
Abstract
We chose an Elaeis interspecific pseudo-backcross of first generation (E. oleifera × E. guineensis) × E. guineensis to identify quantitative trait loci (QTLs) for fatty acid composition of palm oil. A dense microsatellite linkage map of 362 loci spanned 1.485 cM, representing the 16 pairs of homologous chromosomes in the Elaeis genus from which we traced segregating alleles from both E. oleifera and E. guineensis grandparents. The relative linear orders of mapped loci suggested the probable absence of chromosome rearrangements between the E. oleifera and E. guineensis genomes. A total of 19 QTL associated to the palm oil fatty acid composition were evidenced. The QTL positions and the species origin as well as the estimated effects of the QTL marker alleles were in coherence with the knowledge of the oil biosynthesis pathway in plants and with the individual phenotypic correlations between the traits. The mapping of chosen Elaeis key genes related to oleic acid C18:1, using intra-gene SNPs, supported several QTLs underlying notably FATA and SAD enzymes. The high number of hyper-variable SSR loci of known relative linear orders and the QTL information make these resources valuable for such mapping study in other Elaeis breeding materials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The oil palm (Elaeis guineensis Jacq.) is the most productive oil crop in the world, yielding an average of 4.1 t of vegetal oil per hectare per year. According to the United States Department of Agriculture, total world vegetable oil production in 2011 was 155.8 Mt, with the palm oil in first place (50.7 Mt), followed by soybean oil (42.4 Mt) and rapeseed oil (24.3 Mt). The consumption of vegetable oils falls into two major applications: food industry (with over 80 % of the market) and chemical industry for formulation of paints, inks, resins, varnishes, plasticizers, biodiesel production, etc. (Rosillo-Calle et al. 2009).
The oil palm is a monoic monocot, perennial, and allogamous plant (Hartley 1988). Its diploid genome consists of 16 chromosome pairs (Maria et al. 1995), and the content of its nuclear DNA has been estimated by flow cytometry as 3.9 pg/2C (Rival et al. 1997). There are only two species in the genus Elaeis: E. guineensis Jacq. originating from Africa and E. oleifera (H.B.K) from South America. The Elaeis fruit is a drupe, produced in high numbers within tight bunches. Its outer pulp (mesocarp) contains the (red) palm oil, and its kernel (endosperm) contains the kernel oil, minor oil similar to the coconut oil. The species E. oleifera has a very low yield in palm oil compared to commercial oil palm varieties (Tan et al. 1985), but it has several desirable agronomic characteristics: Elaeis oleifera presents a slow growth of the stem (Hardon 1969), shows resistance to the bud rot disease in South America (Meunier 1991), and a more edible palm oil in its fatty acid composition and concentrations of metabolites (carotenes, tocoferols, and tocotrienols) also important for the phytopharmaceutical industry (Choo et al. 1997). Genetic advantageous features from both Elaeis species can be combined by interspecific hybridization for creating more performing oil palm varieties (Ong et al. 1981). The palm oil of E. guineensis contains approximately 50 % of saturated fatty acids, with 44 % of palmitic acid (C16:0), 5 % of stearic acid (C18:0), and trace amounts of myristic acid (C14:0). The unsaturated fatty acids are about 40 % of oleic acid (C18:1) and 10 % of polyunsaturated linoleic acid (C18:2) and linolenic acid (C18:3) (Cottrell 1991; Sambanthamurthi et al. 2000). The species E. oleifera is characterized by its high content of unsaturated fatty acids compared to E. guineensis. Among the E. oleifera palms from the Amazon region, the range for C18:1 is 47 to 52 %, while the range for C16:0 is 20 to 29 % (Meunier 1975; Mohd Din et al. 2000; Rey et al. 2003). The iodine value (IV) is a multi-parameter measure of the global unsaturation degree of the fatty acids in the vegetal oil. The IV for E. oleifera has been reported to be between 70 and 87 % (Meunier 1975; Mohd Din et al. 2000; Sambanthamurthi et al. 2000; Rey et al. 2004), whereas the value for E. guineensis tenera varieties is between 53 and 60 % (Tan et al. 1985; Ekpa et al. 1994; Noh et al. 2002; Rey et al. 2004).
In plants, de novo fatty acid synthesis takes place in the plastids and starting with the carboxylation of acetyl-CoA. Then, a series of condensation reactions catalyzed by the enzyme KAS (β-ketoacyl-ACP synthase) type I, II, and III allow the elongation of carbon chains of C4:0 (butiril acid) until C18:0 (stearic acid) (Ohlrogge and Jaworski 1997; Nishida 2004). Afterward, desaturase enzymes produce carbon chain unsaturation and originate fatty acids like C18:1 (oleic acid) and C18:3 (linolenic acid) (Browse et al. 1993; Okuley et al. 1994; Salas and Ohlrogge 2002). Simultaneously, there are subsequent elongations catalyzed by the fatty acid elongase (FAE) complex in the endoplasmic reticulum (ER), and finally, the fatty acids are incorporated into molecules of triacylglycerols (TAGs) which are assembled through the Kennedy pathway (Guschina and Harwood 2007 and references therein).
The traits controlled by multiple genes such as palm oil composition can be deciphered using modern DNA marker technologies to implement a marker-assisted selection (MAS) for more desirable fatty acid composition, as it is already done for other oil crops like oilseed rape (Brassica napus) (Hu et al. 1999, 2006; Smooker et al. 2011) soybean (Glycine max (L.) Merr.) (Wilson et al. 2001; Monteros et al. 2008; Ha et al. 2010; Li et al. 2011; Chen et al. 2012) and olive (Olea europaea L.) (Poghosyan et al. 1999; Hernández et al. 2005; Banilas et al. 2011). With respect to genes involved in the fatty acid biosynthesis in oil palm, Nakkaew et al. (2008) identified and cloned the gene of biotin carboxylase (accD) from E. guineensis which is involved in the first step of the fatty acid synthesis. More recently, Al-Shanfari et al. (2011) constructed a suppression subtractive hybridization (SSH) library to identify genes involved in fruit ripening in E. guineensis. Bourgis et al. (2011) and Tranbarger et al. (2011) notably analyzed the gene expression and regulatory mechanisms underlying the fatty acid biosynthesis during the oil palm fruit mesocarp maturation and ripening. Singh et al. (2009) identified quantitative trait loci (QTLs) for fatty acid composition using 81 individuals of an Elaeis interspecific cross. However, these QTLs were only detected on the E. guineensis parent and not from the highly homozygous E. oleifera parent. In that way, no E. oleifera QTLs of palm oil composition are known.
A palm oil with a higher proportion of unsaturated fatty acids could mean new market opportunities for oil palm planters. This can be achieved by an introgressive hybridization between E. oleifera and E. guineensis (Tan et al. 1985; Amblard et al. 1995). The identification of QTLs or genes involved in the variation of fatty acid composition in both Elaeis species will allow a MAS. Therefore, the aim of our mapping study was to identify QTLs involved in the fatty acid biosynthesis of palm oil in both E. oleifera and E. guineensis genomes using an interspecific pseudo-backcross (E. oleifera × E. guineensis) × E. guineensis. As hypothesis, this population is suitable in identifying QTLs of palm oil composition as it is possible to distinguish the segregating alleles from E. oleifera and E. guinnensis. We suppose that the proportions of fatty acids are traits involving QTLs with effects strong enough to be detected despite the limited size of our population, while minor QTLs might not be detected. Also, we focused on Elaeis key genes involved in the oleic acid (C18:1) proportion using intra-gene SNPs. The future purpose is to apply an introgression of E. oleifera genes into elite palm oil commercial varieties, by MAS in order to obtain a palm oil more rich in unsaturated fatty acids.
Materials and methods
Plant material
The mapping population consisted of 134 full-sibs derived from an interspecific pseudo-backcross obtained from the interspecific hybrid palm SA65T (E. oleifera SA49D × E. guineensis LM2466P) and an E. guineensis genitor PO3228D. This pseudo-backcross, named SA569, was planted in 1991 at two different locations in Colombia: Hacienda la Cabaña (LC) in the Department of Meta and Indupalma (IP) in the Department of Cesar. Palms were planted in progeny row trials in both sites, where the two agro-climatic environments are similar: alluvial soils, annual rainfall between 2,500 and 3,000 mm, medium temperature of 27 °C, a wet season between April and September, and a dry season between December and March. We assume that the environment effect is the same for all genotypes in both sites and that an additive correction of the phenotypic trait values from the planting site effect is sufficient.
This is a pseudo-backcross because the grandparents and parents are not totally homozygous and the E. guineensis parent is not the same genotype as the grandparent. Three fruit type varieties exist in the Elaeis genus depending on the shell thickness controlled by a major co-dominant gene called Sh (Beirnaert and Vanderweyen 1941): dura (Sh+/Sh+) with thick shell, pisifera (Sh-/Sh-) without shell, and tenera (Sh+/Sh-) with thin shell. The female grandparent (SA49D) is a wild E. oleifera palm (dura) from the Coari region (Brazilian Amazon), and the male grandparent (LM2466P) is a pisifera oil palm obtained by selfing the LM2T oil palm genitor, an elite individual of the La Mé breeding population. The E. guineensis male parent PO3228D is a dura oil palm derived by selfing the DA115D oil palm genitor, an elite individual of the Deli breeding population. The fruit variety, either dura or tenera, was determined for each progeny as a morphological marker in order to map the Sh gene. About half of the progenies were tenera and the other half dura, without significant deviation from the 1:1 segregation ratio expected. Total genomic DNA was extracted for each grandparent, parent, and progeny from freeze-dried leaf samples using the NucleoSpin® 96 Plant II kit (Ref 740663.2) following the manufacturer's (Macherey-Nagel GmbH, Germany) instructions.
Measurements of the fatty acid composition
A total of 115 progenies (90 from LC and 25 from IP), as well as the E. oleifera grandparent SA49D and the interspecific hybrid parent SA65T, were analyzed for fatty acid composition and the iodine value of the palm oil in mature fruits. The number of SA569 individuals planted was initially high (200), but some palms died before sampling. Also, SA569 had abort/sterility problems which affected the bunch development, resulting in no or low bunch production, which is a common problem in interspecific oil palm materials (Obasola et al. 1977; Pamin et al. 1995). The mapping population is quite small and borderline acceptable for QTL mapping, a somewhat inevitable situation considering the limitations that the crop presents for accessing larger families and the phenotyping work involved. An alternative solution could have been to use the multi-parent QTL mapping strategy applied in oil palm by Billotte et al. (2010), using in our case pseudo-backcrosses sharing common parents, but no such mating design exists yet.
For each palm, two ripe bunches were collected, and 30 random fruits were sampled per bunch (two fruit samples/palm), to provide a good estimate of the palm oil quality (Wuidart and Gascon 1975), based on previous experiments of the former “Institut de Recherche des Huiles et des Oléagineux” (I.R.H.O) in France. Palm oil composition analysis was carried out on these two biological replicates per palm, and results are reported as mean values. No phenotypic data were available for the E. guineensis grandparent LM2466P and parent PO3228D. Fruits were boiled (100 °C) in water and then hot-pressed using a screw press to extract the palm oil. The extracted oil and hot water were recovered in an Erlenmeyer and then allowed to settle in the oven. The oil fraction was recovered with a syringe and stored at -20 °C until chemical analysis. Fatty acids extracted by a standard procedure were converted into fatty acid methyl esters (FAMEs), and FAMEs were analyzed by gas chromatography (GC) according to the AFNOR method NF T60-233/1977 (AFNOR 1977) using Tchobo et al. (2007) protocol with minor modifications. Each oil sample (≈40 mg) was added to 3 ml of sodium methylate solution with phenolphthalein in a 25-ml round bottom flask. Reaction medium was refluxed for 10 min. A volume of 3 ml of methanolic HCl was added until phenolphthalein discoloration occurred. The mixture was refluxed again for 10 min and then cooled down to room temperature. A volume of 8 ml of hexane and 10 ml of water were added, and the organic phase was recovered, dried over anhydrous sodium sulfate, and filtered for subsequent GC analysis with an Agilent 6890 series (Bios Analytique, France) using a Supelcowax 10 capillary column (SGE, Courtaboeuf, France) with the following characteristics: length, 30 m; internal diameter, 0.32 mm; film thickness, 0.25 μm; FAMEs (1 μl) directly injected into the GC; carrier gas, helium flow 1.0 ml/min; splitting ratio, 1/20; injector temperature, 250 °C; and FID detector temperature, 270 °C. The temperature settings were as follows: initial temperature 185 °C for 2 min and 185 to 225 °C at 5 °C/min. The final results are expressed as the relative percentage of the peak area. Quantitative phenotypic traits in this study considered the nine main fatty acids: myristic acid (C14:0), palmitic acid (C16:0), palmitoleic acid (C16:1), stearic acid (C18:0), oleic acid (C18:1), linoleic acid (C18:2), linolenic acid (C18:3), arachidic acid (C20:0), and gadoleic acid (C20:1). In addition, the iodine value (IV) was determined by the Wijs method described in the ISO 3961:2009 standard.
Statistical analysis of phenotypic data
An analysis of variance (ANOVA) test (post hoc test of Tukey, α = 0.05) was realized for all traits to evidence eventual differences between the planting sites LC and IP. If significant differences were detected for a given trait, the individual phenotypic data for the IP site were standardized regarding those of the LC site, by a mean correction for further statistical analyses as follows: IPstandardized data = IPraw data + (LCmean − IPmean). The Gauss distribution of the quantitative data was checked by a normality test of Shapiro–Wilk at an α threshold of 5 %. The relationships between phenotypic traits, at the individual palm level, were estimated by calculating the Pearson’s correlation coefficients. The trait heritability h 2 (broad sense) was estimated by ANOVA with one fixed factor (palm) using the two bunches measured per palm assuming that for these oil traits, variability between bunches on the same tree is not far from variability between bunches on separate trees of a clone in the same trial, as follows: model: Y ij = m + A i + E ij , where i (1 to 115) is the palm, j (1 to 2) is the bunch measured, Y ij is the phenotypic value of the palm i for its bunch j, A i is the trait genetic value of the palm, E ij is the error (environment effect on the trait phenotypic value), and \( {h}^2=\frac{\sigma {\mathrm{G}}^2}{\sigma {\mathrm{G}}^2+\sigma {\mathrm{E}}^2}=\frac{M{S}_{\mathrm{P}}-M{S}_{\mathrm{E}}}{M{S}_{\mathrm{P}}+M{S}_{\mathrm{E}}} \), where σ 2G is the genetic variance, σ 2E is the error variance, MS P is the mean square for palm, and MS E is the error mean square.
Microsatellite analyses
SSR genotyping of SA569 along with its grandparents and parents was performed by the commercial laboratory ADNid (Montpellier, France, http://www.adnid.fr). The legitimacy of the progenies was checked using 12 SSR loci according to Durand-Gasselin et al. (2009). An initial set of 700 SSR loci was pre-screened for polymorphism on grandparents and parents. A total of 384 SSR loci were genotyped and consisted of 250 oil palm SSR loci (Billotte et al. 2001, 2005 and unpublished by CIRAD), 21 coconut SSR loci (Billotte et al. 2010), and 113 oil palm EST-SSR loci (Tranbarger et al. 2012). Our choice favored the SSRs with best amplification patterns, a priori segregating from the SA65T and PO3228D parents, and with high polymorphism. The plant material was analyzed by the SSR technique in fluorescent conditions according to Roy et al. (1996) using the capillary electrophoresis and detection systems of a 3500xL Genetic Analyzer (Applied Biosystems, USA). The PCR amplification was performed in a 384-well Eppendorf Mastercycler with 25 ng of DNA in a 10-μl final volume of buffer (10 mM Tris–HCl (pH 8.3), 50 mM KCl, 0.001 % w/v glycerol,) containing 1.50 mM MgCl2, 0.08 μM of the forward M13-tailed primer, 0.1 μM of the reverse primer, 200 μM of dNTP, 1 U of Taq DNA polymerase (Life Technologies, USA), and 0.1 μM of M13 primer-fluorescent dye FAM, NED, PET, or VIC (Applied Biosystems, USA). A touchdown PCR program was performed as follow: initial denaturation at 94 °C for 4 min; 10 cycles at 94 °C for 45 s, primer melting temperature (Tm) (55 to 50 °C, -0.5 °C/cycle) for 1 min, and 72 °C for 1 min 15 s; additional round of 25 cycles of 94 °C for 45 s, 50 °C for 1 min, 72 °C for 1 min, and a final elongation step at 72 °C for 30 min. For post-PCR multiplexing 2.0 μl of FAM, 2.5 μl of NED, 3.5 μl of PET, and 2.0 μl of VIC PCR products were pooled together, and the final volume was adjusted to 20 μl with sterile water. PCR pooled products (2 μl) were mixed with 15 μl of a mixture of GeneScan™-600 LIZ® (Applied Biosystems, USA) and highly deionized Hi-Di™ Formamide (Applied Biosystems, USA). The mixture was prepared by adding 12 μl of size standard GeneScan™-600 LIZ® to 1.5 ml of Hi-Di™ formamide. PCR products were subject to the capillary electrophoresis, detected by the laser system of a 3500xL Genetic Analyzer (Applied Biosystems, USA), and visualized using the GeneMapper® 4.1 software (Applied Biosystems, USA). The presence or absence of each amplified SSR allele in each individual was identified, cross-checked by two different operators, and the genotype configuration of each locus was coded according to the nomenclature of Ritter et al. (1990), with one to four alleles segregating in our cross between two partially heterozygous parents SA65T and PO3228D.
Intra-gene SNP detection and mapping of chosen key genes
Five Elaeis genes of the palm oil biosynthesis were chosen for their mapping and a QTL/gene co-localization study focusing on the oleic acid C18:1 in palm oil: for the enzymes β-ketoacyl-ACP synthase of type I and II (KAS I, KAS II), the Acyl-ACP thioesterases (FATA, FATB) and the Δ9 stearoyl-ACP desaturase (SAD). cDNA sequences of these genes were identified from eight Elaeis full-length cDNA libraries of pulp (unpublished) previously made from developing fruits between 60–75 and 150–180 days after pollination. E. guineensis sequences of these genes are available from Bourgis et al. (2011) at http://www.biomemb.cnrs.fr/contigs.html in file oil-contigs.doc (Table 4). Each Elaeis species was represented by four libraries from different genetic backgrounds: La Mé, Deli, Yangambi, and Nigeria for E. guineensis and Colombia, Brazil, Costa Rica, and Surinam for E. oleifera. These libraries of the International Oil Palm Genome Projects (OPGP) Consortium were sequenced by the Genoscope-CEA (Evry, France) using the 454 Roche/GS-FLX Pyrosequencing technology (1/2 run per library). Each read library was analyzed using the ESTtik tool (Expressed Sequence Tag treatment and investigation kit) (Argout et al. 2008) especially modified for the analysis of 454 sequence data. The 454 reads obtained from the eight pulp libraries were assembled using the TGICL (Pertea et al. 2003) assembler. Contigs were automatically annotated using the standalone BLAST software (Altschul et al. 1990). We searched for similar sequences on the Oryza sativa japonica proteome TIGR6, the Uniprot Knoledgebase Swiss-Prot and TrEMBL (Boeckmann et al. 2003), the GenBank non-redundant protein database NR, and the GenBank nucleotide database NT. We used an e-value cutoff of 1e-5 and retrieve the 10 best hits to annotate each contig. We used the Blast2GO pipeline (Götz et al. 2008) to annotate the contigs with the Gene Ontology terms based on our BLAST results. Also, a BLAST analysis was performed with the E. guineensis cDNA libraries of Bourgis et al. (2011) and Tranbarger et al. (2011). We used the Prot4EST pipeline (Wasmuth and Blaxter 2004) to obtain a protein prediction from our contig sequences. We used the program IDEG6 (Romualdi et al. 2003) for data normalization and statistical analysis of the read libraries (general chi-square test applying the Bonferroni correction, p <0.0001), to test the differential expression of our five genes between the two Elaeis species. Finally, the ACE assembly output from TGICL was treated to obtain a collection of FASTA alignment using the AceToFastaAlignment script from the SNiPlay utilities (Dereeper et al. 2011). We used the SNiPlay pipeline on our FASTA alignment collection for single nucleotid polymorphism (SNP) analysis. SNP loci were identified from cDNA reads for our five targeted genes, with eventually several SNPs for a given gene. Designed SNPs were genotyped by the commercial laboratory ADNid (Montpellier, France, http://www.adnid.fr), combining the GoldenGate Genotyping Assay with the VeraCode technology (Illumina, San Diego, USA) and using the Illumina’s BeadXpress Reader System for detection.
Linkage mapping
A linkage map of SA569 was constructed based on 115 progenies, without missing data for the polymorphic SSRs, SNPs, and the Sh gene. SSR loci showing a significant distortion (p < 0.01) according to the expected Mendelian segregating ratio of their alleles were removed from the analysis. The linkage phases between SSR marker alleles were determined using the JoinMap v.4.0 software (Van Ooijen and Voorrips 2001) in our pseudo-backcross considered to be a double pseudo-test cross (Grattapaglia and Sederoff 1994). Marker loci with estimated allele phases were then analyzed using the CarthaGene software (Schiex and Gaspin 1997) at LOD 7.5 with a maximum recombination threshold of 0.3. The most probable relative linear orders of markers within each linkage group (LG) were estimated using the several multi-point maximum likelihood algorithms of CarthaGene. The Haldane mapping function was used to convert recombination frequencies into map distances (Haldane 1919). The chart of the linkage map was generated using BioMERCATOR v.3.1 (Arcade et al. 2004).
QTL analysis
QTL analyses using the SSR-SNP linkage map and all available genotypic and phenotypic data were performed with the MapQTL®5 software (Van Ooijen 2004). As a first step, the non-parametric Kruskal–Wallis (K-W) test was performed to identify significant marker-trait associations at p < 0.005. Subsequently, the interval mapping (IM) method was performed using the LOD statistic test with a mapping step size of 1 cM and a maximum number of 5 neighboring markers. For each trait, threshold LOD values were estimated at the genome-wide (GW) global risk α of 5 and 1 % for declaring the presence of a QTL by the re-sampling method and permutation of the trait data (1.000 iterations) of the software. Finally, a multiple-QTL model (MQM) method was undertaken along with the automatic selection of cofactors and using the previous threshold LOD values. The confidence interval of each significant QTL by IM or MQM was determined by the LOD–1 method. The phenotypic values of fatty acids in traces or in small amounts (C14:0, C16:1, C18:2, C18:3, C20:0, and C20:1) did not follow a normal distribution. The non-parametric Kruskal–Wallis (KW) test should be considered here as more accurate compared to the IM or MQM methods, as these latter are biased by deviations from normality (Smooker et al. 2011).
To correct the bias in overestimating the phenotypic variances associated with identified QTL using a limited population size, we used the following correction proposed by Luo et al. (2003) and Xu (2003), keeping in mind this only corrects the bias due to the sampling error.
Considering that the variance explained by an identified QTL is, as estimated under MapQTL,
with σ 2a corresponding to the genetic variance due to additive effect and σ 2p corresponding to the phenotypic variance.
The corrected variance explained by this QTL was re-estimated as follows:
where LOD corresponds to the LOD value of the identified QTL.
In addition, the QTL marker loci were used to perform an ANOVA test (type III, post hoc test of Tukey at α = 0.05) to estimate the mean effects of the parent QTL marker alleles on the mean of each phenotypic trait. For the hybrid parent SA65T, the species origin of the QTL marker alleles were identified, and the allelic effects at the QTL were therefore estimated by contrast of E. oleifera (grandparent SA49D) against E. guineensis (grandparent LM2466P). The QTLs (position, confidence interval, variation explained) were visualized on the linkage map using BioMERCATOR v.3.1 (Arcade et al. 2004).
Results
Palm oil fatty acid composition
The principal palm oil fatty acids found on the SA569 progenies were (Table 1) the following: oleic acid C18:1 (mean 45.0 %), palmitic acid C16:0 (mean 38.8 %), and linoleic acid C18:2 (mean 11.0 %), followed by stearic acid C18:0 (mean 3.5 %). Their coefficients of variation were high to medium, with 8.2 % (C18:1), 7.5 % (C16:0), 14.5 % (C18:2), and 38.7 % (C18:0). The other fatty acids (C14:0, C16:1, C18:3, C20:0, and C20:1) were found in trace amounts (<1 %). The interspecific hybrid parent SA65T showed rather similar proportions of C16:0 and C18:1 compared to its E. oleifera parent SA49D. The individual phenotypic values of the SA569 progenies were normally distributed for the four traits C16:0, C18:1, C18:2, and iodine value (IV) while not for C18:0 and fatty acids in traces (C14:0, C16:1, C18:3, C20:0, and C20:1). The data histograms (data not shown) presented a continuous variation in all traits with the exception of C20:1, which was excluded from the QTL analysis. The ANOVA test (data not shown) showed significant mean differences between the two plantation sites LC and IP for the traits C18:2, C20:0, and IV. The individual phenotypic data were therefore corrected as mentioned above for further statistical analyses.
Pearson’s correlations between the individual fatty acid proportions and IV are given in Table 2. The myristic acid C14:0 was positively correlated with the palmitic acid C16:0 and palmitoleic acid C16:1 while negatively correlated with the stearic acid C18:0 and oleic acid C18:1. The palmitic acid C16:0 was negatively correlated with the oleic acid C18:1 and with the iodine value IV, indicative for the total proportion of unsaturated fatty acids. Two other correlations for C16:0 were positive with C14:0 and negative with C18:3. Additional correlations were negative between C16:1 and C18:0, C18:0 and C18:1, and C18:1 and C18:2 and positive between C18:2 and C18:3. The broad sense heritabilities h 2 were high in the pseudo-backcross (0.8 and above) for the principal fatty acid proportions C16:0, C18:0, C18:1, and C18:2 as well as for the iodine value.
Intra-gene SNPs and microsatellite polymorphism
In the eight Elaeis full-length cDNA libraries of pulp, the number of cDNA consensus contigs, i.e., distinct copies in Elaeis for the studied key genes expressed in the pulp, and their related genotyped SNPs are: 3 copies for SAD (15 SNPs), 3 copies for KAS I (10 SNPs), 1 copy for KAS II (3 SNPs), 1 copy for FATA (2 SNPs), and 2 copies for FATB (4 SNPs). In total, 14 out of the 34 intra-gene SNPs were polymorphic on the SA569 parents. Every gene copy had at least one polymorphic SNP, except one copy of KAS I.
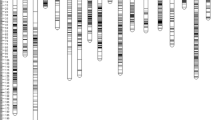
Up to 84 % of the 700 tested SSRs were correctly amplified in the grandparents and parents of SA569. For these amplified SSRs, the number of heterozygous loci was 161 (28 %) in the E. oleifera grandparent SA49D, 147 (25 %) in the E. guineensis grandparent LM2466P, 457 (78 %) in the interspecific hybrid SA65T, and 128 (22 %) for the E. guineensis parent PO3228D (data not shown). About 19 % of the SSRs were homozygous in both parents, and 81 % (471) were mappable on SA569, including 19 % in one parent only and 69 % in both parents. About 12 % of the SSRs showed at least one null allele in the E. oleifera palm SA49D, compared to 1 and 3 %, respectively, in the E. guineensis palms LM2466P and PO3228D. Out of the 384 SSRs genotyped on SA569, 372 (97 %) showed effectively a traceable segregation of their alleles, allowing the mapping of their loci (Fig. 1). The SSR allele patterns of the latter showed six of the nine genotype configurations defined by Ritter et al. (1990) for a cross between two heterozygous parents. These SSR configurations were cases No. 1, 3, 4, 7, 8, and 9. On the pseudo-backcross, 67 % (258) SSR loci belonged to genotype configuration No. 3, where only one parent is heterozygous, which was the hybrid SA65T in most (251) cases. In fact, the large majority of the SSRs was segregating only from the hybrid SA65T (genotype configurations No. 1 and 3) and most others from both parents (genotype configurations No. 7, 8, and 9 with 3 to 4 segregating alleles). In parallel, 30 % of the SSRs were segregating from the recurrent parent PO3228D. Considering all genotype configurations, 365 (95.0 %) genotyped SSRs showed alleles segregating from the interspecific hybrid parent SA65T and 117 (30.5 %) from the E. guineensis parent PO3228D.

Genotype configuration and distribution of segregating marker loci in the pseudo-backcross SA569 between the interspecific hybrid SA65T and E. guineensis PO3228D parents
Microsatellite and intra-gene SNP linkage map
Few SSRs (11) were removed from the genotypic data set because of significant segregation deviation from the Mendelian ratio or due to missing data (12) or locus redundancy (2). A total of 347 segregating SSRs, 14 SNPs of genes, and the Sh locus were finally used to establish the linkage map of SA569. This linkage map consisted of 16 linkage groups corresponding to the 16 pairs of homologous chromosomes in Elaeis and spanned 1.485 cM with an average marker density of 4 cM (Fig. 2). The standard error in estimating the map position of a given co-dominant marker was ±0.5 cM in our backcross, according to Allard (1965). The linkage group length ranged from 49.1 to 175.9 cM, with an average of 92.8 cM. There were 10 to 38 marker loci per linkage group (average of 22), generally uniformly distributed along the chromosomes, except for a few regions lacking markers (max. ± 30 cM) on linkage groups No. 7, 9, 11, and 12. Regarding each map parent, 73 % (253) of the mapped SSRs segregated only from the hybrid parent SA65T, 2 % (7) from PO3228D only, and 27 % (93) were common SSRs segregating from both parents. Out of the 14 mapped SNPs of genes, 11 segregated only from the hybrid parent SA65T and 4 from both parents.



Nineteen QTLs of fatty acid proportion and iodine value of the palm oil in the interspecific Elaeis pseudobackcross SA569. Note: one star (*) or two stars (**): QTL detected by the MQM method at the genome-wide α threshold value of 5 or 1 %, respectively. No star: putative QTL as only detected by the Kruskall–Wallis test at p < 0.005. The linkage map encompasses 347 SSRs, 14 SNPs, and the Sh locus distributed on 16 linkage groups corresponding to the 16 homologous chromosomes of the plant. The names and the positions (cM) of the markers are given on the right side of the linkage groups. mEgCIRxxxx and mEgESTxxxx: E. guineensis SSR loci. E. guineensis sEgOPGPxxxx: SNP loci. mCnCIRxxxx: Cocos nucifera SSR loci. Reference marker loci in the cross LM2T × DA10D (Billotte et al. 2010) are indicated by an extension “_R”. The names, positions, and confidence regions of the QTLs are given on the left side of the linkage groups. In red are figured the QTLs of saturated fatty acid proportion; in blue: the QTLs of unsaturated fatty acid proportion and of iodine value. The names of mapped genes are given on the right side of corresponding SNP names
A total of 156 SSRs (45 %) and the Sh locus were previously mapped on the E. guineensis consensus multi-parent map published in oil palm by Billotte et al. (2010). In general, common loci were co-linear between the interspecific pseudo-backcross SA569 and the E. guineensis multi-parent map, except few cases of closely linked markers. Two new SSR loci (mEgCIR3676 and mEgEST0248) were proved on LG 4 to flank the Sh gene controlling the fruit type, at 12.5 cM for each of them (Fig. 2).
Identified QTLs
A total of 19 QTLs associated with palm oil fatty acid composition were evidenced by the K-W analysis at p < 0.005, with one to three QTLs per fatty acid or iodine value IV (Fig. 2). Subsequently, 13 QTLs were also evidenced by the IM and 11 by MQM analyses at the significant genome-wide threshold α of 1 or 5 % (Table 3). These latter QTLs were located on four linkage groups (LGs) 4, 5, 6, and 10. The other QTLs, only identified by the K-W method and therefore considered as “putative” QTLs, were mapped to LGs 2, 3, 5, and 15. For these putative QTLs, a maximum LOD value was also observed (data not shown) with the IM and/or MQM methods at the same or nearby location, despite being not significant. Some of these putative QTLs were closely linked, such as for C16:0 and IV on LG 15, or co-localized with another significant QTL determined by the IM and/or MQM methods, as for instance a putative QTL of C16:0 with a QTL of C18:1 on LG 5. The confidence interval for the positions of the putative or significant QTLs was 16.3 cM on average (min 6.0 and max 30 cM). Some traits showed common significant QTLs, as for C18:2 and C18:3 sharing the same location at 11.1 cM on LG 10 or for traits C16:1 and C18:0 at 27.4 cM on LG 6. Other significant QTLs were closely linked, such as for C18:1 and C20:0 on LG 6.
The 14 intra-gene SNPs were distributed on five LGs (Fig. 2 and Table 4). They proved by mapping the different copies in Elaeis of the genes KAS I (2 on LGs 1 and 15), SAD (3 on LGs 2, 6, and 7), and FATB (2 on LGs 1 and 6), while the single copy for FATA was mapped on LG 6. Obviously, the third copy of KAS I, monomorphic in SA569, was not mapped. Two SNPs, of SAD (sEgOPGP00038) and FATA (sEgOPGP00050), co-localized on LG 6 with the common QTL for C16:1 and C18:0 and were located within the confidence intervals of the QTLs for C18:1 and C20:0. Two other SNPs of KAS I (sEgOPGP00082) and KAS II (sEgOPGP00037) co-localized on LG 14 with the putative QTL of C14:0. Other SNPs did not co-localize with detected QTLs. Regarding the expression of mapped genes at the species level (Table 5), the statistical analysis of the read libraries showed that five mapped gene copies were differentially expressed between E. oleifera and E. guineensis: FATB (sEgOPGP00017) on LG1, SAD (sEgOPGP00063) on LG 2, FATA (sEgOPGP00050), and SAD (sEgOPGP00038) co-localizing with QTL(s) on LG 6.
The percentage of the phenotypic variation explained by a significant QTL, corrected from the sampling error, was medium to high and ranged between 12 and 50 % (Table 3). The corrected phenotypic variations explained by the QTLs for the principal fatty acids were 16 % (C16:0), 50 % (C18:0), 15 % (C18:1), and 21 % (C18:2). For IV, the explained cumulative variation was 31 % (two QTLs). The ANOVA (data not shown), of the individual phenotypic data depending on the QTL marker alleles for each trait, proved that only the hybrid parent SA65T had a significant parental effect on the trait, due to its inherited E. oleifera and E. guineensis alleles, while no significant allelic effect was found for the E. guineensis parent PO3228D for the few loci (5) heterozygous in that latter (data not shown). The effects associated to the E. oleifera QTL marker alleles were negative for the proportions of saturated fatty acids C14:0, C16:0, C18:0, and C20:0 (Table 3). In parallel, they were positive for the percentage of the unsaturated fatty acids C16:1, C18:1, C18:2, and C18:3 and for the iodine value IV. Only for C18:3, the E. oleifera allele of the QTL locus mEgCIR0801 presented a negative effect.
Discussion
Mapping population and linkage map
No line exists in the allogamous Elaeis species, and subsequently, no true backcross is available for QTL analysis. We chose an Elaeis interspecific pseudo-backcross of first generation (E. oleifera × E. guineensis) × E. guineensis to develop molecular markers for fatty acid composition of Elaeis palm oil. The pseudo-backcross was highly heterozygous and allowed to map a large number of loci based on different segregating E. oleifera and E. guineensis grandparent alleles which were traceable in the progenies. The LM2466P and PO3228D palms were the respective selfings of two unrelated LM2T and DA115D genitors. The homozygosity degrees of LM2T and DA115D were 37 and 59 % based on SSR data (Billotte 2004) and increased beyond 75 % in LM2466P and in PO3228D. Due to this reason, the proportion of heterozygous loci useful for QTL analysis on the SA65T interspecific genome went up to 98 % with traceable E. oleifera alleles. On the other side, this proportion was rather low (28 %) for the highly homozygous PO3228D, leading to reduced power for QTL detection in this E. guineensis parent. Most loci, segregating only from the hybrid SA65T (Fig. 1), improved the estimates for linkage phase and genetic distances. The total map length is about the same with the SSR linkage map of the oil palm LM2T × DA10D cross (1479 cM) published by Billotte et al. (2010) and shorter than the 1815 or 2247 cM of other published maps for Elaeis (Singh et al. 2009; Seng et al. 2011). In addition, only a few (3 %) skewed markers were identified. All loci in Mendelian segregation could be assigned to 1 of the 16 linkage groups. Our linkage map is the first satured one to be published for an Elaeis pseudo-backcross. The co-linearity of loci common to the E. guineensis map in oil palm (Billotte et al. 2010) indicated the probable absence of chromosome rearrangements between the E. oleifera and E. guineensis genomes. This is in coherence with the karyological examination of interspecific meiotic pairing with a balanced distribution of chromosomes in gametes and formation of regular tetrads (Schwendiman et al. 1982).
The size of our mapping population is limited due to the lack of a larger pseudo-backcross in current Elaeis breeding materials. In that way, our experiment is a preliminary study where a certain number of QTLs with small or medium effects might not be detected. Studies showed that by increasing the population size, it is possible to increase the total number of QTLs detected and to reveal those with small effects (Vales et al. 2005; Raghavan and Collard 2012). This is well known that the power to detect QTLs is rather low using a small number of individuals, depending on the trait heritability and QTL effects, with also the non-negligible risk to evidence false QTLs especially for those with weak effects (Beavis 1998). Considering that the best detection of QTLs is observed in highly heritable traits, high heritabilities may provide clues about the accuracy of our QTLs found. The broad sense heritabilities in our interspecific backcross were very high for principal fatty acids and relevant compared to those reported in oil palm by Noiret and Wuidart (1976) or Ollagnier and Olivin (1984), with values of 0.8 for oleic acid and 0.9 for iodine value. In soybean RIL populations, heritabilities between 0.6 and 0.7 for fatty acid composition were reported (Panthee et al 2006). In peanut, values of 0.9 for oleic acid and linoleic acid were observed in RIL population (Sarvamangala et al 2011), and for rapeseed, values between 0.8 and 0.9 were founded for fatty acids, with one exception for stearic acid (0.4) in doubled haploid lines (Zhao et al 2008). The heritability values reported by previous authors for oil palm and other oil crops indicate the reliability of our experimentation. The broad sense heritability is a maximum value of the narrow sense heritability, which is latter being the sum of the phenotypic variances explained by all true QTLs, whatever detected or not. Here, the phenotypic variances explained by detected QTLs (MQM method) were between 10 and 50 % for principal fatty acids or iodine value, to be compared to broad sense heritabilities of 80 to 90 %. Apart from possible QTLs, our experiment was capable of identifying QTLs of medium to high effects, while smaller and undetected QTLs explaining a non-negligible part of the variation. The QTL power detection of our experiment can be precised according to the simulations of Muranty (1996). In our pseudo-backcross of about 100 individuals, for a biallelic QTL of an expected heterozygosity He (Nei 1973) of 0.5, the chance to detect a QTL is about 75 % when the explained phenotypic variance (EPV) by that QTL is 20 % or above. Increasing the number of individuals for a single cross allows such a QTL detection power, even for QTLs with an EPV as low as 5 %, while the power is not increased for QTLs with an EPV of 20 % and over, even with 500 individuals. In the future, a multi-parent QTL mapping design (Muranty 1996) as used in oil palm by Billotte et al. (2010) will be used to detect QTLs of palm oil composition in interspecific materials.
Using a population of a small size leads also to overestimation of the additive variance associated with correctly detected QTL. The bias is the sum of two biases: due to the sampling error (contribution of the environmental variance to the estimate of the additive variance of the QTL) and of the Beavis effect (Beavis 1998). Similar with those proposed by Luo et al. (2003) and Xu (2003), we corrected the variances explained by the QTLs from the bias due to the sampling error, knowing that the major part of the overestimation, which is the Beavis effect itself, cannot be corrected. Notwithstanding, the identified QTLs and mapped homologous genes involved in the palmitic and oleic acids biosynthesis are new information in the Elaeis genus and on a saturated genetic map. The next step will be to expand our population to reach at least 200 individuals for a better detection and effect estimation of the QTLs. Also, in a near future, other single pseudo-backcrosses will be used or again a multi-parent QTL mapping design (Muranty 1997) like that tested in oil palm by Billotte et al. (2010). An association mapping study with unrelated Elaeis accessions in collections could also precise the accuracy of the identified QTLs (associated marker effects) and evidenced those undetected one.
Fatty acid composition
We have analyzed the two classes of saturated (C14:0, C16:0, C18:0, and C20:0) and unsaturated (C16:1, C18:1, C18:2, C18:3, and C20:1) fatty acids in the pulp of mature fruits. An interspecific cross E. oleifera × E. guineensis shows mean values for fatty acid proportions which are intermediate between the mean values of its E. oleifera and E. guineensis parents (Hardon 1969; Macfarlane et al. 1975; Meunier and Boutin 1975; Opute and Obasola 1979; Tan et al. 1985). This is in accordance with the co-dominance theory for genes involved in palm oil biosynthesis and their alleles inherited from each Elaeis parent (Hardon 1969; Ong et al. 1981). Surprisingly, the proportion of oleic acid of the interspecific hybrid parent SA65T was quite similar to the one of its E. oleifera parent of the Brazilian Coari origin (Table 1). This is probably due to equivalent proportions of oleic acid in both E. oleifera and the E. guineensis parent of the La Mé origin, which is known to have a high oleic acid proportion (Gascon and Wuidart 1975; Monde et al. 2009).
Pearson correlations do not determine the cause-and-effect relationships between the phenotypic traits but estimate the strength of association between them, at the individual palm level, which is useful for breeding purposes. The oil palm breeders are using for a long time the iodine value as an indicator and a selection parameter for the degree of unsaturation of palm oil (Wuidart and Gascon 1975; Corley and Tinker 2003). The iodine value showed the well-known positive correlations with the proportions of unsaturated fatty acids and negative one with the saturated fatty acids. These correlations are congruent with those of the study by Singh et al. (2009) in an Elaeis interspecific cross and also with other correlations in E. guineensis (Noh et al. 2002). The positive correlation between C14:0 and C16:0 or C16:1 is in accordance with reports in rice by Ying et al. (2012) and in oil palm by Singh et al. (2009). The negative correlation between C16:0 and C18:1 has been reported in oil palm by several authors (Meunier 1975; Wuidart and Gascon 1975; Noh et al. 2002; Singh et al. 2009). In other crops such as Sesamun indicum L (Were et al. 2006), Brassica napus L (Zhao et al. 2008), or Zea mays (Yang et al. 2010), this negative correlation is frequently justified as pleiotropic effects of single genes or closely linked genes controlling different traits (Yang et al. 2010). The negative correlation between C18:2 and C18:1 had been reported in Sesamum indicum L (Uzun et al. 2008), maize (Yang et al. 2010), and Arachis hypogaea L. (Sarvamangala et al. 2011). However, estimates of genetic (additive) and pure environmental correlations cannot be precisely estimated on our single cross where all offsprings have the same kinship degree. Such estimates are also not available in the literature. Therefore, our conclusions on correlations among fatty acids should be considered with caution as they are just phenotypic correlations.
Mapped QTLs
The hybrid SA65T holds several QTLs with alleles inherited from both Elaeis genomes, while no statistical effect of any E. guineensis allele was evidenced in the E. guineensis parent PO3228D. This is mainly due to the high homozygosity rate of PO3228D and probably to the narrow genetic diversity of its Deli origin descending from an early introduction of only four “historic” palms to Indonesia (Hartley 1988). The confidence interval is rather large for many of the detected QTLs, and such regions represent several Mbp of genomic DNA considering the large genome size of Elaeis species, of about 1.9 Gb in oil palm (Zieler et al. 2010). Many functional genes and regulatory elements may underlie a single QTL.
In our full-length Elaeis cDNA libraries, the number of distinct expressed copies for our five genes KAS I, KAS II, SAD, FATA, and FATB were in accordance with the cDNA data published in E. guineensis by Bourgis et al. (2011) and Tranbarger et al. (2011). They were proved as distinct by mapping (different loci and not allelic variants). They might be the true number of copies in both E. oleifera and E. guineensis genomes for these genes involved in the proportion of C18:1. The intra-gene SNP markers of the Elaeis genes SAD and FATA (respectively, sEgOPGP00038 and sEgOPGP00050) co-localized and were mapped on LG 6 within the confidence intervals overlap of the five QTLs for C16:1, C18:0, C18:1, C20:0, and IV (Fig. 2 and Table 3). The SNP marker associated to SAD could indicate that this mapped gene is involved in the final proportions of C18:0 and C18:1. The E. oleifera versus E. guineensis allele effects of SAD are coherent on the two traits as, respectively, negative and positive. Also, based on its SNP marker, the FATA enzyme, which is known to export C18:1 from plastid to ER, is a good candidate to explain the QTL of C18:1 in that region. Also, the gene KAS I seems to be a good candidate underlined by the putative QTL for C14:0 on LG 15. KAS II (chain lengthening of C16:0 to C18:0) mapped at that same locus could also support the accuracy of this QTL, as it has been demonstrated that KAS II enzyme can use C10:0 to C14:0 as substrates, although substrate C16:0 is preferred (Shimakata and Stumpf 1982). Other gene SNP markers were mapped outside QTLs. This fact might be due to insufficient explained phenotypic variation (true absence of QTL at their positions) and/or again to the limited size of our mapping population for detecting co-localized QTLs (undetected QTL at these positions). Nevertheless, these SNP markers are useful information upon FATB and other gene copies of KAS I and SAD in the Elaeis genomes. We will not discuss neither cannot interpret without complementary data the differential expression of the mapped genes at the species level, only note the absence of clear relation with QTLs detected, except the co-localized SAD and FATA on LG 6.
Conclusion
The dense microsatellite linkage map of our interspecific pseudo-backcross represents the 16 pairs of homologous chromosomes in the Elaeis genus from which we could trace segregating alleles from both E. oleifera and E. guineensis grandparents. The number of mapped SSR loci, with accurate relative linear orders, and their molecular hyper-variability make this resource valuable for other research centers wishing to undertake rapidly any genetic map in Elaeis breeding materials. The probable absence of chromosome rearrangement between the E. oleifera and E. guineensis genomes would ease the identification, validation, and use of QTL markers in both species.
The QTL positions, the E. guineensis or E. oleifera species origin of the QTL marker alleles, and the estimated allelic effects at the QTLs were in good coherence with both the knowledge of the oil biosynthesis pathway in plants and with the individual correlations estimated between the fatty acid proportions in the palm oil. Moreover, one chromosome region on LG 6 presented good candidates for enzyme genes FATA and SAD involved in the variation of the C18:1 proportion in palm oil.
This preliminary study represents a first stone in view to assist a backcross strategy to exploit genetic differences at level of fatty acid composition inside the Elaeis genus. Our results are specific to our mapping population, and QTL information should be confirmed in other independent and larger Elaeis segregating populations. Since species of the Elaeis genus are outcrossing species, QTLs mapped on our genetic background may not hold on other crosses due to loss of polymorphism and to the fact that family linkage disequilibrium structure may not resemble linkage disequilibrium at population levels. Therefore, the markers identified as linked to the QTLs for fatty acid composition may not be useful while screening other crosses in a MAS strategy. For assigning known genes to QTLs and for practical MAS, the next step of our study will be to search all genes known to be involved in fatty acid synthesis, in our Elaeis cDNA libraries of pulp, and to identify and map their polymorphic intra-gene SNPs. On the other hand, it will be possible to anchor the genetic map(s) to a whole-genome sequence of Elaeis guineensis, as soon as this latter will be available, in order to search SSRs and SNPs for a QTL fine-mapping strategy. Also, such anchoring allows us to identify candidate genes or genes known to be involved in fatty acid synthesis among the genes underlying the fined-mapped QTL regions, for which intra-gene markers may be developed for practical use of MAS (once candidate genes are validated). At the phenotypic level, the individual palms of the mapping population(s) would be characterized for the kinetic of the palm oil biosynthesis, by fatty acid analysis all along the development of ripening fruits, and for the transcript profiles of the related genes, by parallel RT-QPR analysis.
The current reference method of MAS for quantitative traits is the genomic selection (GS) developed by Meuwissen et al. (2001). In GS, all marker effects are estimated simultaneously in the training population and used to estimate the value of selection candidates, without testing the significance of individual marker effects. This minimizes the bias of marker effects and is allows capturing small effects. Odegard et al. (2009) showed that GS could be efficient in introgressing a polygenic trait from an inferior to a superior population by backcrossing. As our pseudo-backcross used E. guineensis individuals from elite families, this is a relevant starting point to introgress the fatty acid profile of E. oleifera into breeding populations of E. guineeenis. The progeny SA569 could be used as training population, while the test population would be produced by backcrossing its best individuals (in terms of fatty acid profile) to elite E. guineensis genitors. The availability of intra-gene SNPs should also prove useful, as Meuwissen and Goddard (2010) showed that including causative mutations increased the accuracy of GS. However, the size of the training population is a key parameter of accuracy. A study is therefore required to estimate the accuracy that GS could achieve with our dataset. This could be done by cross-validation.
Abbreviations
- ACCase:
-
Acetyl-CoA carboxylase
- ACP:
-
Acyl carrier protein
- AFLP:
-
Amplified fragment length polymorphism
- ANOVA:
-
Analysis of variance
- C14:0:
-
Myristic acid
- C16:0:
-
Palmitic acid
- C16:1:
-
Palmitoleic acid C16:1Δ9
- C18:0:
-
Stearic acid
- C18:1:
-
Oleic acid C18:1Δ9
- C18:2:
-
Linoleic acid C18:2Δ9, Δ12
- C18:3:
-
Linolenic acid C18:3Δ9, Δ12, Δ15
- C20:0:
-
Arachidic acid
- C20:1:
-
Gadoleic acid C20:1Δ11
- ER:
-
Endoplasmic reticulum
- FATA:
-
Acyl-ACP thioesterase type A
- FATB:
-
Acyl-ACP thioesterase type B
- IM:
-
Interval mapping
- IV:
-
Iodine value
- LG:
-
Linkage group
- KAS (I II, III):
-
β-Ketoacyl-ACP synthase (I, II, III)
- K-W:
-
Kruskal–Wallis
- MAS:
-
Marker-assisted selection
- MQM:
-
Multiple-QTL model
- QTL:
-
Quantitative trait loci
- RFLP:
-
Restriction fragment length polymorphism
- SAD:
-
Δ9 stearoyl-ACP desaturase
- SNP:
-
Single nucleotid polymorphism
- SSH:
-
Suppression substractive hybridization
- SSR:
-
Simple sequence repeat
- TAG:
-
Triacylglycerol
References
AFNOR (1977) Norme Francaise NFT 60–233, “Préparation des esters méthyliques d’acides gras”, edited by Association Française de Normalisation (AFNOR)
Allard RW (1965) Genetic systems associated with colonizing ability in predominantly self-pollinated species. In: Baker HG, Stebbins GL (eds) The genetics of colonizing species. Academic, New York, pp 49–75
Al-Shanfari AB, Abdullah SNA, Saud HM, Omidvar V, Napis S (2011) Differential gene expression identified by suppression subtractive hybridization during late ripening of fruit in oil palm (Elaeis guineensis Jacq.). Plant Mol Biol Report 30(3):768–779
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basical local alignment search tool. J Mol Biol 215:403–410
Amblard P, Noiret JM, Kouamé B, Potier F, Adon B (1995) Performances comparées des hybrides interspécifiques et du matériel commercial E. guineensis. OCL Oléagineux, Corps Gras, Lipides 2:335–340
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J (2004) BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20(14):2321–2326
Argout X, Fouet O, Wincker P, Gramacho K, Legavre T, Sabau X, Risterucci AM, Da Silva C, Cascardo J, Allegre M, Kuhn D, Verica J, Courtois B, Loor G, Babin R, Sounigo O, Ducamp M, Guiltinan MJ, Ruiz M, Alemanno L, Machado R, Phillips W, Schnell R, Gilmour M, Rosenquist E, Butler D, Maximova S, Lanaud C (2008) Towards the understanding of the cocoa transcriptome: production and analysis of an exhaustive dataset of ESTs of Theobroma cacao L. generated from various tissues and under various conditions. BMC Genomics 9:512
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7:986–995
Banilas G, Karampelias M, Makariti I, Kourti A, Hatzopoulos P (2011) The olive DGAT2 gene is developmentally regulated and shares overlapping but distinct expression patterns with DGAT1. J Exp Bot 62:521–532
Beavis WD (1998) QTL analyses: power, precision, and accuracy. In: Paterson AH (ed) Molecular issection of complex traits. CRC Press, New York, pp 145–162
Beirnaert A, Vanderweyen R (1941) Contribution a l’étude génétique et biométrique des variétés d’Elaeis guineensis Jacq. Publ Inst Nat Etude agron Congo Belge Ser Sci 27:1–101
Billotte N (2004) Recherche et étude des locus contrôlant les caractères à déterminisme génétique complexe (QTL) du palmier à huile (Elaeis guineensis Jacq.), par cartographie génétique multiparentale. Thèse Ecole Nationale Supérieure Agronomique de Montpellier. pp 103
Billotte N, Risterucci AM, Barcelos E, Noyer JL, Amblard P, Baurens FC (2001) Development, characterisation, and across-taxa utility of oil palm (Elais guineensis Jacq.) microsatellite markers. Genome 44:413–425
Billotte N, Marseillac N, Risterucci AM, Adon B, Brottier P, Baurens FC, Singh R, Herran A, Asmady H, Billot C, Amblard P, Durand-Gasselin T, Courtois B, Asmono D, Cheah SC, Rohde W, Ritter E, Charrier A (2005) Microsatellite-based high density linkage map in oil palm (Elaeis guineensis Jacq.). Theor Appl Genet 110:754–765
Billotte N, Jourjon MF, Marseillac N, Berger A, Flori A, Asmady H, Adon B, Singh R, Nouy B, Potier F, Cheah SC, Rohde W, Ritter E, Courtois B, Charrier A, Mangin B (2010) QTL detection by multi-parent linkage mapping in oil palm (Elaeis guineensis Jacq.). Theor Appl Genet 120:1673–1687
Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31:365–370
Bourgis F, Kilaru A, Cao X, Ngando-Ebongue GF, Drira N, Ohlrogge JB, Arondel V (2011) Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc Natl Acad Sci USA 108:12527–12532
Browse J, McConn M, James D Jr, Miquel M (1993) Mutants of Arabidopsis deficient in the synthesis of α-linolenate: biochemical and genetic characterization of the endoplasmic reticulum linoleoyl desaturase. J Biol Chem 268:16345–16351
Chen H, Wang FW, Dong YY, Wang N, Sun YP, Li XY, Liu L, Fan XD, Yin HL, Jing YY, Zhang XY, Li Y, Chen G, Li HY (2012) Sequence mining and transcript profiling to explore differentially expressed genes associated with lipid biosynthesis during soybean seed development. BMC Plant Biol 12:122
Choo YM, Ma AN, Yap SC (1997) Carotenes, vitamin E and sterols in oils from Elaeis guineensis, Elaeis oleifera and their hybrids. Palm Oil Dev 27:1–9
Corley RHV, Tinker PB (2003) The oil palm, 4th edn. Blackwell Publishing, Oxford
Cottrell RC (1991) Introduction: nutritional aspects of palm oil. Am J Clin Nutr 53:989S–1009S
Dereeper A, Nicolas S, Le Cunff L, Bacilieri R, Doligez A, Peros JP, Ruiz M, This P (2011) SNiPlay: a web-based tool for detection, management and analysis of SNPs. Application to grapevine diversity projects. BMC Bioinforma 12:134
Durand-Gasselin T, Billotte N, Pomies V, Mastin G, Potier F, Amblard P, Flori A, Cochard B (2009) ID checking by microsatellite type markers (SSR) during the oil palm variety selection and production processes. “International seminar on Oil Palm Genomics and its application to Oil Palm Breeding.” ISOPB (The International Society for Oil Palm Breeders), conferences, Kuala Lumpur Convention Centre Malaysia
Ekpa OD, Fubara EP, Morah FNI (1994) Variation in fatty acid composition of palm oils from two varieties of the oil palm (Elaeis guineensis). J Sci Food Agric 64:483–486
Gascon JP, Wuidart W (1975) Amélioration de la production et de la qualité de l’huile d’Elaeis guineensis Jacq. Oléagineux 30(1):1–4
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-test cross mapping strategy and RAPD markers. Genetics 137:1121–1137
Guschina IA, Harwood JL (2007) Complex lipid biosynthesis and its manipulation in plants. In: Ranalli P (ed) Improvement of crop plants for industrial end uses. Springer, Dordrecht, pp 253–279
Ha BK, Monteros MJ, Boerma HR (2010) Development of SNP assays associated with oleic acid QTLs in N00-3350 soybean. Euphytica 176:403–415
Haldane JBS (1919) The combination of linkage values and the calculation of distance between the loci of linked factors. J Genet 8:299–309
Hardon JJ (1969) Interspecific hybrids in the genus Elaeis II. Vegetative growth and yield of F1 hybrids E. guineensis × E. oleifera. Euphytica 18:380–388
Hartley CWS (1988) The oil palm, 2nd edn. Longman, London
Hernández ML, Mancha M, Martínez-Rivas JM (2005) Molecular cloning and characterization of genes encoding two microsomal oleate desaturases (FAD2) from olive. Phytochemistry 66:1417–1426
Hu J, Li G, Struss D, Quiros C (1999) SCAR and RAPD markers associated with 18-carbon fatty acids in rapeseed, Brassica napus. Plant Breed 118:145–150
Hu X, Sullivan-Gilbert M, Gupta M, Thompson SA (2006) Mapping of the loci controlling oleic and linolenic acid contents and development of fad2 and fad3 allele-specific markers in canola (Brassica napus L.). Theor Appl Genet 113:497–507
ISO 3961:2009. Animal and vegetable fats and oils—determination of iodine value. International Standard. Fourth edition. Switzerland
Lespinasse D, Rodier-Goud M, Grivet L, Leconte A, Legnate H, Seguin M (2000) A saturated genetic linkage map of rubber tree (Hevea spp.) based on RFLP, AFLP, microsatellite, and isozyme markers. Theor Appl Genet 100:127–138
Li H, Zhao T, Wang Y, Yu D, Chen S, Zhou R, Gai J (2011) Genetic structure composed of additive QTL, epistatic QTL pairs and collective unmapped minor QTL conferring oil content and fatty acid components of soybeans. Euphytica 182:117–132
Luo L, Mao Y, Xu S (2003) Correcting the bias in estimation of genetic variances contributed by individual QTL. Genetica 119:107–113
Macfarlane N, Swetman T, Cornelius JA (1975) Analysis of mesocarp and kernel oils from the american oil palm and F1 hybrids with the west african oil palm. J Sci Food Agric 26:1293–1298
Maria M, Clyde MM, Cheah SC (1995) Cytological analysis of Elaeis guineensis (tenera) chromosomes. Elaeis 7:122–134
Meunier J (1975) Le « palmier à huile » américain Elaeis melanococca. Oléagineaux 30(2):51–61
Meunier J (1991) Una posible solución genética para el control de la pudrición de cogollo en la palma aceitera. Hibrido interespecifico Elaeis oleifera × Elaeis guineensis. Palmas 12(2):39–42
Meunier J, Boutin D (1975) L’Elaeis malanococca et l’hybride Elaeis melanococca × Elaeis guineensis Premières données. Oléagineaux 30(1):5–8
Meuwissen THE, Goddard ME (2010) Accurate prediction of genetic value for complex traits by whole-genome resequencing. Genetics 185:623–631
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Mohd Din A, Rajanaidu N, Jalani B (2000) Performance of Elaeis oleifera from Panama, Costa Rica, Colombia and Honduras in Malaysia. J Oil Palm Res 12(1):71–80
Monde AA, Michel F, Carbonneau MA, Tiahou G, Vernet MH, Eymard-Duvernay S, Badiou S, Adon B, Konan E, Sessa D, Cristol JP (2009) Comparative study of fatty acid composition, vitamin E and carotenoid contents of palm oils from four varieties of oil palm from Côte d’Ivoire. J Sci Food Agric 89:2535–2540
Monteros MJ, Burton JW, Boerma HR (2008) Molecular mapping and confirmation of QTLs associated with oleic acid content in N00–3350 soybean. Crop Sci 48:2223–2234
Muranty H (1996) Power of test for quantitative trait loci detection using full-sibs families in different schemes. Heredity 76:156–165
Muranty H (1997) Valorisation de plans de croisements pour la recherche de QTL chez les arbres forestiers, exemple d’un diallèle merisier. Thèse. Centre d’Orsay, Univerité Paris XI, France. 131 p
Nakkaew A, Chotigeat W, Eksomtramage T, Phongdara A (2008) Cloning and expression of a plastid-encoded subunit, beta-carboxyltransferase gene (accD) and a nuclear encoded subunit, biotin carboxylase of acetyl-CoA carboxylase from oil palm (Elaeis guineensis Jacq.). Plant Sci 175:497–504
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nishida I (2004) Plastid metabolic pathways for fatty acid metabolism. In: Daniell H, Chase C (eds) Molecular biology and biotechnology of plant organelles. Springer, Netherlands, pp 543–564
Noh A, Rajanaidu N, Kushairi A, Mohd Rafil Y, Mohd Din A, Mohd Isa ZA, Saleh G (2002) Variability in the fatty acid composition, iodine value and carotene content in the MPOB oil palm germoplasm collection from Angola. J Oil Palm Res 14:18–23
Noiret JM, Wuidart W (1976) Possibilités d’amélioration de la composition en acides gras de l'huile de palme. Resultats et perspectives. Oléagineux 31:465–474
Obasola C, Obisesan I, Opute F (1977) Breeding for short-stemmed oil palm in Nigeria III. Morphological characters, fruit form segregation and bunch quality of the first backcross population of interspecific hybrids of Elaeis oleifera × Elaeis guineensis to E. guineensis. In: Earp D, Newall W (eds) International developments in oil palm. Malaysian International Agricultural Oil Palm Conference, Kuala Lumpur, pp 68–94
Odegard J, Yazdi MH, Sonesson AK, Meuwissen THE (2009) Incorporating desirable genetic characteristics from an inferior into a superior population using genomic selection. Genetics 181(2):737–745
Ohlrogge JB, Jaworski JG (1997) Regulation of fatty acid synthesis. Annu Rev Plant Physiol Plant Mol Biol 48:109–137
Okuley J, Lightner J, Feldmann K, Yadav N, Lark E, Browse J (1994) Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid synthesis. Plant Cell 6:147–158
Ollagnier M, Olivin J (1984) Effects of nutrition on yield. Genetic progress and effects of nutrition on the quality of palm oil. Oléagineux 39:401–407
Ong SH, Chua CC, Sow HP (1981) The co-dominance theory: genetic interpretations of analyses of mesocarp oils from Elaeis guineensis, Elaeis oleifera and their hybrids. J Am Oil Chem Soc 58:1032–1038
Opute FI, Obasola CO (1979) Breeding for short-stemmed oil palm in Nigeria: fatty acids, their significance and characteristics. Ann Bot 43:677–681
Pamin K, Hutomo T, Purba AR (1995) Performance of growth, yield and oil quality of E. guineensis × E. oleifera insterspecific hybrids and their backcross. In: Seminar on worldwide performance of DXP oil palm planting materials, clones and interspecific hybrids. Proceedings. PORIM, Barranquilla, pp 101-107
Panthee DR, Pantalone VR, Saxton AM (2006) Modifier QTL for fatty acid composition in soybean oil. Euphytica 152:67–73
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J (2003) TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19:651–652
Poghosyan ZP, Haralampidis K, Martsinkovskaya AI, Murphy DJ, Hatzopoulos P (1999) Developmental regulation and spatial expression of a plastidial fatty acid desaturase from Olea europaea. Plant Physiol Biochem 37(2):109–119
Raghavan C, Collard BCY (2012) Effect of small mapping population sizes on reliability of quantitative trait locus (QTL) mapping. Afr J Biotechnol 4:10661–10674
Rey L, Ayala I, Delgado W (2003) Colecta de material genético de la palma americana Nolí Elaeis oleífera (H.B.K) Cortés en el trapecio Amazonico. Ceniavances 101:1–4
Rey L, Gómez PL, Ayala I, Delgado W, Rocha P (2004) Colecciones genéticas de palma de aceite Elaeis guineensis (Jacq.) y Elaeis oleífera (H.B.K) de Cenipalma: Características de importancia en el sector palmicultor. Palmas 25:39–48
Ritter E, Gebhardt C, Salamini F (1990) Estimation of recombination frequencies and constructions of RFLP linkage maps in plants from crosses between heterozygous parents. Genetics 224:645–654
Rival A, Beule T, Barre P, Hamon S, Duval Y, Noirot M (1997) Comparative flow cytometric estimation of nuclear DNA content in oil palm (Elaeis guineensis, Jacq.) tissue cultures and seed derived plants. Plant Cell Rep 16:884–887
Romualdi C, Bortoluzzi S, D'Alessi F, Danieli GA (2003) IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics 12(2):159–162
Rosillo-Calle F, Pelkmans L, Walter A (2009) A global overview of vegetable oils, with reference to biodiesel. A report for the IEA Bioenergy Task 40
Roy R, Steffens DL, Gratside B, Jang GY, Brumbaugh JA (1996) Producing STR locus patterns from bloodstains and other forensic samples using an infrared fluorescent automated DNA sequencer. J Forensic Sci 41:418–424
Salas JJ, Ohlrogge JB (2002) Characterization of substrate specificity of plant FatA and FatB acyl-ACP thioesterases. Arch Biochem Biophys 403:25–34
Sambanthamurthi R, Sundram K, Tan Y (2000) Chemistry and biochemistry of palm oil. Prog Lipid Res 39:507–558
Sarvamangala C, Gowda MVC, Varshney RK (2011) Identification of quantitative trait loci for protein content, oil content and oil quality for groundnut (Arachis hypogaea L.). Field Crops Res 122:49–59
Schiex T, Gaspin C (1997) CarthaGene: constructing and joining maximum likelihood genetic maps. Proc Int Conf Intell Syst Mol Biol 5:258–267
Schwendiman J, Pallares P, Amblard P (1982) Premiers examens des accidents de fertilité chez l’hybride interspécifique de palmier à huile Elaeis melanococca x E. guineensis. Oléagineux 37(7):331–341
Seng T-Y, Mohamed Saad SH, Chin C-W, Ting N-C, Singh R, Singh H, Zaman FQ, Tan S-G, Syed Alwee SSR (2011) Genetic linkage map of a high yielding FELDA DelixYangambi oil palm cross. PLoS One 6(11):e26593. doi:10.1371/journal.pone.0026593
Shimakata T, Stumpf PK (1982) Isolation and function of spinach leaf beta-ketoacyl-[acyl-carrier-protein] synthases. Proc Natl Acad Sci USA 79:5808–5812
Singh R, Tan SG, Panandam JM, Rahman RA, Ooi LCL, Low EL, Sharma M, Jansen J, Cheah S (2009) Mapping quantitative trait loci (QTLs) for fatty acid composition in an interspecific cross of oil palm. BMC Plant Biol 9:114
Smooker AM, Wells R, Morgan C, Beaudoin F, Cho K, Fraser F, Bancroft I (2011) The identification and mapping of candidate genes and QTL involved in the fatty acid desaturation pathway in Brassica napus. Theor Appl Genet 122:1075–1090
Stekel DJ, Git Y, Falciani F (2000) The comparison of gene expression from multiple cDNA libraries. Genome Res 10:2055–2061
Tan BK, Ong SH, Rajanaidu N, Rao V (1985) Biological modification of oil composition. J Am Oil Chem Soc 62:230–236
Tchobo FP, Natta AK, Barea B, Barouh N, Piombo G, Pina M, Villeneuve P, Soumanou MM, Sohounhloue DCK (2007) Characterization of Pentadesma butyracea Sabine butters of different production regions in Benin. J Am Oil Chem Soc 84:755–760
Tranbarger TJ, Dussert S, Joët T, Argout X, Summo M, Champion A, Cros D, Omore A, Nouy B, Morcillo F (2011) Regulatory mechanisms underlying oil palm fruit mesocarp maturation, ripening and functional specialization in lipid and carotenoid metabolism. Plant Physiol 156:564–584
Tranbarger TJ, Kluabmongko W, Sangsrakru D, Morcillo F, Tregear JW, Tragoonrung S, Billotte N (2012) SSR markers in transcripts of genes linked to post-transcriptional and transcriptional regulatory functions during vegetative and reproductive development of Elaeis guineensis. BMC Plant Biol 12:1. doi:10.1186/1471-2229-12-1
Uzun B, Arslan C, Furat S (2008) Variation in fatty acid compositions, oil content and oil yield in a germplasm collection of sesame (Sesamum indicum L.). J Am Oil Chem Soc 85:1135–1142
Vales MI, Schön CC, Capettini F, Chen XM, Corey AE, Mather DE, Mundt CC, Richardson KL, Sandoval-Islas JS, Utz HF, Hayes PM (2005) Effect of population size on the estimation of QTL: a test using resistance to barley stripe rust. Theor Appl Genet 111:1260–1270
Van Ooijen JW (2004) MapQTL5, software for mapping of quantitative loci in experimental populations. Kyazma BV. Wageningen, the Netherlands
Van Ooijen JW, Voorrips RE (2001) JoinMap® 4.0. Software for the calculation of genetic linkage maps. Kyazma BV. Wageningen, the Netherlands
Wasmuth JD, Blaxter ML (2004) prot4EST: translating expressed sequence tags from neglected genomes. BMC Bioinforma 5:187
Were BA, Onkware AO, Gudu S, Welander M, Carlsson AS (2006) Seed oil content and fatty acid composition in East African sesame (Sesamum indicum L.) accessions evaluated over 3 years. Field Crop Res 97:254–260
Wilson RF, Marquardt TC, Novitzky WP, Burton JW, Wilcox JR, Dewey RE (2001) Effect of alleles governing 16:0 concentration on glycerolipid composition in developing soybeans. J Am Oil Chem Soc 78:329–334
Wuidart W, Gascon JP (1975) Etude de la composition de l’huile d’Elaeis guineensis Jacq. Oléagineux 30:401–408
Xu S (2003) Theoretical basis of the Beavis effect. Genetics 165:2259–2268
Yang X, Guo Y, Yan J, Zhang J, Song T, Rocheford T, J-S LI (2010) Major and minor QTL and epistasis contribute to fatty acid compositions and oil concentration in high-oil maize. Theor Appl Genet 120:665–678
Ying J, Shan J, Gao J, Zhu M, Shi M, Lin H (2012) Identification of quantitative trait loci for lipid metabolism in rice seeds. Mol Plant 5(4):865–875
Zhao J, Dimov Z, Becker HC, Ecke W, Möllers C (2008) Mapping QTL controlling fatty acid composition in a doubled haploid rapeseed population segregating for oil content. Mol Breed 21:115–125
Zieler H, Richardson T, Schwartz A, Herrgard M, Lomelin D, Mathur E, Cheah SC, Tee TS, Lee WW, Chua KL, Kwan BH, Miller J, Koren S, Chan AP, Maiti R, Amedeo P, Ferriera S, Rogers Y-H, Sutton G, CD, Venter JC (2010) Whole-genome shotgun sequencing of the oil palm and Jatropha genomes. Plant & Animal Genomes XVIII Conference. January 9-13, 2010. San Diego, CA, USA
Acknowledgments
The experiment was part of a Ph.D. fellowship granted by the Centro de Investigaciones en Palma de Aceite (CENIPALMA), Colombia. This study was funded by the International Consortium Oil Palm Genome Projects (OPGP) and its members: Centre de Coopération Internationale en Recherche Agronomique pour le Développement (CIRAD), France; Instituto Vasco de Investigación y Desarrollo Agrario (NEIKER), Spain; Advanced Agriecological Research Sdn. Bhd (AAR), Malaysia; PT Astra Agro Lestari Tbk, Indonesia; PT Inti Indosawit Subur and its affiliates (Asian Agri Group), Indonesia; PT Bakrie Sumatera Plantations Tbk, Indonesia; Centro de Investigaciones en Palma de Aceite (CENIPALMA), Colombia; Empresa Brasileira de Pesquisa Agropecuária (EMBRAPA), Brazil; Felda Agricultural Services Sdn Bhd, Malaysia; Genting Plantations Berhad, Malaysia; IOI Corporation Berhad, Malaysia; Indonesian Oil Palm Research Institute (IOPRI), Indonesia; PT Matahari Kahuripan Indonesia, Indonesia; PT SMART Tbk, Indonesia; PT Sampoerna Agro Tbk, Indonesia; and SOCFIN Group, Luxemburg. Authors are thankful to Plantation Hacienda La Cabaña S.A. (Colombia), Indupalma LTAD (Colombia), the Centre de Recherche Agronomique des Plantes Pérennes (CRA-PP) of the Institut National de Recherche Agronomique du Bénin (INRAB), the Indonesian Oil Palm Research Institute (IOPRI), and the Centre National de Recherche Agronomique (CNRA) in Côte d’Ivoire for providing vegetal samples and/or phenotypic data. We would like to thank the Genoscope-CEA (Evry, France) for its support and the 454 Roche/GS-FLX Pyrosequencing of the Elaeis cDNA libraries. We wish to thank Dr. Timothy Tranbarger for his early provision of the PCR primer sequence information of the EST-SSR loci used in this study. We are also grateful to Dr. Brigitte Courtois, Dr. Jean-Marc Gion, Dr. Brigitte Mangin, and Dr. Dominique This for their scientific advice. Finally, we would like to thank the reviewers of this journal for their corrections and kind help to improve this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. Grattapaglia
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Montoya, C., Lopes, R., Flori, A. et al. Quantitative trait loci (QTLs) analysis of palm oil fatty acid composition in an interspecific pseudo-backcross from Elaeis oleifera (H.B.K.) Cortés and oil palm (Elaeis guineensis Jacq.). Tree Genetics & Genomes 9, 1207–1225 (2013). https://doi.org/10.1007/s11295-013-0629-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-013-0629-5