessels in tumors are not only disorganized due to abnormal development but also have structural and functional defects due to the lack of tissue hierarchy of normal vascular beds [1]. As a result, the tumor has uneven gaps between vascular endothelial cells, and poor perfusion makes the tumor hypoxic, thereby promoting metastatic escape of cancer cells [40, 41]. In turn, the hypoxic environment stimulates EC proliferation and angiogenesis [42, 43], which creates a vicious cycle of hypoxia and tumor development.
The effects of hypoxia on cancer cell metabolism have become clearer. Briefly, restricted and intermittent blood supply in tumor tissues leads to periodic hypoxia and re-oxygenation, resulting in oxidative stress and hypoxia in the tumor microenvironment. Hypoxic cancer cell metabolism under hypoxic conditions upregulates the expression of genes that promote glycolysis, such as GLUT1, GLUT3, PFKFB3, and HK1-3, while it decreases FAO, promotes FA synthesis, and decreases glutamine carboxylation; all of which contribute to increased glycolytic flux and FA synthesis while downregulating aerobic respiration in hypoxic cancer cells. However, the role of hypoxia and HIF signaling in EC metabolism is still not fully elucidated [44].
Classification of hypoxic conditions has identified acute hypoxia as selective splicing and upregulation of genes involved in pyruvate metabolism and glucose transport [45]. In contrast, the effect of chronic hypoxia on EC metabolism involves the regulation of the activity of multiple metabolic pathways, such as glycolysis, amino acid biosynthesis, carbon metabolism, PPP, and cysteine/methionine metabolism. However, this chronic hypoxia may be different from the intermittent hypoxic conditions of the tumor hypoxic response described previously. Therefore, the changes in EC metabolism observed during chronic hypoxia need to be confirmed in an intermittent hypoxic environment.
Hypoxia and HIF signaling in angiogenic function of TECs
The prolyl hydroxylase family (PHD) hydroxylates the alpha subunit of HIF. The enzymatic activity of PHD requires oxygen for activation, and the hydroxylated HIF alpha subunit is targeted by the von Hippel‒Lindau (VHL) factor and undergoes ubiquitination and proteasomal degradation [46]. In a hypoxic environment, the enzymatic activity of PHD is inhibited, and VHL factors are unable to degrade HIF-α, leading to the activation of HIF-related pathways and functions, including the induction of angiogenesis, glucose metabolism, and tumor growth, invasion, and metastasis [43]. Among the HIFα subunit family, HIF-1α remains in a stable state during acute hypoxia and is the major subunit that induces glycolysis and other metabolic changes [47].
By focusing on HIF signaling in the study of the tumor hypoxic microenvironment, it has been found that HIF directly induces an increase in VEGF levels in ECs [48], VEGF contains a hypoxia response element (HRE) that can be activated in response to reduced oxygen, causing hypoxic tissues and macrophages to express VEGF and stimulate angiogenesis to restore oxygen and nutrient supply to hypoxic regions. VEGF directly promotes angiogenesis while inducing subsequent activation of other pro-angiogenic growth factors, such as placental growth factor (PlGF) and FGF [49]. Thus, the induction of angiogenesis by hypoxia and HIF is largely dependent on VEGF. Hypoxia and HIF signaling affect EC function and angiogenesis in multiple ways. First, hypoxia induces transcriptional activation of multiple angiogenic factors, including VEGF, PlGF, and angiopoietin (ANGPT1 and ANGPT2) [50]. These vascular growth factors modulate proangiogenic chemokines and receptors that induce EC stem cells to migrate to angiogenic sites [51]. In addition, hypoxia stabilizes these vascular growth factors and promotes PFKFB3 expression, thereby increasing glycolytic flux in ECs [12]. Hypoxia also directly stimulates EC proliferation and neovascular germination as well as promotes neovascular stabilization by remodeling the extracellular matrix [48, 52,53,54]. In addition, hypoxia and HIF signaling also modulate vascular function by increasing the production and release of NO to promote vasodilation [55].
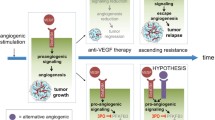
Stabilization of HIF-1α and HIF-2α has different regulatory effects on angiogenesis and normalization of blood vessels. Specifically, HIF1α deficiency in the tumor endothelium reduces the number of tumor vessels, which reduces tumor growth but increases tumor necrosis; roxarsenical (Rox), an organoarsenic compound, has a significant pro-tumor effect in vivo due to its ability to increase HIF-1α [56, 57]. In contrast, the lack of tumor endothelial HIF2α leads to reduced vascular integrity in tumor models, increasing the likelihood of tumor metastasis but reducing tumor growth [58, 59]. Functional and structural damage to the tumor vasculature increases the likelihood of metastasis, and poor local blood supply also hinders the full efficacy of chemotherapy and immunotherapy. Defective PHD2 mediates the stabilization of HIF2α in ECs, normalizes TECs, reverses tumor hypoxia, and reduces cancer cell metastasis [60]. Here, we draw a schematic diagram to summarize this chapter. (Fig. 1). Thus, targeting the PHD2-HIF2α signaling pathway may be a future tumor treatment strategy based on EC metabolism with antiangiogenesis as the goal.

Hypoxia and HIF signaling in angiogenic function of TECs
Glycolytic characteristics of TECs
Endothelial cells in tumor vasculature have higher glycolytic flux to produce ATP compared to normal endothelial cells, and single-cell RNA sequencing has shown that the glycolytic flux of TECs is 2–4 times higher than that of normal ECs [61]. TECs exhibit a highly glycolytic cellular phenotype, such as enhanced expression of the GLUT1 glucose transporter and the PFKFB3 glycolytic activator [62, 63], which may be attributed to the hypoxia-dependent altered expression of glycolytic enzymes and increased secretion of proangiogenic growth factors, such as VEGF. In response to the high glycolytic phenotype of TECs, the hypoxic tumor microenvironment (TME) and the stimulation of inflammatory factors may be critical for the upregulation of PKFBF3 expression [63]. Reducing the glycolysis of tumor endothelial cells by inhibiting the function of PFKFB3 inhibits their proliferation and thus normalizes the tumor vasculature as evidenced by regular TEC alignment, a dense tumor vascular barrier, and unobstructed blood perfusion. The aim of this therapeutic strategy is not to completely remove the glycolytic flux, which would lead to endothelial cell death and tumor vascular disintegration, but rather to reduce the glycolytic level to that of normal endothelial cells, inhibit their metastasis, and prevent the development of advanced cancer [64, 65]. In addition, the pentose phosphate and serine biosynthetic pathways for biosynthesis are highly activated in tumor endothelial cells compared to healthy endothelial cells [63].
The expression of PKM2, one of the key enzymes of the glycolytic pathway, is traditionally thought to be associated with the proliferation of endothelial cells, especially TECs; however, it has been shown that PKM2 is not required for EC proliferation as PKM2-deficient ECs do not significantly differ from controls in vitro or in vivo, and cell cycle arrest in the absence of PKM2 may be driven by compensatory PKM1 upregulation [17]. PKM2 accumulates at the junctions of VE-cadherin-expressing endothelial cells and the area near F-actin-rich filopodium and lamellipodium, and VE-cadherin is a major regulator of EC junction formation and stability. After knockdown of PKM2, ECs exhibit unstable intercellular junctions and decreased migration distance, demonstrating that the migration ability of ECs and intercellular junction ability are related to the expression of PKM2, which is closely related to the migration ability of ECs and intercellular connectivity. Although the importance of the glycolytic pathway in maintaining EC function and normalizing TECs is well established, only the PFKBF3 glycolytic enzyme has been shown to regulate inter-EC connections by some unknown mechanism [63]. In addition to glycolytic enzymes, extracellular vesicles (H-EVs) have recently been found to promote adhesion of cancer cells to endothelial cells in triple-negative breast cancer, and Circulating galectin-3(cirGal-3) enhances this proadhesive effect by a mechanism that may be related to cirGal-3-induced increased expression of ICAM-1, leading to upregulation of glycolysis in endothelial cells [66]. This special function of PKM2 in maintaining the integrity of the endothelial barrier during migration and thus reducing tumor metastasis provides a new therapeutic strategy for antitumor treatment, namely, targeting PKM2 to inhibit tumor metastasis [67].
As mentioned previously, the PPP is one of the bypasses of glycolysis and achieves redox homeostasis in ECs by synthesizing NADPH, and G6PD is an important rate-limiting enzyme for the PPP. Due to the hypoxic conditions in tumors, high levels of reactive oxygen species (ROS) in the TME stimulate endoplasmic reticulum (ER) stress due to insufficient reduction flux generated by the pentose phosphate pathway (PPP), which activates the IRE-1 and PERK signaling pathways, thereby increasing autophagy, while in turn, the PPP can also play a role in autophagy regulation due to changes in G6PD activity [68]. ROS play a role in cancer suppression, but it has been shown that autophagy promotes tumor angiogenesis by activating the JAK2/STAT3 pathway and targeting VEGF; in addition, autophagy regulates the ability of cancer stem cells (CSCs) to differentiate into TECs [69,70,71,72].
Although the glycolytic processes in TEC proliferation and tumor angiogenesis are important, it has recently been reported that microvascular endothelial cells may not be glucose-dependent in their early growth phase as they exhibit good growth capacity even in a glucose-deficient environment. Evidence suggests that glutamine metabolism plays a more important role in the early growth of microvascular endothelial cells [73]. This phenomenon suggests that the metabolism of TECs may be different from one species to another, and tumor treatment strategies targeting TEC metabolism may need to change accordingly with their different metabolic profiles.
Lactic acid metabolism
In addition to having a direct effect on ECs, the hypoxic microenvironment of tumors can also lead to EC uptake of lactate through lactate accumulation, and the effect of lactate on TECs varies. First, TECs increase the expression of lactate dehydrogenase B, which converts lactate taken up by TECs into pyruvate to enter the TCA cycle to promote biosynthesis and energy supply, thus promoting TEC proliferation and angiogenesis. Second, lactate uptake by ECs induces ROS-mediated activation of the NF-kappaB/IL-8 pathway, promoting angiogenesis [74]. In addition, lactate directly regulates tyrosine kinase receptors in ECs and stabilizes N-Myc downstream regulatory gene 3 (NDRG3), thus promoting angiogenesis under hypoxic conditions [75, 76]. Finally, lactate, as a signaling molecule, also enhances angiogenesis by activating the HIF-1a and PI3K/AKT pathways, thereby promoting angiogenesis [75, 77, 78]. Overall, lactate independently stimulates angiogenesis, and lactate accumulation is associated with disease progression in tumors.
Although the acidic environment due to high lactic acid is not conducive to cell survival, TECs rapidly proliferate in a high lactic acid environment, allowing tumor angiogenesis and survival. The reason for this phenomenon is that TECs upregulate the expression of carbonic anhydrase 2 (CAII). Knockdown of CAII reduces the survival of TECs under lactic acidosis and nutrient-adequate conditions [79]. In normal ECs, vascular endothelial growth factor A (VEGFA) induces CAII expression, which is an indirect proangiogenic mechanism of VEGFA. Interestingly, the carbonic anhydrase inhibitor, acetazolamide, does not significantly reduce tumor angiogenesis but instead promotes vascular maturation in tumors, thereby reducing metastasis [79].
Monocarboxylate transporter (MCT) protein, as a lactate transporter protein, is responsible for transporting lactate inside and outside the cell. MCT4 is the predominant regulator of lactate transport in tumor cells and endothelial cells, which are high glycolytic donors; MCT4 promotes EC migration as well as tumor proliferation and invasion under tumor cell and EC coculture conditions, while the MCT-specific blocker, 7ACC1, reverses the tumor-promoting effect of MCT. However, these findings have only been demonstrated in vitro [80].
Interactions between different cell types in TME
Based on recent studies, we understand that the level of glycolysis in TECs is inter-cellularly regulated by other cell types in the tumor microenvironment as demonstrated by a 2016 report on tumor-associated macrophages (TAMs) affecting EC glycolytic function. Hypoxia upregulates the expression of regulated in development and DNA damage responses-1 (REDD1), which is an inhibitor of mTOR activation. Knockdown of REDD1 in mice results in an mTOR-dependent enhancement of glycolysis levels in TAMs, and competition between TAMs and TECs for extracellular glucose results in a reduction in glycolytic flux in TECs, indirectly promoting normalization of tumor vasculature [81]. Because HIF1α deficiency reduces CXCL1-mediated macrophage recruitment, hypoxia in TECs affects TAM behavior and recruitment, thus altering the interaction between TAMs and TECs in the tumor microenvironment [82].
Cancer associated fibroblasts (CAFs) in the TME maintain a relatively high glycolytic flux in the resting state to maintain basal cell function, and their glycolysis levels are doubled when proliferating [83]. The reason for this is that the activity of PHD proteins in CAFs is inhibited by high levels of ROS from neighboring cancer cells, which subsequently cause autophagic degradation of caveolin-1 by stabilizing HIF-1α. Caveolin-1 is an NO inhibitory protein whose degradation leads to excessive NO production. These high levels of NO lead to mitochondrial dysfunction of CAFs, resulting in the removal of CAFs by mitochondrial autophagy; thus, CAFs require high levels of glycolysis to produce energy and thus supply lactate, which cancer cells use in the TCA cycle along with converted pyruvate for ATP production [84, 85]. CAFs increase glycolytic flux to maintain a specific link with cancer cells. This phenomenon contradicts the mainstream view of the “Warburg effect” in tumors and has been termed the “reverse Warburg effect”. It is reasonable to speculate that highly glycolytic TECs, similar to CAFs, have this special relationship with cancer cells in terms of lactic acid supply.
TECs preserve functional mitochondria
Although TECs have a highly glycolytic profile similar to that of CAFs, the mitochondria within TECs are not as dysfunctional as those in CAFs; instead, TECs retain functional mitochondria [86]. The majority of pyruvate is converted to lactate at the end of glycolysis and transported out of the TECs and less than 1% of glucose-derived pyruvate is transported to the mitochondria for the subsequent TCA cycle [87]. However, this oxidative phosphorylation process within the mitochondria not only makes the TEC energy supply more flexible but also maintains TEC proliferation and promotes neovascular sprouting by increasing the amount of biosynthesis. Therefore, the oxidative phosphorylation process in mitochondria is crucial for highly proliferating tumor endothelial cells. Although inhibition of mitochondrial respiration induces death of highly proliferating tumor endothelial cells, there is no significant damage to quiescent normal endothelial cells [88,89,90,91,92]. If the oxidative respiratory chain is interrupted by inactivating the ubiqui-none-binding protein, QPC, a subunit of mitochondrial complex III, then amino acid levels in ECs will not be maintained; although the migratory function of ECs is not affected, their ability to proliferate will be greatly reduced, which further emphasizes the importance of mitochondrial function for EC proliferation [93]. This mechanism of cell death through inhibition of mitochondrial respiration may be related to ROS production and uncoupling of mitochondrial membrane potential. Thus, inhibition of mitochondrial function is an antitumor strategy that has not been explored in depth and deserves further investigation.
Translational implications
Many current targeted antiangiogenic therapies aim to inhibit the VEGF signaling pathway in ECs, which is clinically associated with a high rate of drug resistance. In addition, single antiangiogenic therapies may also increase the potential for tumor metastasis due to hypoxia in the tumor microenvironment [94]. Compared to the genetic stability of normal cells, tumor cells are genetically unstable and have a high mutation rate, making it difficult to advance therapies targeting tumor cell metabolism. It is reasonable to assume that combination therapy would not only benefit the long-term survival of cancer patients but also attenuate the side effects caused by targeted therapies and improve the quality of life. For example, the PFKFB3 inhibitor, PFK15, has been shown to have significant antitumor proliferation and proapoptotic effects in mice [93], and it synergistically inhibits the proliferation and migration of human umbilical vein endothelial cells (HUVECs) when combined with the tyrosine kinase inhibitor, sunitinib [95, 96]. Regrettably, there is little research related to TEC metabolism, and further understanding of the mechanisms involved in the role and effects of TEC glycolysis will facilitate the development of new therapeutic approaches to treat cancer. Here, we present in table form a list of potentially promising therapeutic agents or substances that currently target the TEC glycolytic pathway (Table 1) [63, 64, 97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116].
Conclusions
The regulation of glycolytic metabolism in endothelial cells (ECs) has gained significant attention, particularly in the context of pathological angiogenesis within the tumor microenvironment. This review provides a comprehensive summary of the differences in glycolytic features between normal ECs and tumor endothelial cells (TECs), as well as the factors that affect their regulation. A promising approach for cancer treatment could be targeting the regulation of glycolytic metabolism in TECs, in combination with classical antiangiogenic therapies. Although many agents targeting aerobic glycolysis of ECs are still in the preclinical or early clinical trial stages, with the continuous exploration in this field, it is optimistic that these emerging drugs may provide new options for cancer treatment in the future.
Data availability
No datasets were generated or analysed during the current study.
References
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473(7347):298–307 Epub 2011/05/20. doi: 10.1038/nature10144. PubMed PMID: 21593862; PubMed Central PMCID: PMCPMC4049445
Potente M, Gerhardt H, Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis. Cell 146(6):873–887 Epub 2011/09/20. https://doi.org/10.1016/j.cell.2011.08.039
Phng LK, Gerhardt H (2009) Angiogenesis: a team effort coordinated by notch. Dev Cell 16(2):196–208 Epub 2009/02/17. https://doi.org/10.1016/j.devcel.2009.01.015
Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM et al (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12(10):943–953 Epub 2010/09/28. https://doi.org/10.1038/ncb2103
Förstermann U, Sessa WC (2012) Nitric oxide synthases: regulation and function. Eur Heart J 33(7):829–837 837a-837d. Epub 2011/09/06. https://doi.org/10.1093/eurheartj/ehr304
Leo F, Suvorava T, Heuser SK, Li J, LoBue A, Barbarino F et al (2021) Red blood cell and endothelial eNOS independently Reg-Ulate circulating nitric oxide metabolites and blood pressure. Circulation 144(11):870–889 Epub 2021/07/08. https://doi.org/10.1161/circulationaha.120.049606
Pober JS, Sessa WC (2007) Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. ;7(10):803–815. Epub 2007/09/26. https://doi.org/10.1038/nri2171. PubMed PMID: 17893694
Opitz B, Förster S, Hocke AC, Maass M, Schmeck B, Hippenstiel S et al (2005) Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ Res 96(3):319–326 Epub 2005/01/18. https://doi.org/10.1161/01.RES.0000155721.83594.2c
Wang J, Alexanian A, Ying R, Kizhakekuttu TJ, Dharmashankar K, Vasquez-Vivar J et al (2012) Acute exposure to low glucose rapidly induces endothelial dysfunction and mitochondrial oxidative stress: role for AMP kinase. Arterioscler Thromb Vasc Biol 32(3):712–720 Epub 2011/12/31. https://doi.org/10.1161/atvbaha.111.227389
Wang Q, Liang B, Shirwany NA, Zou MH (2011) 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS ONE 6(2):e17234 Epub 2011/03/10. https://doi.org/10.1371/journal.pone.0017234
Uldry M, Thorens B (2004) The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch 447(5):480–489 Epub 2003/05/17. https://doi.org/10.1007/s00424-003-1085-0
De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR et al (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154(3):651–663 Epub 2013/08/06. https://doi.org/10.1016/j.cell.2013.06.037
Duffy MJ, O’Grady S, Tang M, Crown J (2021) MYC as a target for cancer treatment. Cancer Treat Rev 94:102154 Epub 2021/02/02. https://doi.org/10.1016/j.ctrv.2021.102154
Han X, Ren C, Lu C, Qiao P, Yang T, Yu Z (2022) Deubiquitination of MYC by OTUB1 contributes to HK2 mediated glycolysis and breast tumorigenesis. Cell Death Differ 29(9):1864–1873 Epub 2022/03/18. https://doi.org/10.1038/s41418-022-00971-8
Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC (2008) Pyruvate kinase M2 is a phosphotyrosine-binding pro-tein. Nature 452(7184):181–186 Epub 2008/03/14. https://doi.org/10.1038/nature06667
Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K et al (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2(97):ra73 Epub 2009/11/19. https://doi.org/10.1126/scisignal.2000431
Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G et al (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155(2):397–409 Epub 2013/10/15. https://doi.org/10.1016/j.cell.2013.09.025
Boeckel JN, Derlet A, Glaser SF, Luczak A, Lucas T, Heumüller AW et al (2016) JMJD8 regulates angiogenic sprouting and cel-lular metabolism by interacting with pyruvate kinase M2 in endothelial cells. Arterioscler Thromb Vasc Biol 36(7):1425–1433 Epub 2016/05/21. https://doi.org/10.1161/atvbaha.116.307695
Doddaballapur A, Michalik KM, Manavski Y, Lucas T, Houtkooper RH, You X et al (2015) Laminar shear stress inhibits endothe-lial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler Thromb Vasc Biol 35(1):137–145 Epub 2014/11/02. https://doi.org/10.1161/atvbaha.114.304277
Niimi K, Ueda M, Fukumoto M, Kohara M, Sawano T, Tsuchihashi R et al (2017) Transcription factor FOXO1 promotes cell Mi-Gration toward exogenous ATP via controlling P2Y1 receptor expression in lymphatic endothelial cells. Biochem Biophys Res Commun 489(4):413–419 Epub 2017/06/01. https://doi.org/10.1016/j.bbrc.2017.05.156
Wilhelm K, Happel K, Eelen G, Schoors S, Oellerich MF, Lim R et al (2016) FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529(7585):216–220 Epub 2016/01/07. https://doi.org/10.1038/nature16498
Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS et al (2017) FGF-dependent metabolic control of vascular development. Nature 545(7653):224–228 Epub 2017/05/04. https://doi.org/10.1038/nature22322
Vizán P, Sánchez-Tena S, Alcarraz-Vizán G, Soler M, Messeguer R, Pujol MD et al (2009) Characterization of the metabolic changes underlying growth factor angiogenic activation: identification of new potential therapeutic targets. Carcinogenesis 30(6):946–952 Epub 2009/04/17. https://doi.org/10.1093/carcin/bgp083
Jongkind JF, Verkerk A, Baggen RG (1989) Glutathione metabolism of human vascular endothelial cells under peroxidative stress. Free Radic Biol Med 7(5):507–512 Epub 1989/01/01. https://doi.org/10.1016/0891-5849(89)90026-9
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20 Epub 2008/01/08. https://doi.org/10.1016/j.cmet.2007.10.002
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033 Epub 2009/05/23. https://doi.org/10.1126/science.1160809
Ghesquière B, Wong BW, Kuchnio A, Carmeliet P (2014) Metabolism of stromal and immune cells in health and disease. Nature 511(7508):167–176 Epub 2014/07/11. https://doi.org/10.1038/nature13312
Lorenzi M (2007) The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res 2007:61038 Epub 2008/01/29. https://doi.org/10.1155/2007/61038
Wells L, Vosseller K, Hart GW (2001) Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science 291(5512):2376–2378 Epub 2001/03/28. https://doi.org/10.1126/science.1058714
Vosseller K, Sakabe K, Wells L, Hart GW (2002) Diverse regulation of protein function by O-GlcNAc: a nuclear and cytoplasmic carbohydrate post-translational modification. Curr Opin Chem Biol 6(6):851–857 Epub 2002/12/10. https://doi.org/10.1016/s1367-5931(02)00384-8
Croci DO, Cerliani JP, Dalotto-Moreno T, Méndez-Huergo SP, Mascanfroni ID, Dergan-Dylon S et al (2014) Glycosyla-tion-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156(4):744–758 Epub 2014/02/18. https://doi.org/10.1016/j.cell.2014.01.043
Rahimi N, Costello CE (2015) Emerging roles of post-translational modifications in signal transduction and angiogenesis. Pro-teomics 15(2–3):300–309 Epub 2014/08/28. https://doi.org/10.1002/pmic.201400183
Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I et al (2015) Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520(7546):192–197 Epub 2015/04/02. https://doi.org/10.1038/nature14362
Kalucka J, Bierhansl L, Conchinha NV, Missiaen R, Elia I, Brüning U et al (2018) Quiescent endothelial cells upregulate fatty acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab 28(6):881–894 .e813. Epub 2018/08/28. https://doi.org/10.1016/j.cmet.2018.07.016
Kim B, Li J, Jang C, Arany Z (2017) Glutamine fuels proliferation but not migration of endothelial cells. Embo j 36(16):2321–2333 Epub 2017/07/01. https://doi.org/10.15252/embj.201796436
Huang H, Vandekeere S, Kalucka J, Bierhansl L, Zecchin A, Brüning U et al (2017) Role of glutamine and interlinked asparagine metabolism in vessel formation. Embo j 36(16):2334–2352 Epub 2017/07/01. https://doi.org/10.15252/embj.201695518
Eelen G, Dubois C, Cantelmo AR, Goveia J, Brüning U, DeRan M et al (2018) Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 561(7721):63–69 Epub 2018/08/31. https://doi.org/10.1038/s41586-018-0466-7
Brown JM, Wilson WR (2004) Exploiting tumor hypoxia in cancer treatment. Nat Rev Cancer. ;4(6):437–447. Epub 2004/06/02. https://doi.org/10.1038/nrc1367. PubMed PMID: 15170446
Bache M, Kappler M, Said HM, Staab A, Vordermark D (2008) Detection and specific targeting of hypoxic regions within solid tumors: current preclinical and clinical strategies. Curr Med Chem 15(4):322–338 Epub 2008/02/22. doi: 10.2174/092986708783497391. PubMed PMID: 18288988
DeClerck K, Elble RC (2010) The role of hypoxia and acidosis in promoting metastasis and resistance to chemotherapy. Front Biosci (Landmark Ed) 15(1):213–225 Epub 2009/12/29. doi: 10.2741/3616. PubMed PMID: 20036816
Jain RK (2014) Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26(5):605–622 Epub 2014/12/18. https://doi.org/10.1016/j.ccell.2014.10.006
Clementi E, Brown GC, Feelisch M, Moncada S (1998) Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A 95(13):7631–7636 Epub 1998/06/24. https://doi.org/10.1073/pnas.95.13.7631
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer. ;3(10):721–732. Epub 2003/09/18. https://doi.org/10.1038/nrc1187. PubMed PMID: 13130303
Wong BW, Marsch E, Treps L, Baes M, Carmeliet P (2017) Endothelial cell metabolism in health and disease: impact of hypoxia. Embo j 36(15):2187–2203 Epub 2017/06/24. https://doi.org/10.15252/embj.201696150
Weigand JE, Boeckel JN, Gellert P, Dimmeler S (2012) Hypoxia-induced alternative splicing in endothelial cells. PLoS ONE 7(8):e42697 Epub 2012/08/10. https://doi.org/10.1371/journal.pone.0042697
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR et al (2001) C. Elegans EGL-9 and mammalian homo-logs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107(1):43–54 Epub 2001/10/12. https://doi.org/10.1016/s0092-8674(01)00507-4
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC (2003) Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 23(24):9361–9374 Epub 2003/12/04. https://doi.org/10.1128/mcb.23.24.9361-9374.2003
Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ et al (2005) Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105(2):659–669 Epub 2004/09/18. https://doi.org/10.1182/blood-2004-07-2958
Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD et al (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16(9):4604–4613 Epub 1996/09/01. https://doi.org/10.1128/mcb.16.9.4604
Semenza GL (2003) Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med 54:17–28 Epub 2002/10/03. https://doi.org/10.1146/annurev.med.54.101601.152418
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME et al (2004) Progenitor cell trafficking is regu-lated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10(8):858–864 Epub 2004/07/06. https://doi.org/10.1038/nm1075
Faller DV, Weng H, Choi SY (1997) Activation of collagenase IV gene expression and enzymatic activity by the Moloney murine leukemia virus long terminal repeat. Virology 227(2):331–342 Epub 1997/01/20. https://doi.org/10.1006/viro.1996.8345
Abidia A (2000) Endothelial cell responses to hypoxic stress. Clin Exp Pharmacol Physiol. ;27(8):630. Epub 2000/07/20. https://doi.org/10.1046/j.1440-1681.2000.03310.x. PubMed PMID: 10901394
Li W, Petrimpol M, Molle KD, Hall MN, Battegay EJ, Humar R (2007) Hypoxia-induced endothelial proliferation requires both mTORC1 and mTORC2. Circ Res. ;100(1):79–87. Epub 2006/11/18. https://doi.org/10.1161/01.RES.0000253094.03023.3f. PubMed PMID: 17110594
Chan CK, Vanhoutte PM (2013) Hypoxia, vascular smooth muscles and endothelium. Acta Pharm Sinica B 3(1):1–7. https://doi.org/10.1016/j.apsb.2012.12.007
Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP et al (2004) Loss of HIF-1alpha in endothelial cells disrupts a hypox-ia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 6(5):485–495 Epub 2004/11/16. https://doi.org/10.1016/j.ccr.2004.09.026
Chen X, Zhang M, Chen L, Zhou Z, Chen B, Wang C et al (2021) Roxarsone Promotes Glycolysis and Angiogenesis by Inducing Hypoxia-Inducible Factor-1α In Vitro and In Vivo. ACS Omega. ;6(14):9559–9566. Epub 2021/04/20. https://doi.org/10.1021/acsomega.1c00072. PubMed PMID: 33869936; PubMed Central PMCID: PMCPMC8047655
Skuli N, Liu L, Runge A, Wang T, Yuan L, Patel S et al (2009) Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood 114(2):469–477 Epub 2009/05/15. https://doi.org/10.1182/blood-2008-12-193581
Skuli N, Majmundar AJ, Krock BL, Mesquita RC, Mathew LK, Quinn ZL et al (2012) Endothelial HIF-2α regulates murine patho-logical angiogenesis and revascularization processes. J Clin Invest 122(4):1427–1443 Epub 2012/03/20. https://doi.org/10.1172/jci57322
Mazzone M, Dettori D, de Oliveira RL, Loges S, Schmidt T, Jonckx B et al (2009) Heterozygous deficiency of PHD2 restores Tu-mor oxygenation and inhibits metastasis via endothelial normalization. Cell 136(5):839–851 Epub 2009/02/17. https://doi.org/10.1016/j.cell.2009.01.020
Rohlenova K, Goveia J, García-Caballero M, Subramanian A, Kalucka J, Treps L et al (2020) Single-cell RNA sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab 31(4):862–877 .e814. Epub 2020/04/09. https://doi.org/10.1016/j.cmet.2020.03.009
Yeh WL, Lin CJ, Fu WM (2008) Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol Pharmacol 73(1):170–177 Epub 2007/10/19. https://doi.org/10.1124/mol.107.038851
Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A et al (2016) Inhibition of the glycolytic activator PFKFB3 in Endothelium induces Tumor Vessel normalization, Impairs Metastasis, and improves chemotherapy. Cancer Cell 30(6):968–985 Epub 2016/11/22. https://doi.org/10.1016/j.ccell.2016.10.006
Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquière B, Cauwenberghs S et al (2014) Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab 19(1):37–48 PubMed PMID: 24332967
Conradi LC, Brajic A, Cantelmo AR, Bouché A, Kalucka J, Pircher A et al (2017) Tumor vessel disintegration by maximum tol-erable PFKFB3 blockade. Angiogenesis 20(4):599–613 Epub 2017/09/07. https://doi.org/10.1007/s10456-017-9573-6
Wang L, Du DD, Zheng ZX, Shang PF, Yang XX, Sun C et al (2022) Circulating galectin-3 promotes tumor-endothelium-adhesion by upregulating ICAM-1 in endothelium-derived extracellular vesicles. Front Pharmacol 13:979474 Epub 2022/11/18. https://doi.org/10.3389/fphar.2022.979474
Gómez-Escudero J, Clemente C, García-Weber D, Acín-Pérez R, Millán J, Enríquez JA et al (2019) PKM2 regulates endothelial cell junction dynamics and angiogenesis via ATP production. Sci Rep 9(1):15022 Epub 2019/10/23. https://doi.org/10.1038/s41598-019-50866-x
Mele L, la Noce M, Paino F, Regad T, Wagner S, Liccardo D et al (2019) Glucose-6-phosphate dehydrogenase blockade potenti-ates tyrosine kinase inhibitor effect on breast cancer cells through autophagy perturbation. J Exp Clin Cancer Res 38(1):160 Epub 2019/04/17. https://doi.org/10.1186/s13046-019-1164-5
Tsai HC, Tzeng HE, Huang CY, Huang YL, Tsai CH, Wang SW et al (2017) WISP-1 positively regulates angiogenesis by control-Ling VEGF-A expression in human osteosarcoma. Cell Death Dis 8(4):e2750 Epub 2017/04/14. https://doi.org/10.1038/cddis.2016.421
Carbajo-Pescador S, Ordoñez R, Benet M, Jover R, García-Palomo A, Mauriz JL et al (2013) Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br J Cancer 109(1):83–91 Epub 2013/06/13. https://doi.org/10.1038/bjc.2013.285
Huynh J, Etemadi N, Hollande F, Ernst M, Buchert M (2017) The JAK/STAT3 axis: a comprehensive drug target for solid malig-nancies. Semin Cancer Biol 45:13–22 Epub 2017/06/26. https://doi.org/10.1016/j.semcancer.2017.06.001
Hassanpour M, Rezabakhsh A, Pezeshkian M, Rahbarghazi R, Nouri M (2018) Distinct role of autophagy on angiogenesis: high-lights on the effect of autophagy in endothelial lineage and progenitor cells. Stem Cell Res Ther 9(1):305. https://doi.org/10.1186/s13287-018-1060-5PubMed PMID: 30409213; PubMed Central PMCID: PMCPMC6225658 Epub 2018/11/10
Ocaña MC, Martínez-Poveda B, Quesada AR, Medina M (2019) Highly Glycolytic Immortalized Human Dermal Microvascular Endothelial Cells are Able to Grow in Glucose-Starved Conditions. Biomolecules. ;9(8). Epub 2019/08/04. https://doi.org/10.3390/biom9080332. PubMed PMID: 31374952; PubMed Central PMCID: PMCPMC6723428
Végran F, Boidot R, Michiels C, Sonveaux P, Feron O (2011) Lactate influx through the endothelial cell monocarboxylate trans-porter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res 71(7):2550–2560 Epub 2011/02/09. https://doi.org/10.1158/0008-5472.Can-10-2828
Ruan GX, Kazlauskas A (2013) Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor re-ceptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem 288(29):21161–21172 Epub 2013/06/12. https://doi.org/10.1074/jbc.M113.474619
Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM et al (2015) A lactate-induced response to hypoxia. Cell 161(3):595–609 PubMed PMID: 25892225
Hunt TK, Aslam RS, Beckert S, Wagner S, Ghani QP, Hussain MZ et al (2007) Aerobically derived lactate stimulates Revasculari-Zation and tissue repair via redox mechanisms. Antioxid Redox Signal 9(8):1115–1124 Epub 2007/06/15. https://doi.org/10.1089/ars.2007.1674
Porporato PE, Payen VL, De Saedeleer CJ, Préat V, Thissen JP, Feron O et al (2012) Lactate stimulates angiogenesis and acceler-ates the healing of superficial and ischemic wounds in mice. Angiogenesis 15(4):581–592 Epub 2012/06/05. https://doi.org/10.1007/s10456-012-9282-0
Annan DA, Maishi N, Soga T, Dawood R, Li C, Kikuchi H et al (2019) Carbonic anhydrase 2 (CAII) supports tumor blood endo-thelial cell survival under lactic acidosis in the tumor microenvironment. Cell Commun Signal 17(1):169 Epub 2019/12/19. https://doi.org/10.1186/s12964-019-0478-4
Guo C, Huang T, Wang QH, Li H, Khanal A, Kang EH et al (2019) Monocarboxylate transporter 1 and monocarboxylate trans-porter 4 in cancer-endothelial co-culturing microenvironments promote proliferation, migration, and invasion of renal cancer cells. Cancer Cell Int 19:170 Epub 2019/07/13. https://doi.org/10.1186/s12935-019-0889-8
Wenes M, Shang M, Di Matteo M, Goveia J, Martín-Pérez R, Serneels J et al (2016) Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab 24(5):701–715 Epub 2016/10/25. https://doi.org/10.1016/j.cmet.2016.09.008
Akhtar S, Hartmann P, Karshovska E, Rinderknecht FA, Subramanian P, Gremse F et al (2015) Endothelial hypoxia-inducible Factor-1α promotes atherosclerosis and monocyte recruitment by upregulating MicroRNA-19a. Hypertension 66(6):1220–1226 Epub 2015/10/21. https://doi.org/10.1161/hypertensionaha.115.05886
Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I et al (2010) Quiescent fibroblasts exhibit high meta-bolic activity. PLoS Biol 8(10):e1000514 Epub 2010/11/05. https://doi.org/10.1371/journal.pbio.1000514
Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I et al (2012) Warburg meets autophagy: cancer-associated fibro-blasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal 16(11):1264–1284 Epub 2011/09/03. https://doi.org/10.1089/ars.2011.4243
Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A et al (2010) Understanding the lethal drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol Ther 10(6):537–542 Epub 2010/09/24. https://doi.org/10.4161/cbt.10.6.13370
Koziel A, Woyda-Ploszczyca A, Kicinska A, Jarmuszkiewicz W (2012) The influence of high glucose on the aerobic metabolism of endothelial EA.hy926 cells. Pflugers Arch 464(6):657–669 Epub 2012/10/12. https://doi.org/10.1007/s00424-012-1156-1
De Bock K, Georgiadou M, Carmeliet P (2013) Role of endothelial cell metabolism in vessel sprouting. Cell Metab 18(5):634–647 Epub 2013/08/27. https://doi.org/10.1016/j.cmet.2013.08
Blecha J, Novais SM, Rohlenova K, Novotna E, Lettlova S, Schmitt S et al (2017) Antioxidant defense in quiescent cells deter-mines selectivity of electron transport chain inhibition-induced cell death. Free Radic Biol Med 112:253–266 Epub 2017/08/05. https://doi.org/10.1016/j.freeradbiomed.2017.07.033
Coutelle O, Hornig-Do HT, Witt A, Andree M, Schiffmann LM, Piekarek M et al (2014) Embelin inhibits endothelial mitochon-drial respiration and impairs neoangiogenesis during tumor growth and wound healing. EMBO Mol Med 6(5):624–639 Epub 2014/03/22. https://doi.org/10.1002/emmm.201303016
Rohlena J, Dong LF, Kluckova K, Zobalova R, Goodwin J, Tilly D et al (2011) Mitochondrially targeted α-tocopheryl succinate is antiangiogenic: potential benefit against tumor angiogenesis but caution against wound healing. Antioxid Redox Signal 15(12):2923–2935 Epub 2011/09/10. https://doi.org/10.1089/ars.2011.4192
Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S et al (2003) A peptide trivalent arsenical inhibits tumor angio-genesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell 3(5):497–509 Epub 2003/06/05. https://doi.org/10.1016/s1535-6108(03)00109-0
Orecchioni S, Reggiani F, Talarico G, Mancuso P, Calleri A, Gregato G et al (2015) The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int J Cancer 136(6):E534–544 Epub 2014/09/10. https://doi.org/10.1002/ijc.29193
Diebold LP, Gil HJ, Gao P, Martinez CA, Weinberg SE, Chandel NS (2019) Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat Metab 1(1):158–171 Epub 2019/05/21. https://doi.org/10.1038/s42255-018-0011-x
Ribatti D, Annese T, Ruggieri S, Tamma R, Crivellato E (2019) Limitations of anti-angiogenic treatment of tumors. Transl On-col 12(7):981–986 Epub 2019/05/24. https://doi.org/10.1016/j.tranon.2019.04.022
Horváthová J, Moravčík R, Matúšková M, Šišovský V, Boháč A, Zeman M (2021) Inhibition of Glycolysis Suppresses Cell Prolif-eration and Tumor Progression In Vivo: Perspectives for Chronotherapy. Int J Mol Sci. ;22(9). Epub 2021/05/01. https://doi.org/10.3390/ijms22094390. PubMed PMID: 33922320; PubMed Central PMCID: PMCPMC8122821
Zlacká J, Murár M, Addová G, Moravčík R, Boháč A, Zeman M (2022) Synthesis of glycolysis inhibitor PFK15 and its synergistic action with an approved Multikinase Antiangiogenic Drug on Human Endothelial Cell Migration and Proliferation. Int J Mol Sci 23(22). https://doi.org/10.3390/ijms232214295PubMed PMID: 36430773; PubMed Central PMCID: PMCPMC9697023 Epub 2022/11/27
Kaji K, Nishimura N, Seki K, Sato S, Saikawa S, Nakanishi K et al (2018) Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int J Cancer 142(8):1712–1722 Epub 2017/12/06. https://doi.org/10.1002/ijc.31193
Ocaña MC, Martínez-Poveda B, Marí-Beffa M, Quesada AR, Medina M (2020) Fasentin diminishes endothelial cell proliferation, differentiation and invasion in a glucose metabolism-independent manner. Sci Rep 10(1):6132 Epub 2020/04/11. https://doi.org/10.1038/s41598-020-63232-z
Wu CH, Ho YS, Tsai CY, Wang YJ, Tseng H, Wei PL et al (2009) In vitro and in vivo study of phloretin-induced apoptosis in human liver cancer cells involving inhibition of type II glucose transporter. Int J Cancer 124(9):2210–2219 Epub 2009/01/07. https://doi.org/10.1002/ijc.24189
Fu Z, Chen X, Guan S, Yan Y, Lin H, Hua ZC (2015) Curcumin inhibits angiogenesis and improves defective hematopoiesis in-duced by tumor-derived VEGF in tumor model through modulating VEGF-VEGFR2 signaling pathway. Oncotarget 6(23):19469–19482 Epub 2015/08/09. https://doi.org/10.18632/oncotarget.3625
Zheng L, Li D, Xiang X, Tong L, Qi M, Pu J et al (2013) Methyl jasmonate abolishes the migration, invasion and angiogenesis of gastric cancer cells through down-regulation of matrix metalloproteinase 14. BMC Cancer 13:74 Epub 2013/02/12. https://doi.org/10.1186/1471-2407-13-74
Huang CC, Wang SY, Lin LL, Wang PW, Chen TY, Hsu WM et al (2015) Glycolytic inhibitor 2-deoxyglucose simultaneously targets cancer and endothelial cells to suppress neuroblastoma growth in mice. Dis Model Mech 8(10):1247–1254 Epub 2015/09/24. https://doi.org/10.1242/dmm.021667
Singh S, Pandey S, Chawla AS, Bhatt AN, Roy BG, Saluja D et al (2019) Dietary 2-deoxy-D-glucose impairs tumor growth and metastasis by inhibiting angiogenesis. Eur J Cancer 123:11–24 Epub 2019/11/02. https://doi.org/10.1016/j.ejca.2019.09.005
Merchan JR, Kovács K, Railsback JW, Kurtoglu M, Jing Y, Piña Y et al (2010) Antiangiogenic activity of 2-deoxy-D-glucose. PLoS ONE 5(10):e13699. https://doi.org/10.1371/journal.pone.0013699PubMed PMID: 21060881; PubMed Central PMCID: PMCPMC2965179 Epub 2010/11/10
Agnihotri S, Mansouri S, Burrell K, Li M, Mamatjan Y, Liu J et al (2019) Ketoconazole and Posaconazole selectively Target HK2-expressing Glioblastoma cells. Clin Cancer Res 25(2):844–855 Epub 2018/10/17. https://doi.org/10.1158/1078-0432.Ccr-18-1854
Clem BF, O’Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA 2 et al (2013) Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther 12(8):1461–1470 Epub 2013/05/16. https://doi.org/10.1158/1535-7163.Mct-13-0097
Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A et al (2008) Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther 7(1):110–120 Epub 2008/01/19. https://doi.org/10.1158/1535-7163.Mct-07-0482
Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P (2011) Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol 2:49 Epub 2011/09/10. https://doi.org/10.3389/fphar.2011.00049
Lin H, Ma X, Yang X, Chen Q, Wen Z, Yang M et al (2022) Natural shikonin and acetyl-shikonin improve intestinal microbial and protein composition to alleviate colitis-associated colorectal cancer. Int Immunopharmacol 111:109097 Epub 2022/08/12. https://doi.org/10.1016/j.intimp.2022.109097
Dai Y, Liu Y, Li J, Jin M, Yang H, Huang G (2022) Shikonin inhibited glycolysis and sensitized cisplatin treatment in non-small cell lung cancer cells via the exosomal pyruvate kinase M2 pathway. Bioengineered 13(5):13906–13918 PubMed PMID: 35706397; PubMed Central PMCID: PMCPMC9275963
Wang Y, Hao F, Nan Y, Qu L, Na W, Jia C et al (2018) PKM2 inhibitor shikonin overcomes the cisplatin resistance in bladder Cancer by inducing necroptosis. Int J Biol Sci 14(13):1883–1891 Epub 2018/11/18. https://doi.org/10.7150/ijbs.27854
Yang W, Liu J, Hou L, Chen Q, Liu Y (2021) Shikonin differentially regulates glucose metabolism via PKM2 and HIF1α to over-come apoptosis in a refractory HCC cell line. Life Sci 265:118796 Epub 2020/11/22. https://doi.org/10.1016/j.lfs.2020.118796
Zhou S, Li D, Xiao D, Wu T, Hu X, Zhang Y et al (2022) Inhibition of PKM2 enhances sensitivity of Olaparib to Ovarian Cancer cells and induces DNA damage. Int J Biol Sci 18(4):1555–1568 Epub 2022/03/15. https://doi.org/10.7150/ijbs.62947
Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Trarbach T, Folprecht G et al (2011) Prognostic and predictive role of lactate dehydrogenase 5 expression in colorectal cancer patients treated with PTK787/ZK 222584 (vatalanib) antiangiogenic ther-apy. Clin Cancer Res 17(14):4892–4900 Epub 2011/06/03. https://doi.org/10.1158/1078-0432.Ccr-10-2918
Pang X, Wu Y, Wu Y, Lu B, Chen J, Wang J et al (2011) (-)-Gossypol suppresses the growth of human prostate cancer xenografts via modulating VEGF signaling-mediated angiogenesis. Mol Cancer Ther 10(5):795–805 Epub 2011/03/05. https://doi.org/10.1158/1535-7163.Mct-10-0936
El-Sisi AE, Sokar SS, Abu-Risha SE, El-Mahrouk SR (2017) Oxamate potentiates taxol chemotherapeutic efficacy in experimental-ly-induced solid ehrlich carcinoma (SEC) in mice. Biomed Pharmacother 95:1565–1573 Epub 2017/09/28. https://doi.org/10.1016/j.biopha.2017.09.090
Funding
This study was supported by grants from the National Natural Science Foundation of China (82172597), Zhejiang Provincial Natural Science Foundation (LY22H160027), and Beijing Bethune Charitable Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision, C.Z. and X.X.; writing—original draft preparation, S.X., J.L. and B.L.; writing—review and editing, S.X.; funding acquisition, X.X. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval
The authors declare no conflict of interest. This study did not involve Human Participants and/or Animals. All authors have read and agreed to the published version of the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, S., Liao, J., Liu, B. et al. Aerobic glycolysis of vascular endothelial cells: a novel perspective in cancer therapy. Mol Biol Rep 51, 717 (2024). https://doi.org/10.1007/s11033-024-09588-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09588-1