
Abstract
Peanut is an important source of edible vegetable oils worldwide. Saturated fatty acid content is a key factor that affects the quality of peanut oil; thus, reducing its content is a key objective in peanut breeding. In this study, we constructed a dense genetic map of recombinant inbred line (RIL) population (Zhonghua 10 × ICG 12625) with 1443 SSR loci, covering a total length of 2279.10 cM. Using phenotypic data from three environments, we have identified 59 additive QTLs including eight stable ones and nine epistatic QTLs for various fatty acids. Combination of conditional and unconditional QTL analyses further revealed that there was a high genetic correlation between stearic acid and very long-chain saturated fatty acids. In addition, four QTL clusters (CLB04-1, CLB04-2, CLB04-3, and CLB06) harboring major and stable QTLs for saturated fatty acids were identified, which explained 10.77–36.73%, 35.10–41.89%, 10.79–39.18%, and 4.85–20.32% phenotypic variations, respectively. The physical interval of four QTL clusters was located at pseudomolecule B04 and B06 of Arachis ipaensis, containing putative genes that encode enzymes and transcription factors involved in fatty acid biosynthesis pathways. This study comprehensively analyzed the genetic components that controlled saturated fatty acid profiles and identified four QTL clusters containing stable QTLs with large effects. Our results could provide a platform for further gene discovery and guide the development of markers for genome-wide assisted selection of varieties with desired components of saturated fatty acids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Peanut (Arachis hypogaea L., 2n = 4x = 40) is one of the most important oilseed crops, that is widely cultivated in over 100 countries. In 2016, global production of peanut was 43.98 m tons (in the shell) harvested from an area of 27.66 m ha (FAO 2016). Peanut seeds contain approximately 50% oil that has been used as edible oil. The annual global consumption of peanut oil was more than 5.50 m tons (USDA 2017).
Peanut oil is composed of eight fatty acids, i.e., palmitic acid (C16:0), stearic acid (C18:0), oleic acid (C18:1), linoleic acid (C18:2), arachidic acid (C20:0), gadoleic acid (C20:1), behenic acid (C22:0), and lignoceric acid (C24:0). Unsaturated fatty acids, such as oleic acid and linoleic acid, accounted for 80% of peanut oil, influencing oil quality (Pandey et al. 2014; Rizzo et al. 1986; Vassiliou et al. 2009). Another unsaturated fatty acid (gadoleic acid) accounted for a tiny proportion of peanut oil. Remaining 20% of fatty acids are saturated fatty acids that have been proved to be essential for the nutrient value of peanut oil (Wang et al. 2015). Palmitic acid (saturated fatty acid) has been reported to impair insulin sensitivity and increase risks of atrial fibrillation (Fretts et al. 2014; Kato et al. 2008). Other saturated fatty acids, such as stearic acid, arachidic acid, behenic acid, and lignoceric acid, have also been associated with high risk of cardiovascular and multiple sclerosis diseases (Fretts et al. 2014; Hon et al. 2009; Siri-Tarino et al. 2010). Therefore, it is imperative to manipulate recombination of alleles associated with contents of saturated fatty acids to achieve optimized fatty acid composition in peanut.
Various studies have been conducted to elucidate fatty acid biosynthetic pathway in many crops (Barker et al. 2007; Basnet et al. 2016; Ha et al. 2014; Montoya et al. 2013; Singh et al. 2013). In peanut, two mutants of ahFAD2 genes were isolated and characterized from genotypes with high oleic/linoleic acid (O/L) ratio, showing that ahFAD2 genes encode enzymes to facilitate the conversion of oleic acid into linoleic acid (López et al. 2000). Later, two different genetic maps with 206 loci for S-population (SunOleic 97R × NC94022) and 378 loci for T-population (Tifrunner ×GT-C20) were constructed to evaluate the contribution of FAD2 genes/loci in oil quality through QTL analyses (Pandey et al. 2014). Using a high-density genetic map based on SNP and SSR markers, large effect of FAD2 genes/loci on contents of oleic acid and linoleic acid has also been estimated again in different genetic background (Hu et al. 2018). Except for FAD2 genes/loci, four QTLs for oleic acid, linoleic acid, and O/L ratio with 1.4–9.7% phenotypic variation explained (PVE) were identified using a genetic map with 45 loci (Sarvamangala et al. 2011). Further, QTL analysis for saturated fatty acids and gadoleic acid was performed in two RIL populations (SunOleic 97R × NC94022 and Tifrunner ×GT-C20), and 164 additives and 27 epistatic QTLs were identified for six fatty acids (Wang et al. 2015). Based on the RIL population (Zhonghua 10 × ICG12625), our lab previously constructed a genetic map with 470 SSR markers. In total, ten QTLs for six fatty acids with 1.72–20.20% PVE have been detected in a single environment (Huang et al. 2015). Recently, a dense genetic map with 1435 loci was constructed to explore the loci associated with saturated and unsaturated fatty acids using phenotypic data from an F2 population. In total, 21 QTLs with 7.6–78.6% PVE were identified in a single environment (Shasidhar et al. 2017). Except for linkage analysis, association analysis for fatty acids was conducted in the mutant population consisted of 50 genotypes. In total, 12 loci associated with eight fatty acids have been identified (Hake et al. 2017). Although various QTLs have been identified for fatty acids in previous studies, only two FAD2 genes/loci showed large and stable effects for two unsaturated fatty acids (oleic acid and linoleic acid). However, large and stable QTLs with tightly linked markers are still rare for saturated fatty acids in peanut.
In this study, we constructed a highly dense and quality map with 1443 SSR loci to explore stable QTLs controlling contents of saturated fatty acids across multiple environments. The markers tightly linked to stable QTLs would be potentially used in genome-assisted breeding for the desirable saturated fatty acid profile in peanut.
Materials and methods
Development of mapping population
In this study, a mapping population with 140 inbred lines was developed from a cross between Zhonghua 10 and ICG12625 using single seed descent method at the Oil Crops Research Institute of Chinese Academy of Agricultural Sciences (OCRI-CAAS), Wuhan, China. The female parent, Zhonghua 10 (A. hypogaea var. vulgaris), was developed by OCRI-CAAS and the male parent, ICG12625 (A. hypogaea var. aequatoriana), was a germplasm line from International Crop Research Institute for Semi-Arid Tropic (ICRISAT), India. Both parents contained wild-type alleles of FAD2A and FAD2B. The F7 generation of RIL population was utilized for generating genotypic data, and F8–F10 generations were used for analysis of fatty acids.
Phenotyping of fatty acids in the mapping population
RIL population and two parental lines were cultivated in experimental fields with two replications in Wuhan, China, during 2015–2017. The field trial in each year was considered as one environment. Thus, three trials were designated as three environments named as Wuhan2015, Wuhan2016, and Wuhan2017. The peanut diversity panel consisting of 99 accessions from Chinese peanut mini-mini core collection was planted in the experimental fields in 2016, Wuhan. The seeds of RIL lines and two parents were harvested and used for chemical analysis of oleic acid (C18:1), linoleic acid (C18:2), palmitic acid (C16:0), stearic acid (C18:0), arachidic acid (C20:0), behenic acid (C22:0), and lignoceric acid (C24:0).
Fatty acid profile was generated as described previously (Pandey et al. 2014). Approximately 20 mg of finely grounded seed tissue was used for extraction with 1 mL petroleum ether. Then 400 μL of 0.5 mol/L sodium methoxide (NaOCH3) in methanol solution was added to the tube to convert fatty acids to methyl esters. After 1 h, the organic layer containing the methyl esters was collected and subsequently transferred to an autosampler vial for gas chromatograph analysis. An Agilent 7890B gas chromatograph equipped with a flame ionization detector (FID) was used to analyze the fatty acid profile of the samples (Agilent Technologies, USA). Peak separation was carried out on DB-23 capillary column (30 m × 0.25 mm I.D., 0.25 μm film thickness, Agilent Technologies, USA). At split ratio of 60:1, 1 μL of sample was injected into the column at 220 °C isothermally. The temperature of the inlet and detector were set at 260 °C and 280 °C, respectively. Nitrogen was used as carrier gas at a flow rate of ~ 1 mL/min. Fatty acid composition was determined by calculating relative peak areas.
Phenotypic data analysis
Statistical analyses for phenotyping data of fatty acid composition in three environments were performed using IBM SPSS Statistics software (version 21). Analysis of variance was performed to evaluate the significant difference among RILs, environments, and RILs × environments interaction. Correlation analysis among different fatty acids was also performed on IBM SPSS Statistics software (version 21).
Genotyping of RILs and construction of an improved genetic map
Genomic DNA was extracted from young leaves using a modified CTAB method. PCR was performed according to the previous study in our lab (Luo et al. 2017b). Total 2240 SSR markers developed from genomes of A. duranensis and A. ipaensis were utilized to screen polymorphism between two parents (Luo et al. 2017c). Further, polymorphic SSR markers were used to genotype the RIL population to improve the previous genetic map (Huang et al. 2016). The improved genetic map was constructed using JoinMap software with minimum logarithm of odds (LOD) of 4.0 (Stam 1993). Kosambi mapping function was used to transform recombinant ratio into genetic distance. Linkage groups (LGs) were assigned to the chromosome through aligning to the integrated consensus genetic map (Shirasawa et al. 2013) and genome sequences of A. duranensis and A. ipaensis (Bertioli et al. 2016). Pearson’s chi-squared test was carried out to assess the goodness of fit to expected segregation ratio 1:1 for each locus (P < 0.05).
QTL analysis of fatty acids
The improved genetic map along with phenotyping data of saturated fatty acid contents was used to identify QTLs. IciMapping was performed to detect additive QTLs (Meng et al. 2015), and QTLNetwork was carried out to identify additive and epistatic QTLs (Yang et al. 2008). For IciMapping software, inclusive composite interval mapping method with 1D scanning was employed in additive QTL analysis. The mapping parameters were set as 1.0 cM step, 0.001 probability, and LOD threshold score was 3.0. Mixed liner model and 2D genome scan from QTLNetwork were used to identify additive and epistatic QTLs. Thresholds of QTL detection were calculated by 1000 permutations, and Monte Carlo Markov chain method with 20,000 Gibbs sample size was used to estimate QTL effect. The parameters, such as walk speed and testing window size, were set at 1.0 cM and 10 cM, respectively. In addition, conditional phenotypic values, i.e., y(C20:0|C18:0), y(C22:0|C18:0), and y(C24:0|C18:0), generated by QGA Station software (Zhu 1995) were utilized to identify conditional QTLs using QTLNetwork. C20:0|C18:0, C22:0|C18:0, and C24:0|C18:0 represent the traits of arachidic acid, behenic acid, and lignoceric acid, respectively, conditioning on the trait of stearic acid.
Annotation of putative genes in physical intervals of four QTL clusters
Markers linked to QTL clusters were aligned on the genome of A. ipaensis to locate physical interval of four QTL clusters. Sequences and annotations of putative genes in physical intervals were downloaded from PeanutBase database (Bertioli et al. 2016). Lipid metabolism-related genes and transcription factor genes were obtained from previous studies (Bates et al. 2014; Wilson et al. 2008) and searched against homologous genes in Arachis duranensis (A. duranensis) and Arachis ipaensis (A. ipaensis) using Blast tool with an E value cutoff of 1e−5 (https://blast.ncbi.nlm.nih.gov).
Results
Phenotypic variation of fatty acids in peanut
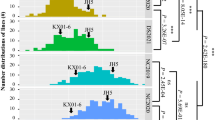
Significant phenotypic variations of fatty acid composition were observed in the RIL population and two parents across three environments (Table S1). Continuous distributions with transgressive segregation of phenotypic values for seven fatty acids in RILs are presented in Fig. 1. Further, the Shapiro-Wilk test demonstrated that phenotypic data were normally distributed for palmitic acid, oleic acid, linoleic acid, behenic acid, and lignoceric acid; however, they were skewed towards lower values for stearic acid and arachidic acid (Table S1, Fig. 1). Variance analyses of three environments indicated that genetic and environmental effects significantly influenced the phenotypes of seven fatty acids (P < 0.05) (Table S2).

Phenotypic distribution of seven fatty acids in the RIL population. x-axis represented average percentage range of 3 years (2015 to 2017) of fatty acids and y-axis showed the number of individuals in the RIL population. Red and blue arrows indicated phenotype of female and male parent, respectively
Correlation analysis demonstrated that palmitic acid was negatively correlated with stearic acid (r = − 0.485, P < 0.001), oleic acid (r = − 0.599, P < 0.001), and arachidic acid (r = − 0.522, P < 0.001). However, it was positively correlated with linoleic acid (r = 0.562, P < 0.001). Similarly, stearic acid was positively correlated with arachidic acid (r = 0.903, P < 0.001) and arachidic acid was positively correlated with behenic acid (r = 0.540, P < 0.001). Further, lignoceric acid was also positively correlated with behenic acid (r = 0.581, P < 0.001) (Table S3). Thus, five saturated fatty acids were found to be correlated with each other.
Improved genetic map construction
The RIL population derived from the cross of Zhonghua 10 and ICG 12625 has been previously used to construct a genetic map with 1272 loci (Huang et al. 2016). In this study, additional polymorphic SSR markers developed from genomes of A. duranensis and A. ipaensis (Luo et al. 2017c) were utilized to improve the genetic map. Finally, 1399 polymorphic SSR markers generated 1443 loci for the RIL population. One SSR marker amplified three loci, 42 markers amplified two loci, and the remaining 1356 markers amplified a single locus. Of 1443 genetic loci, 1387 loci were co-dominant, while others were dominant. The improved genetic map of 1443 loci covered a total length of 2279.10 cM with an average inter-marker distance of 1.58 cM (Table 1). All loci were grouped into 20 LGs, designated as A01–A10 for A subgenome and B01–B10 for B subgenome through alignment of the markers to the integrated consensus genetic map (Shirasawa et al. 2013) and genome of A. duranensis and A. ipaensis (Bertioli et al. 2016). The number of mapped loci varied from 30 (A06) to 106 (A05) and length of LGs ranged from 70.88 cM (B05) to 164.47 cM (A02). In total, 687 loci with a length of 1213.62 cM were assigned to A subgenome and 756 loci with a length of 1065.48 cm were assigned to B subgenome (Table 1, Fig. S1). Chi-squared analysis indicated that 154 loci (10.67%) exhibited significantly distorted segregation (P < 0.05). Of the 154 loci, 110 skewed towards Zhonghua 10 and remaining 44 loci skewed towards ICG 12625. The percent of skewed loci in LGs varied from 1.89% (A05) to 36.96% (A03). The skewed loci in A03, A09, B07, and B10 favored Zhonghua 10 allele, while the skewed loci in A02 and B01 favored ICG 12625. Besides, there were 10 LGs that harbored less than five skewed loci (Table 1, Table S4).
Identification of additive QTLs for seven fatty acids
Genotype and phenotype data of the RIL population across three environments (Wuhan2015, Wuhan2016, and Wuhan2017) were used to perform genome-wide QTL analysis for seven fatty acids. Results showed that there were 18 additive QTLs on four LGs and 41 additive QTLs on nine LGs for seven fatty acids detected by QTLNetwork and IciMapping, respectively (Table 2). In total of 59 QTLs, 10 QTLs for palmitic acid, 11 QTLs for stearic acid, four QTLs for oleic acid, three QTLs for linoleic acid, 13 QTLs for arachidic acid, 10 QTLs for behenic acid, and eight QTLs for lignoceric acid showed corresponding phenotypic variations of 7.68–21.52%, 4.68–41.89%, 9.28–21.53%, 10.61–16.42%, 3.63–36.90%, 7.90–19.26%, and 6.54–43.44%, respectively. Among the additive QTLs, eight QTLs for four saturated fatty acids (one for palmitic acid, three for stearic acid, three for arachidic acid, and one for behenic acid) were repeatedly detected in multiple environments. In particular, four major and stable QTLs located on LG B04 explained phenotypic variations of 13.84–41.89% for stearic acid, arachidic acid, and behenic acid in different environments. In addition, two stable QTLs for stearic acid and arachidic acid were mapped on LG B06. Although there were nine LGs harboring 52 additive QTLs for saturated fatty acids, LG B04 held the highest number of 23 QTLs followed by 12 QTLs on LG B06. It indicated that chromosomes B04 and B06 are rich in genes controlling contents of saturated fatty acids.
Detection of epistatic QTLs for fatty acid in the RIL population
Epistatic QTL analysis using QTLNetwork resulted in detection of nine epistatic QTLs (E-QTLs) with two-locus interactions for palmitic acid (1), stearic acid (2), oleic acid (2), linoleic acid (1), and arachidic acid (3) (Table S5, Fig. 2). The effect of additive by additive interaction varied from − 0.37 to 1.11 and the PVE of two-locus interaction ranged from 0.87 to 9.44%. Besides, one epistasis-by-environment interaction with 1.64% PVE was detected for oleic acid. It was observed that nine two-locus interactions were found to be mapped on eight LGs, and four interactions with positive effect for saturated fatty acids were observed between B04 and B06. In particular, one epistatic locus between two markers (AGGS1854 and AHGS1943) on LG B06 was able to interact with three different loci on LG B04 (two for arachidic acid and one for stearic acid). In addition, there were nine epistatic loci which did not have individual additive effect but showed epistasis effect on interaction with other loci, suggesting that these loci might affect fatty acid contents by epistatic interaction and not by individual effect.

Diagram of epistatic interaction for multiple fatty acids. a–e represent palmitic acid, stearic acid, oleic acid, linoleic acid, and arachidic acid, respectively. Red balls indicated epistatic QTL with individual effect; black balls represented epistatic QTLs without individual effect. Red lines indicated two loci have additive × additive interaction, and blue dashed line indicates two loci have additive × (additive × environment) interaction
Localization of QTL clusters on chromosomes B04 and B06
After plotting QTLs on LGs, it was observed that many QTLs for saturated fatty acids clustered together. And four QTL clusters (CLB04-1, CLB04-2, CLB04-3, and CLB06) were found to be mapped on LG B04 and B06 (Table 2, Fig. 3). CLB04-1 was located between AHGS2017 and AGGS1601 on LG B04, which contained six QTLs with 10.77–36.73% PVE for palmitic acid, stearic acid, arachidic acid, and behenic acid. CLB04-2 was found to be in between Ai04B9397 and AHGS2100 harboring two major and stable QTLs for stearic acid and arachidic acid explaining 35.10–41.89% of phenotypic variations. CLB04-3, located between HAS0556 and AGGS1236, consisted of 12 QTLs for five saturated fatty acids, including two stable and major QTLs with 10.79–39.18% PVE. CLB06 located between Ai06B271120 and Ai06B28485 on LG B06 contained six QTLs with 4.85–20.32% PVE, including two stable QTLs for stearic acid, arachidic acid, and behenic acid. Altogether, four QTL clusters harbored half of the total additive QTLs for five saturated fatty acids (26 of 52), including six stable QTLs with up to 41.89% PVE for stearic acid, arachidic acid, and behenic acid.

Position of QTL clusters on physical maps. a–d represent QTL clusters CLB04-1, CLB04-2, CLB04-3, and CLB06, respectively. Left bars represented genetic maps and right bars indicated corresponding physical maps. Boxes with dashed line indicated clusters of QTLs for different saturated fatty acids. The markers linked to QTL clusters are highlighted in red color
After mapping the linked markers into the genome of A. ipaensis, it was observed that the QTL clusters, CLB04-1, CLB04-2, CLB04-3, and CLB06, were located at 105.0–107.2 Mb, 27.4–31.7 Mb, 12.8–20.7 Mb of pseudomolecule B04, and 115.4–121.1 Mb of pseudomolecule B06, containing 112, 83, 381, and 294 putative genes, respectively (Fig. 3). Gene ontology (GO) analysis indicated that binding and catalytic activities were most frequently observed among various molecular functions in four QTL clusters. Among all biological processes, cellular process and metabolic process were most frequently presented (Table S6 and Fig. S2). A total of 20 genes were found in four QTL clusters, which were homologous to the genes involved in lipid metabolic pathways. These pathways include plastidial fatty acid synthesis, TAG synthesis, plastidial glycerolipid, galactolipid, and sulfolipid synthesis, cuticular wax synthesis, lipid trafficking, beta-oxidation, lipase, eukaryotic phospholipid synthesis, mitochondrial fatty acid, and lipoic acid synthesis (Table S6). In addition, 41 genes in four QTL clusters were annotated as transcription factors, belonging to 12 families (AP2, B3, MYB, WRKY, LBD, CAMTA, FAR1, ARR-B, ERF, bHLH, NAC, and MYB) (Table S6). It was noteworthy that several members in MYB, B3, and AP2 transcription factor families could regulate fatty acid synthesis, fatty acid elongation, and TAG synthesis (Raffaele et al. 2008; Seo et al. 2011; To et al. 2012).
Validation of four QTL clusters for saturated fatty acids in peanut diversity panel
Since four QTL clusters were found to harbor major and stable QTLs for saturated fatty acids, 99 accessions from Chinese peanut mini-mini core collection were employed to verify that these loci are associated with the content of saturated fatty acids in different genetic background. Fatty acid composition of 99 accessions is presented in Table S7. Genotyping of 554 SSR markers in 99 accessions with Zhonghua 10 and ICG12625 has been reported previously (Zhao et al. 2017). SSR markers, such as AHGS1579, AHGS0288, AHGA17736, AGGS1039 adjoining to CLB04-1, CLB04-2, CLB04-3, and CLB06, respectively, and marker AGGS0186 within CLB04-3, were found to be polymorphic in 99 accessions (Fig. 2, Table S7). Accessions containing Zhonghua 10 allele of AHGS1579 were significantly different from accessions containing ICG12625 allele of AHGS1579 in terms of palmitic acid, stearic acid, arachidic acid, and behenic acid (Table S8). Moreover, stearic acid and arachidic acid were found to be significantly different between accessions with Zhonghua 10 allele of AHGS0288 and accessions with ICG12625 allele of AHGS0288. For marker AHGA17736, accessions containing Zhonghua 10 allele were significantly different from accessions with ICG12625 allele in terms of palmitic acid, stearic acid, and arachidic acid content. In addition, for marker AGGS0186 within CLB04-3, behenic acid and lignoceric acid in accessions with Zhonghua 10 allele were significantly lower than that of accessions with ICG12625 allele (Table S8). For marker AGGS1039, stearic acid and arachidic acid in accessions containing Zhonghua 10 allele were significantly higher than that of accessions with ICG12625 allele (Table S8).
Conditional QTL analysis
Results of unconditional QTL analysis showed that additive QTLs for stearic acid clustered together with QTLs for arachidic acid, behenic acid, and lignoceric acid (Fig. 3). To investigate the role of stearic acid on very long-chain saturated fatty acids at QTL level, we conducted conditional QTL analysis using conditional phenotypic values y(C20:0|C18:0), y(C22:0|C18:0), and y(C24:0|C18:0). Six conditional QTLs with 7.98–11.97% PVE were identified, including three additional QTLs for arachidic acid and behenic acid (only detected in conditional QTL analysis) (Table S9).
Although two additional QTLs with 7.98–11.09% PVE for arachidic acid were detected in conditional QTL mapping, five QTLs, including those with 32.79–36.90% PVE, were missing when compared with unconditional QTL analysis (Table 2, Table S9). Similarly, QTL analysis for behenic acid conditioned on stearic acid showed one additional QTL on LG A06; however, there was a loss of two QTLs on LG B04 identified in unconditional QTL analysis. Nevertheless, it did not influence the QTLs for lignoceric acid irrespective of QTL analyses conditioned on stearic acid or not. These results suggested that stearic acid exhibited significant influence on detection of QTLs for very long-chain saturated fatty acids, except for lignoceric acid.
Discussion
Identification of QTLs for target trait and development of linked markers are prerequisite for genomics-assisted breeding in peanut (Janila et al. 2016). The efficacy and resolution of QTL mapping greatly depend on marker density of genetic map (Asins 2002). In 2009, first genetic map based on SSRs was constructed with 135 loci in peanut (Varshney et al. 2009). Since SSRs are abundantly found in genome, many researchers have developed SSR markers and used them to construct genetic map for QTL analysis (Chen et al. 2016; Cuc et al. 2008; Gautami et al. 2012; Huang et al. 2015; Li et al. 2016; Ferguson et al. 2004; Qin et al. 2012; Shirasawa et al. 2012; Sujay et al. 2012; Zhang et al. 2012). In this study, more than 2000 SSR markers developed from A. duranensis and A. ipaensis genomes were screened (Luo et al. 2017c), and polymorphic markers between parents and RILs were used to improve previously constructed genetic map of the RIL population derived from Zhonghua 10 × ICG 12625. In comparison with the previous genetic map of same RIL population (Huang et al. 2016), total loci and length of linkage groups of improved genetic map in this study increased from 1219 to 1443 and 2038.75 to 2279.10 cM, respectively. As a result, the final average marker interval of improved genetic map decreased to 1.58 cm. In this study, it is worth mentioning that additional SSR markers with known genomic position were selected to specifically increase density of LGs with fewer loci in previous genetic map, such as A02 (from 41 to 69) and B08 (from 45 to 76). The number of loci and density of genetic map in this study were higher than that of recent studies (Chen et al. 2017; Li et al. 2017; Luo et al. 2017a; Luo et al. 2017c) with the exception for integrated consensus map (Shirasawa et al. 2013). Moreover, almost 90% of mapped loci segregated in RILs at the expected ratio of 1:1, which was also relatively higher than other studies (Chen et al. 2017; Luo et al. 2017a; Luo et al. 2017c; Shirasawa et al. 2013). Therefore, it can be ascertained that a high-quality and dense genetic map was generated in this study. Using the same RIL population, our lab previously constructed a genetic map with 470 loci to identify QTLs for fatty acids (Huang et al. 2015). Ten QTLs, including four major QTLs with less than 21% PVE, were mapped on the genetic map for six fatty acids. Intervals of QTLs varied from 2.4 to 15.1 cm, with average length reaching up to 6.92 cM. In this study, more major QTLs (41 of 59) were identified for seven fatty acids. Moreover, average length of QTL interval decreased to 2.07 cM when compared with that in previous study (Table 2). Thus, it can be concluded that high density of genetic map improved the resolution and efficacy of QTL mapping.
QTL mapping on the dense genetic map revealed four QTL clusters on LG B04 and LG B06 for saturated fatty acids (Table 2, Fig. 3). Three QTL clusters (CLB04-1, CLB04-2, and CLB04-3) on LG B04 contained 20 major QTLs, including four stable ones in multiple environments, showing phenotypic variations of 10.77–41.89% for palmitic acid, stearic acid, arachidic acid, behenic acid, and lignoceric acid. A 36.4-cM interval harboring large effect QTLs for saturated fatty acids has been identified in previous study (Wang et al. 2015). The physical region of this interval was overlapped with CLB04-2 and CLB04-3 at chromosome B04, indicating that chromosome B04 is likely to contain key loci mainly controlling fatty acid profile. Another QTL cluster CLB06 harbored six QTLs, including two stable QTLs with up to 20.32% PVE for three saturated fatty acids (stearic acid, arachidic acid, and behenic acid). Moreover, CLB06 was located at 115.4–121.1 Mb of pseudomolecule B06, on which few QTLs for saturated fatty acids (stearic acid, arachidic acid, and behenic acid) have been previously mapped (Huang et al. 2015; Pandey et al. 2014; Shasidhar et al. 2017; Wang et al. 2015). These results indicated that CLB06 was a novel and reliable QTL cluster for various saturated fatty acids. To confirm that these loci are associated with saturated fatty acid content in different genetic backgrounds, markers within or adjoining to QTL clusters were screened in 99 accessions of Chinese peanut mini-mini core collection. Genotypes of five markers from four QTL clusters were found to be polymorphic in 99 accessions. After classifying 99 accessions based on these five markers, significant phenotypic differences between accessions with Zhonghua 10 allele and accessions with ICG12625 allele were observed in different saturated fatty acids (Fig. 3; Tables S7 and S8). These results indicated that four QTL clusters harboring major and stable QTLs could potentially improve saturated fatty acid profile through selecting the markers linked to the cluster in genome-wide assisted breeding.
Moreover, targeting the four QTL clusters provided an opportunity to explore key loci or genes regulating fatty acid biosynthesis pathway in peanut. The physical intervals of four QTL clusters in genome of Arachis ipaensis contained 20 putative genes that are involved in 11 lipid metabolic pathways (Table S6). Of these genes, Araip.RI4NB encodes stearoyl-ACP desaturase, which could directly transform stearic acid to oleic acid. Mutation of homologous gene (AT1G43800.1) in Arabidopsis led to higher accumulation of stearic acid with lower content of oleic acid (Jin et al. 2017). In addition, 41 genes in the four clusters were grouped into 12 transcription factor families (Table S6). Among them, Araip.L6IES is a member of AP2 transcription factor family. The homolog of Araip.L6IES is WRI1 in Arabidopsis, which is known to regulate fatty acid synthesis, fatty acid elongation, and triacylglycerol synthesis (To et al. 2012). There are another five genes in four QTL clusters, such as Araip.H0SLK, Araip.VFC9U, Araip.P03KX, and Araip.T0DQ5, encoding MYB transcript factors. Several members of MYB family in Arabidopsis have been reported to regulate acyl-CoA elongase complex-related genes to control the synthesis of very long-chain saturated fatty acids (Raffaele et al. 2008; Seo et al. 2011). Therefore, these transcription factors located in QTL clusters could affect the content of various fatty acids that are in the same biosynthesis pathway. However, this needs to be further confirmed.
In this study, a high-density genetic map with 1443 loci was constructed to explore additive and epistatic QTLs for comprehensive evaluation of genetic components that control contents of seven fatty acids. Four QTL clusters harboring several major and stable QTLs were identified, which could explain up to 41.89% of phenotypic variation for saturated fatty acids (palmitic acid, stearic acid, arachidic acid, behenic acid, and lignoceric acid). Targeting these QTL clusters for future studies would help understand the fatty acid metabolism in peanut and provide diagnostic markers for improvement of cultivars with desired fatty acid composition.
References
Asins MJ (2002) Present and future of quantitative trait locus analysis in plant breeding. Plant Breed 121:281–291
Barker GC, Larson TR, Graham IA, Lynn JR, King GJ (2007) Novel insights into seed fatty acid synthesis and modification pathways from genetic diversity and quantitative trait loci analysis of the Brassica C genome. Plant Physiol 144:1827–1842
Basnet RK, Del Carpio DP, Xiao D et al (2016) A systems genetics approach identifies gene regulatory networks associated with fatty acid composition in Brassica rapa seed. Plant Physiol 170:568–585
Bates PD, Johnson SR, Cao X, Li J, Nam JW, Jaworski JG, Ohlrogge JB, Browse J (2014) Fatty acid synthesis is inhibited by inefficient utilization of unusual fatty acids for glycerolipid assembly. Proc Natl Acad Sci U S A 111:1204–1209
Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EKS, Liu X, Gao D, Clevenger J, Dash S, Ren L, Moretzsohn MC, Shirasawa K, Huang W, Vidigal B, Abernathy B, Chu Y, Niederhuth CE, Umale P, Araújo ACG, Kozik A, Do Kim K, Burow MD, Varshney RK, Wang X, Zhang X, Barkley N, Guimarães PM, Isobe S, Guo B, Liao B, Stalker HT, Schmitz RJ, Scheffler BE, Leal-Bertioli SCM, Xun X, Jackson SA, Michelmore R, Ozias-Akins P (2016) The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet 48:438–446
Chen W, Jiao Y, Cheng L, Huang L, Liao B, Mei T, Ren X, Zhou X, Chen Y, Jiang H (2016) Quantitative trait locus analysis for pod- and kernel-related traits in the cultivated peanut (Arachis hypogaea L.). BMC Genet 17:1–9
Chen Y, Ren X, Zheng Y, Zhou X, Huang L, Yan L, Jiao Y, Chen W, Huang S, Wan L, Lei Y, Liao B, Huai D, Wei W, Jiang H (2017) Genetic mapping of yield traits using RIL population derived from Fuchuan Dahuasheng and ICG6375 of peanut (Arachis hypogaea L.). Mol Breed 37:17
Cuc LM, Mace ES, Crouch JH, Quang VD, Long TD, Varshney RK (2008) Solation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol 8:1–11
Ferguson ME, Burow MD, Schulze SR, Bramel PJ, Paterson AH, Kresovich S, Mitchell S (2004) Microsatellite identification and characterization in peanut (A. hypogaea L.). Theor Appl Genet 108:1064–1070
Fretts AM, Mozaffarian D, Siscovick DS, Djousse L, Heckbert SR, King IB, Mcknight B, Sitlani C, Sacks FM, Song X (2014) Plasma phospholipid saturated fatty acids and incident atrial fibrillation: the cardiovascular health study. J Am Heart Assoc 3:e000889
Gautami B, Foncéka D, Pandey MK, Moretzsohn MC, Sujay V, Qin H, Hong Y, Faye I, Chen X, Bhanuprakash A (2012) An international reference consensus genetic map with 897 marker loci based on 11 mapping populations for tetraploid groundnut (Arachis hypogaea L.). PLoS One 7:e41213
Ha BK, Kim HJ, Velusamy V, Vuong TD, Nguyen HT, Shannon JG, Lee JD (2014) Identification of quantitative trait loci controlling linolenic acid concentration in PI483463 (Glycine soja). Theor Appl Genet 127:1501–1512
Hake AA, Shirasawa K, Yadawad A, Nayak SN, Mondal S, Badigannavar AM, Nadaf HL, Gowda MVC, Bhat RS (2017) Identification of transposable element markers associated with yield and quality traits from an association panel of independent mutants in peanut (Arachis hypogaea L.). Euphytica 213(12):283
Hon GM, Hassan MS, Rensburg SJV, Abel S, Erasmus RT, Matsha T (2009) Membrane saturated fatty acids and disease progression in multiple sclerosis patients. Metab Brain Dis 24:561–568
Hu XH, Zhang SZ, Miao HR, Cui FG, Shen Y, Yang WQ, Xu TT, Chen N, Chi XY, Zhang ZM, Chen J (2018) High-density genetic map construction and identification of QTLs controlling oleic and linoleic acid in peanut using SLAF-seq and SSRs. Sci Rep 8(1):5479
Huang L, He H, Chen W, Ren X, Chen Y, Zhou X, Xia Y, Wang X, Jiang X, Liao B (2015) Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (Arachis hypogaea L.). Theor Appl Genet 128:1103–1115
Huang L, Ren X, Wu B, Li X, Chen W, Zhou X, Chen Y, Pandey MK, Jiao Y, Luo H, Lei Y, Varshney RK, Liao B, Jiang H (2016) Development and deployment of a high-density linkage map identified quantitative trait loci for plant height in peanut (Arachis hypogaea L.). Sci Rep 6:39478
Janila P, Variath MT, Pandey MK et al (2016) Genomic tools in groundnut breeding program: status and perspectives. Front Plant Sci 7:289
Jin C, Li D, Gao C, Liu K, Qi S, Duan S, Li Z, Gong J, Wang J, Hai J, Chen M (2017) Conserved function of ACYL–ACYL carrier protein desaturase 5 on seed oil and oleic acid biosynthesis between Arabidopsis thaliana and Brassica napus. Front Plant Sci 8:1319
Kato T, Shimano H, Yamamoto T, Ishikawa M, Kumadaki S, Matsuzaka T, Nakagawa Y, Yahagi N, Nakakuki M, Hasty AH, Takeuchi Y, Kobayashi K, Takahashi A, Yatoh S, Suzuki H, Sone H, Yamada N (2008) Palmitate impairs and eicosapentaenoate restores insulin secretion through regulation of srebp-1c in pancreatic islets. Diabetes 57(9):2382–2392
Li H, Wu B, Zhao J, Li H, Chen W, Zheng Y, Ren X, Chen Y, Zhou X, Lei Y (2016) Characterization and transferable utility of microsatellite markers in the wild and cultivated Arachis species. PLoS One 11:e0156633
Li Y, Li L, Zhang X, Zhang K, Ma D, Liu J, Wang X, Liu F, Wan Y (2017) QTL mapping and marker analysis of main stem height and the first lateral branch length in peanut (Arachis hypogaea L.). Euphytica 213:57
López Y, Nadaf HL, Smith OD, Connell JP, Reddy AS, Fritz AK (2000) Isolation and characterization of the Δ12-fatty acid desaturase in peanut (Arachis hypogaea L.) and search for polymorphisms for the high oleate trait in Spanish market-type lines. Theor Appl Genet 101:1131–1138
Luo H, Guo J, Ren X, Chen W, Huang L, Zhou X, Chen Y, Liu N, Xiong F, Lei Y, Liao B, Jiang H (2017a) Chromosomes A07 and A05 associated with stable and major QTLs for pod weight and size in cultivated peanut (Arachis hypogaea L.). Theor Appl Genet 131:267–282. https://doi.org/10.1007/s00122-017-3000-7
Luo H, Ren X, Li Z, Xu Z, Li X, Huang L, Zhou X, Chen Y, Chen W, Lei Y, Liao B, Pandey MK, Varshney RK, Guo B, Jiang X, Liu F, Jiang H (2017b) Co-localization of major quantitative trait loci for pod size and weight to a 3.7 cM interval on chromosome A05 in cultivated peanut (Arachis hypogaea L.). BMC Genomics 18:58
Luo H, Xu Z, Li Z, Li X, Lv J, Ren X, Huang L, Zhou X, Chen Y, Yu J, Chen W, Lei Y, Liao B, Jiang H (2017c) Development of SSR markers and identification of major quantitative trait loci controlling shelling percentage in cultivated peanut (Arachis hypogaea L.). Theor Appl Genet 130:1635–1648
Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3:269–283
Montoya C, Lopes R, Flori A, Cros D, Cuellar T, Summo M, Espeout S, Rivallan R, Risterucci A-M, Bittencourt D (2013) Quantitative trait loci (QTLs) analysis of palm oil fatty acid composition in an interspecific pseudo-backcross from Elaeis oleifera (H.B.K.) Cortés and oil palm (Elaeis guineensis Jacq.). Tree Genet Genomes 9:1207–1225
Pandey MK, Wang ML, Qiao L, Feng S, Khera P, Wang H, Tonnis B, Barkley NA, Wang J, Holbrook CC, Culbreath AK, Varshney RK, Guo B (2014) Identification of QTLs associated with oil content and mapping FAD2 genes and their relative contribution to oil quality in peanut (Arachis hypogaea L.). BMC Genet 15:133
Qin H, Feng S, Chen C, Guo Y, Knapp S, Culbreath A, He G, Wang ML, Zhang X, Holbrook CC, Ozias-Akins P, Guo B (2012) An integrated genetic linkage map of cultivated peanut (Arachis hypogaea L.) constructed from two RIL populations. Theor Appl Genet 124:653–664
Raffaele S, Vailleau F, Léger A, Joubès J, Miersch O, Huard C, Blée E, Mongrand S, Domergue F, Roby D (2008) A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell 20:752–767
Rizzo WB, Watkins PA, Phillips MW, Cranin D, Campbell B, Avigan J (1986) Adrenoleukodystrophy: oleic acid lowers fibroblast saturated C22-26 fatty acids. Neurology 36:357–361
Sarvamangala C, Gowda MVC, Varshney RK (2011) Identification of quantitative trait loci for protein content, oil content and oil quality for groundnut (Arachis hypogaea L.). Field Crops Res 122:49–59
Seo PJ, Lee SB, Suh MC, Park MJ, Go YS, Park CM (2011) The MYB96 transcription factor regulates cuticular wax biosynthesis under drought conditions in Arabidopsis. Plant Cell 23:1138–1152
Shasidhar Y, Vishwakarma MK, Pandey MK, Janila P, Variath MT, Manohar SS, Nigam SN, Guo B, Varshney RK (2017) Molecular mapping of oil content and fatty acids using dense genetic maps in groundnut (Arachis hypogaea L.). Front Plant Sci 8:794
Shirasawa K, Koilkonda P, Aoki K, Hirakawa H, Tabata S, Watanabe M, Hasegawa M, Kiyoshima H, Suzuki S, Kuwata C, Naito Y, Kuboyama T, Nakaya A, Sasamoto S, Watanabe A, Kato M, Kawashima K, Kishida Y, Kohara M, Kurabayashi A, Takahashi C, Tsuruoka H, Wada T, Isobe S (2012) In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut. BMC Plant Biol 12:80
Shirasawa K, Bertioli DJ, Varshney RK, Moretzsohn MC, Leal-Bertioli SCM, Thudi M, Pandey MK, Rami JF, Fonceka D, Gowda MVC, Qin H, Guo B, Hong Y, Liang X, Hirakawa H, Tabata S, Isobe S (2013) Integrated consensus map of cultivated peanut and wild relatives reveals structures of the A and B genomes of Arachis and divergence of the legume genomes. DNA Res 20:173–184
Singh S, Mohapatra T, Singh R, Hussain Z (2013) Mapping of QTLs for oil content and fatty acid composition in Indian mustard [Brassica juncea (L.) Czern. and Coss.]. J Plant Biochem Biotechnol 22:80–89
Siri-Tarino PW, Qi S, Hu FB, Krauss RM (2010) Saturated fatty acids and risk of coronary heart disease: modulation by replacement nutrients. Curr Atheroscler Rep 12:384–390
Stam P (1993) Construction of integrated genetic linkage maps by means of a new computer package: Join Map. Plant J 3:739–744
Sujay V, Gowda MVC, Pandey MK, Bhat RS, Khedikar YP, Nadaf HL, Gautami B, Sarvamangala C, Lingaraju S, Radhakrishan T, Knapp SJ, Varshney RK (2012) Quantitative trait locus analysis and construction of consensus genetic map for foliar disease resistance based on two recombinant inbred line populations in cultivated groundnut (Arachis hypogaea L.). Mol Breed 30:773–788
To A, Joubès J, Barthole G, Lécureuil A, Scagnelli A, Jasinski S, Lepiniec L, Baud S (2012) WRINKLED transcription factors orchestrate tissue-specific regulation of fatty acid biosynthesis in Arabidopsis. Plant Cell 24:5007–5023
Varshney RK, Bertioli DJ, Moretzsohn MC, Vadez V, Krishnamurthy L, Aruna R, Nigam SN, Moss BJ, Seetha K, Ravi K, He G, Knapp SJ, Hoisington DA (2009) The first SSR-based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor Appl Genet 118:729–739
Vassiliou EK, Gonzalez A, Garcia C, Tadros JH, Chakraborty G, Toney JH (2009) Oleic acid and peanut oil high in oleic acid reverse the inhibitory effect of insulin production of the inflammatory cytokine TNF-α both in vitro and in vivo systems. Lipids Health Dis 8:25
Wang ML, Khera P, Pandey MK, Wang H, Qiao L, Feng S, Tonnis B, Barkley NA, Pinnow D, Holbrook CC, Culbreath AK, Varshney RK, Guo B (2015) Genetic mapping of QTLs controlling fatty acids provided insights into the genetic control of fatty acid synthesis pathway in peanut (Arachis hypogaea L.). PLoS One 10(4):e0119454
Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA (2008) DBD––taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res 36:88–92
Yang J, Hu C, Hu H, Yu R, Xia Z, Ye X, Zhu J (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24:721–723
Zhang J, Shan L, Duan J, Jin W, Chen S, Cheng Z, Qiang Z, Liang X, Li Y (2012) De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 13:90
Zhao J, Huang L, Ren X, Pandey Manish, Wu B, Chen Y, Zhou X, Chen W, Xia Y, Li Z, Luo H, Lei Y, Varshney R, Liao B, Jiang H (2017) Genetic variation and association mapping of seed-related traits in cultivated peanut (Arachis hypogaea L.) using single-locus simple sequence repeat markers. Front Plant Sci 8. https://doi.org/10.3389/fpls.2017.02105
Zhu J (1995) Analysis of conditional genetic effects and variance components in developmental genetics. Genetics 141:1633–1639
Funding
This work was financially supported by the National Natural Science Foundation of China (no. 31471534 and no. 31761143005), the National Peanut Industry Technology System Construction (CARS-13), the Plant Germplasm Resources Sharing Platform (NICGR2017-36), the National Program for Crop Germplasm Protection of China (2017NWB033), and the Fundamental Research Funds for Central Non-profit Scientific Institution (Y2017PT55).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
{kind=link}
Cite this article
Liu, N., Chen, H., Huai, D. et al. Four QTL clusters containing major and stable QTLs for saturated fatty acid contents in a dense genetic map of cultivated peanut (Arachis hypogaea L.). Mol Breeding 39, 23 (2019). https://doi.org/10.1007/s11032-019-0934-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-019-0934-2