
Abstract
Brain edema is considered as a common feature associated with hepatic encephalopathy (HE). However, its central role as cause or consequence of HE and its implication in the development of the neurological alterations linked to HE are still under debate. It is now well accepted that type A and type C HE are biologically and clinically different, leading to different manifestations of brain edema. As a result, the findings on brain edema/swelling in type C HE are variable and sometimes controversial. In the light of the changing natural history of liver disease, better description of the clinical trajectory of cirrhosis and understanding of molecular mechanisms of HE, and the role of brain edema as a central component in the pathogenesis of HE is revisited in the current review. Furthermore, this review highlights the main techniques to measure brain edema and their advantages/disadvantages together with an in-depth description of the main ex-vivo/in-vivo findings using cell cultures, animal models and humans with HE. These findings are instrumental in elucidating the role of brain edema in HE and also in designing new multimodal studies by performing in-vivo combined with ex-vivo experiments for a better characterization of brain edema longitudinally and of its role in HE, especially in type C HE where water content changes are small.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brain edema is considered as a common feature associated with hepatic encephalopathy (HE). However, its central role as cause or consequence of HE, as well as its implication in the development of the neurological alterations linked to HE are still under debate. Moreover, it is still unclear whether HE and brain edema are the manifestations of the same pathophysiological mechanism or two different cerebral manifestations of brain dysfunction in liver disease, especially in the context of type C (cirrhosis) HE (Bosoi and Rose 2013; Cudalbu and Taylor-Robinson 2019). It is now well accepted that type A (acute liver failure (ALF)) and type C HE are biologically and clinically different, leading to different manifestations of brain edema (Bosoi and Rose 2013). For instance, in type C HE, the levels of ammonia in the blood are lower, variable and there is sufficient time for effective compensation and stabilization of the osmolyte shift to counteract the osmotic imbalance induced by the astrocytic accumulation of glutamine (Gln). As a result, the findings on net brain edema and type of edema in type C HE are variable and sometimes controversial (Cudalbu and Taylor-Robinson 2019). On the other hand, in type A HE, the natural history of the syndrome is rapid with higher ammonia levels in the blood, thus not allowing the system to compensate for metabolic changes.
As such, the role of brain edema as a central component in the pathogenesis of HE should be revisited in the light of the changing natural history of liver disease, better description of the clinical trajectory of cirrhosis and understanding of molecular pathogenesis of HE (Rose et al. 2020). The role of brain edema as the central mechanism of HE was driven by two observations. First, the seminal observation that astrocytes expressed glutamine synthase (GS), and that the production of Gln would lead to cytosolic hypertonicity and astrocytes would swell (Martinez-Hernandez et al. 1977). Second, the first description of a swollen astrocyte in post-mortem brain biopsies from patients dying due to acute liver failure (Type A HE) (Kato et al. 1992). These early findings should be examined in the light of subsequent developments in our understanding of the pathobiology of HE and its clinical manifestations.
Brain edema represents a net accumulation of fluid, mainly water, in the intracellular or extracellular spaces (ECS) of the brain, occurring on the background of an osmotic gradient. Cerebral edema is commonly observed in a variety of brain injuries. It can appear due to cytotoxic (alterations in cellular metabolism causing accumulation of osmotic molecules followed by entry of water to re-establish the osmotic equilibrium; and sometimes an increase in blood-brain-barrier (BBB) permeability) and/or vasogenic (physical breakdown of BBB) mechanisms (Bosoi and Rose 2013). It is important to emphasize that it is unusual for only vasogenic or cytotoxic edema to exist in isolation, therefore labeling a particular case of edema as “vasogenic” or “cytotoxic” cannot be rigidly applied. Overall, the development of one type of edema will gradually lead to the development of the other type, which also seems to be the case in HE where both types of edema might coexist, thus sometimes rendering it difficult to clearly distinguish them. It is important to mention that the terminology used is sometimes misleading, as edema should always refer to a net increase in water content in the brain. Sometimes edema is also referred to as a shift of water from one compartment to the other without a net increase of water amount in the brain, even though it is suspected that such a shift would always be accompanied by some degree of net brain edema through other mechanisms, such as osmotic gradient across the BBB (Bosoi and Rose 2013).
Role of astrocyte swelling
Although many lines of investigation over the years have confirmed the pathological role of astrocytes in animal models and humans of severe hyperammonemia and liver failure, it has also become clear that additional cell types such as the endothelial cells, neurons, microglia and pericytes in the brain are also involved in the pathogenesis of HE (Flatt et al. 2021; Hadjihambi et al. 2022; Haussinger et al. 1994, 2022; Mosso et al. 2022b; Pelle et al. 2022; Pierzchala et al. 2022; Rackayova et al. 2020, 2021; Rose et al. 2020; Simicic et al. 2022). Subsequent investigations have indicated that the hypothesis of ammonia-Gln-brain swelling is perhaps too simplistic. Indeed, mechanisms are much more complex and clinical observations, imaging studies and postmortem brain biopsies from patients dying with HE have indicated evidence of structural brain injury, presence of neuroinflammation, altered brain metabolism with Gln increase and gene expression affecting multiple pathways, endothelial dysfunction, alterations of blood and glymphatic flow, altered cellular bioenergetics, deposition of metals, mitochondrial dysfunction, lactate transport and senescence (Braissant et al. 2019; Haussinger et al. 2022; Pierzchala et al. 2022; Rose et al. 2020; Simicic et al. 2022). The most important learning from these advances is the associated clinical implications. An example is the observation that in a proportion of patients with HE who undergo liver transplantation, recovery from HE is incomplete, whist the brain continues to show loss of volume, arguing that HE is associated with neuronal loss (Garcia-Martinez et al. 2011; Jalan and Rose 2022; Ochoa-Sanchez et al. 2021). Another example is the lack of a direct correlation between therapies such as lactulose and rifaximin that are routinely used in clinical practice and either ammonia levels or brain swelling (Haussinger et al. 2022; Rose et al. 2020).
Clinical relevance of cerebral edema in HE
In reviewing the paper by Kato et al. (1992), in addition to demonstrating astrocyte swelling in type A HE, their main observations were regarding endothelial cell vacuolization, enlargement and vacuolation of the basement membrane and expansion of the extracellular processes. Derangements of the pericytes were also observed. Although they suggested that cytotoxic edema may be operative, they were not convinced that this was not due to post-mortem effects. It appears that what they were describing was indeed vasogenic brain edema, loss of pericytes, mitochondrial disruption and loss of integrity of the BBB. It is tempting to hypothesize that these early findings might have driven the studies that followed (Martinez-Hernandez et al. 1977) to support the ammonia-Gln-brain swelling hypothesis and partly overlooked the other observations made. Indeed, subsequent studies went on to show the importance of alterations in cerebral blood flow (CBF) (both hyperemia and ischemia) (Jalan et al. 2004; Wendon et al. 1994), systemic and neuroinflammation (Rolando et al. 2000) and, evidence that in the sickest patients with ALF with uncontrolled intracranial hypertension the BBB breaks down and the brain becomes a net cytokine producer (Wright et al. 2007b). Further experimental studies in animal models of ALF showed that intracranial pressure (ICP) could be modulated using albumin dialysis without any impact on the ammonia-Gln-brain swelling pathway, arguing for the limited involvement of cytotoxic edema in the pathogenesis of HE in ALF (Sen et al. 2006).
The link to cerebral edema being the central operative mechanism of HE in cirrhosis is challenging as type C HE develops differently from type A HE and presents lower blood ammonia levels and water content increase, with a longer disease time course. Moreover, these patients show a very wide spectrum of clinical and pathophysiological phenotypes that manifest with varying severities of HE (Rose et al. 2020). It is difficult to conceive how cerebral edema can be pathogenetically important in mobile patients with minimal or Grade 1 HE. Nonetheless these patients show an increased brain Gln and decreased brain osmolytes, signs of osmotic stress which cannot be neglected and which were interpreted as low-grade edema as in type C HE (Haussinger 2006; Haussinger et al. 1994). Indeed, although some of the early imaging studies have measured increased brain Gln and altered brain water, the relationship between one and the other is still not fully elucidated (Cudalbu and Taylor-Robinson 2019). Several studies using magnetic resonance imaging (MRI) and positron emission tomography (PET) scanning have pointed to additional features such as deposition of metal, altered CBF, bioenergetics, oxygen consumption and evidence of neuroinflammation (Haussinger 2006; Haussinger et al. 1994; Mosso et al. 2022b). In patients with Grades 2–4 HE, cerebral edema is observed (Oeltzschner et al. 2016; Shah et al. 2008; Winterdahl et al. 2019). This is unlikely to be due to cytotoxic brain edema alone and more likely it is attributed to multiple related factors, such as neuroinflammation, altered metabolism and CBF and neuronal injury. Studies in animal models of type C HE have, however, showed an increase in brain water content (Bosoi et al. 2012), Gln, decrease of brain osmolytes and structural changes in astrocytes, neurons and microglia (Bosoi et al. 2014; Braissant et al. 2019; Jaeger et al. 2019; Pierzchala et al. 2022; Rackayova et al. 2016, 2020, 2021; Rose 2010; Simicic et al. 2022). An increase in brain Gln will eventually lead to cellular microstructural changes despite the osmoregulation (i.e. release of other brain osmolytes) (Mosso et al. 2021, 2022a). Whether this is involved in patients with covert HE or overt HE remains to be investigated. Therefore, additional longitudinal studies are needed to follow brain Gln, water, microstructural changes, oxidative stress (OS), neuroinflammation, bioenergetics, metabolism together with blood ammonia changes in patients with type C HE.
Acute-on-chronic liver failure (ACLF) is a syndrome that occurs in patients with cirrhosis who are hospitalized with acute decompensation. It is characterized by the failure of hepatic and extrahepatic organs and high rates of mortality (Arroyo et al. 2020). The syndrome is characterized pathophysiologically by systemic inflammation (Claria et al. 2016) consequent on accumulation of pathogen and damage associated molecular patterns, as well as altered signaling that culminates in mitochondrial dysfunction (Moreau et al. 2021). The occurrence of HE in patients with ACLF significantly increases the risk of death (Cordoba et al. 2014). From the pathophysiologic perspective, cerebral edema is observed in about 5% of patients (Joshi et al. 2014). In animal models of ACLF, cerebral edema is frequent and is associated with hyperammonemia, neuroinflammation, altered brain oxygenation, neuronal loss and disturbances of the BBB (Rose et al. 2020; Sawhney et al. 2016). The severity of hyperammonemia is an independent predictor of mortality and a reduction in the ammonia levels is associated with improved survival. Taken together, the brain dysfunction of ACLF is clinically, pathophysiologically and prognostically distinct from both types A and C HE. At present, no special category has been allocated to HE in ACLF patients and continues to be classified under Type C HE (Jalan and Rose 2022). This issue should be revisited in future consensus discussions.
Of note, the current review is not questioning the presence of brain edema in HE. However it is highlighting the fact that it is now accepted that HE is a multifactorial disease where brain edema might not be the only main event. Therefore, we believe that the role of cytotoxic astrocytic brain edema as being central to the pathogenesis of HE should be revisited considering the current understanding of the syndrome (Fig. 1).

Summary of the current knowledge related to edema formation from in-vitro, ex-vivo brain tissue, and in-vivo studies in different HE animal models and patients
Current technics to measure brain edema
Several ex-vivo/invasive or in-vivo/non-invasive methods have been used in the past to assess brain water content and consequently brain edema in both animal models and patients with HE. A detailed description of these methods can be found elsewhere (Cudalbu and Taylor-Robinson 2019). Table 1 presents a summary of the advantages and disadvantages of these methods, while the following tables (Tables 2, 3 and 4) show the main results obtained using these techniques in cell cultures, as well as different animal models and patients with HE (summarized in Fig. 1).
In rodent studies, the most frequently used methods for brain water measurements include the dry/wet weight technique (Marmarou et al. 1978) and the specific gravity method (Hayazaki et al. 1995). While no specific skills or state-of-the-art equipment are required for these methods, just as with the current imaging techniques, the extra- and intracellular water content cannot be distinguished (Bemeur et al. 2016; Cudalbu and Taylor-Robinson 2019). Electron microscopy (EM) offers extracellular space images with nanoscale resolution (Kasthuri et al. 2015). This technique offered the first evidence of a potentially cytotoxic edema, characterized by Alzheimer’s Type II astrocytes, in animals (Norenberg and Lapham 1974) and patients with chronic liver disease (CLD) and HE (Agarwal and Mais 2019). However, the major disadvantage of EM is the need for tissue fixation, making it incompatible with live tissue and real time recordings. Classical chemical fixation dehydrates the sample and effectively disturbs the water distribution in the tissue, leading to cell swelling and shrinkage of the extracellular space (Korogod et al. 2015). While cryofixation immobilises molecules in their hydrated state (reducing structural fixation artifacts), only the outermost few microns of the tissue sample are reportedly artifact-free (Studer et al. 2008), making EM suboptimal for studying brain edema.
Brain water mapping using MRI (Shah et al. 2008; Winterdahl et al. 2019) has been shown to be the most precise non-invasive method that can be applied to patients with HE for absolute quantification of cerebral hydration status in all brain regions (e.g., 1% change in water content have been detected in HE patients) (Oeltzschner et al. 2016; Shah et al. 2008). However, it lacks specificity regarding the etiology of water accumulation (Bemeur et al. 2016). Some other in-vivo and non-invasive methods like diffusion-weighted MRI (DW-MRI), a powerful tool to probe brain microstructure, allow the non-invasive assessment of the microstructural changes in the brain during HE (e.g., extra- and intra-cellular water diffusivity and fraction, soma and processes radius, orientation dispersion of fiber bundles) without a firm conclusion on the absolute water content or on the cell-specificity of these changes (Fig. 2). Water diffusion is hindered or restricted by the brain cellular environment, resulting in a deviation of the MR signal diffusion attenuation from the one expected for free diffusion. With appropriate modelling, the study of these diffusion properties can inform on brain tissue morphology. In particular, the technique is known to be sensitive to subtypes of brain edema especially in stroke, where DW-MRI findings were validated against complementary ex-vivo methods (Benveniste et al. 1992; Moseley et al. 1990): cytotoxic edema (associated with a decreased apparent diffusion coefficient (ADC)) versus vasogenic edema (associated with increased ADC) (Ebisu et al. 1993; Ito et al. 1996). This interpretation of DW-MRI metrics requires validations by specific ex-vivo/histological measures when using new animal models. Moreover, deciphering the underlying mechanism resulting in a decreased ADC remains challenging: it could be due to greater contribution of the slowly-diffusing intracellular space (Benveniste et al. 1992), increased tortuosity of the extracellular space (Latour et al. 1994), neurite beading (Budde and Frank 2010), increased cytoplasmic viscosity (Goodman et al. 2008) or increased fraction of membrane-bound water (Jelescu et al. 2014). These multiple causes contribute to some limitations of the technique to inform on edema. Recently, several biophysical models have been developed in the attempt to overcome these limitations (Jelescu et al. 2020). Most literature in cirrhosis patients with or without HE reports an increased water ADC or mean diffusivity (MD) (Chavarria et al. 2011, 2013a; Kale et al. 2006; Lodi et al. 2004; Mardini et al. 2011). These findings are supported by a recent study in bile-duct ligated (BDL) rats where an increased water MD was also observed at week 6 post-surgery, with respect to Sham-operated rats (Mosso et al. 2022a). In-vivo proton magnetic resonance spectroscopy studies (1H MRS) were among the first in-vivo studied used to shed light on the presence of brain Gln increase leading to the presence of osmotic stress (decreased brain myo-inositol (mIns), total choline (tCho), taurine (Tau), creatine (Cr) in type C HE (Braissant et al. 2019; Rackayova et al. 2020; Kreis et al. 1992) which was linked to the development of low-grade brain edema (Haussinger 2006; Haussinger et al. 1994) (Fig. 2). Diffusion-weighted MR spectroscopy (DW-MRS), which studies the diffusion properties of metabolites that are generally observed in 1H MRS, offers a greater cell-specificity than DW-MRI (Fig. 2). Some metabolites are known to be mostly astrocytic (mIns, Gln) while others mostly neuronal (N-acetylaspartate (NAA), glutamate (Glu)). DW-MRS could thus allow to study edema at the cellular level. The first DW-MRS study on type C HE showed an increased ADC of some glial (mIns) and neuronal metabolites (Glu) as well as of some osmolytes (mIns, Tau) in adult (Mosso et al. 2021) andyoung (BDL surgery at 21 days after birth) BDL rats at week 6 post-surgery (Mosso et al. 2022a).

In-vivo MRS, DW-MRS and DW-MRI in the study of HE. Left panel – brain metabolism: In-vivo 1H MRS - Representative neurometabolic profiles in one BDL and one Sham rat acquired at 14.1T in the cerebellum. Arrows show significant changes in the BDL group (increase in Gln, decrease in osmolytes). Right panel - microstructure: DW-MRS - Representative spectra at different b-values and intra-cellular diffusion attenuation of mIns in the BDL and Sham groups, showing faster mIns diffusivity in BDL rats (as measured from, for e.g., the randomly oriented sticks model (Callaghan et al. 1979)). DW-MRI - Color coded fractional anisotropy map of an axial slice through the body of cerebellum. Water diffusivity is also increased in BDL rats.
In summary, to date there is no “absolute” method to assess brain edema and directly quantify the absolute water content at the cellular level. Overall, current techniques are either only available post-mortem or lack the required resolution and specificity to directly answer the pending question of whether edema in type C HE is of cytotoxic and/or vasogenic origin. In order to address this, both the extracellular space and cell volume/morphology need to be assessed, while methods with increased specificity, resolution and precision are required especially in type C HE where water content changes are small. To overcome these limitations, one solution could be the usage of a multimodal approach by performing in-vivo combined with ex-vivo experiments for brain water content measurements and for structural cellular changes. This approach would allow monitoring of the progression of the disease longitudinally, providing additional information on the temporal resolution of the onset of brain edema. The assessment of brain regional differences in HE is also crucial since some studies pointed towards a brain regional difference (Cudalbu and Taylor-Robinson 2019), which might explain some of the neurological dysfunction in HE, especially in type C HE.
What we learned from cell studies: ammonia toxicity in-vitro
Although the cell disease models are intrinsically limited, i.e., they lack the complex structure, cell type heterogeneity / in-situ environment (lack of vascular delivery system and BBB) and physical history of diseased tissues, astrocytic cultures are extensively utilized to investigate the primary effects of ammonia (hyperammonemia) on brain (Gorg et al. 2008; Gregorios et al. 1985a, b; Häussinger et al. 1997; Lu et al. 2019; Murthy and Norenberg 2002; Norenberg 1987; Norenberg et al. 1991; Rao et al. 2010; Schliess et al. 1996). It is known that astrocytes protect neurons from ammonia toxicity and the importance of structural complexity in in-vitro studies as a model of HE was demonstrated in studies on astrocytes-neurons co-culture (Rao et al. 2005).
In in-vitro studies of ammonia treated astrocytes, intracellular water content varies between reports and ammonia concentrations. Treatment with 5 mM ammonia increased cell volume by 12–43% (Murthy and Norenberg 2002; Norenberg et al. 1991; Rao et al. 2010; Sinke et al. 2008). Difference in cellular edema was also related to the applied serum. Astrocytic cultures maintained in fetal calf serum did show some swelling upon pathological treatment with ammonia, but the effect was smaller and not as consistent as that seen in horse serum-treated cells (Norenberg et al. 1991). Studies have shown that co-treatment with 0.1 mM aspartate suppressed the ammonia-induced swelling of astrocytes by 68%, while 1 mM ornithine had no significant effect on cell morphology (Murthy and Norenberg 2002).
In ex-vivo of brain slices exposed to ammonia, water content increased in a concentration dependent manner, with 5 mM ammonia increasing the water load by 3% and 10 mM ammonia by 6% (Ganz et al. 1989). Swelling effect differed also between species (Benjamin et al. 1978). Guinea pig brain slices exposed to both 5- and 10-mM ammonia did not show significant swelling, while rat brain slices showed significant swelling only when treated with 10 mM ammonia (Benjamin et al. 1978).
Other studies of ex-vivo brain slices exposed to 10 mM ammonia have shown even more pronounced swelling, with slice thickness increasing by ~ 30% (441 ± 40 μm vs. control: 345 ± 33 μm) (Back et al. 2011). Among the 30% increase, only 8% was associated with astrocytic volume changes. Tissue stained with glial fibrillary acidic protein (GFAP) antibody as an astrocyte cytoplasmic fractional volume marker, have shown an increase of immunoreactive voxels (~ 8%) in z-axis (depth), with no change in GFAP content detected (Back et al. 2011).
Increased water uptake in ex-vivo brain slices exposed to toxic levels of ammonia (5–10 mM) was accompanied by an increased tissue content of Na+ and Cl− and loss of K+ (Benjamin et al. 1978), which was suggested to be related to sodium pump impairment as a result of energy failure (Gregorios et al. 1985a).
In-vitro ammonia induced cell swelling and elevation of intracellular osmolarity may be related to the accumulation of Gln and Ca2+ content reduction (Norenberg et al. 1991). Treatment with extracellular adenosine triphosphate (ATP) showed increased Ca2+ influx and accumulation and resulted in reduction of ammonia induced astrocyte swelling (Norenberg et al. 1991). Cell swelling has been also linked to a variety of mechanisms, among which are defects in: ion pumps/channels (Na+/K+), cotransporters (Na+/K+/Cl−), release of osmotically active amino acids and the buildup of osmotically active compounds (Norenberg et al. 1991). Increased Gln production in hyperammonemia deprives the cytosolic glutamate pool, compromising the activity of the malate–aspartate shuttle (MAS), which transports reducing equivalents from the cytosol into the mitochondria (Murthy and Norenberg 2002). Furthermore, cytosolic Glu depletion in the presence of ammonia inhibits pyruvate oxidation and its further entry into the citric acid cycle, resulting in pyruvate to lactate conversion for nicotinamide adenine dinucleotide (NAD) from NADH regeneration and glycolysis continuation (Murthy and Hertz 1988; Murthy and Norenberg 2002).
In-vitro findings indicated activation of mitogen-activated protein kinase (MAPKs) in a Rat Sarcoma Virus (Ras) / Rapidly Accelerated Fibrosarcoma (Raf)- and phosphatidylinositol 3-kinases (PI3-kinase)-dependent manner, triggered by swelling-induced Ca2+ signal in astrocytes exposed to a hypoosmotic environment (Häussinger et al. 1997). In addition, hypoosmotic shock leads to a rapid release of osmolytes (mIns and Tau) most likely via the opening of unspecific channels in the plasma membrane (Häussinger et al. 1997).
The synchronous treatment of astrocytes with hazardous doses of ammonia and cytokines (tumor necrosis factor-α (TNF-α), interleukin 1β (IL-1β), interleukin 6 (IL-6) and interferon-γ (IFN-γ)) had no additive or synergistic impact on swelling. On the other hand, astrocytic swelling increased after a 24-hour pretreatment with ammonia followed by a 24-hour exposure to cytokines (Rao et al. 2010). In addition, exposure to both, ammonia and cytokines, separately or simultaneously, induce the nuclear factor-κB (NF-κB) activation (Rao et al. 2010), which thereafter will influence cell survival in a complex way (neuroprotective or proinflammatory), depending on the pathological stage (Back et al. 2011). NF-κB activation suppression by NF-κB and MAPKs inhibitors, and antioxidants (superoxide dismutase (SOD), vit. E) prevented astrocytic swelling in cultures pre-treated with ammonia and followed by cytokines (Rao et al. 2010; Sinke et al. 2008). These studies indicate a pivotal role of NF-κB in the potentiation of cellular edema. NF-κB activation blockage was also associated with a reduction in the ammonia-induced increase in oxidative/nitrosative stress, inducible nitric oxide synthase (iNOS) protein expression and nitric oxide (NO) generation, plausibly one of the main factor of the ammonia-triggered astrocytic swelling mechanism (Rao et al. 2010; Sinke et al. 2008).
A recent in-vitro and ex-vivo study has indicated that even low concentrations of ammonia (1–5 µM) can induce reactive oxygen species (ROS) thus leading to neuronal cell death (Angelova et al. 2022).
In-vivo longitudinal antioxidant system impairment indicated the presence of OS (Braissant et al. 2019). The observed ascorbate (Asc) decrease (Braissant et al. 2019) can be related to the increased CNS and liver ROS levels (Pierzchala et al. 2022; Simicic et al. 2022), compromised Asc synthesis due to diseased liver (Linster and Van Schaftingen 2007), and decreased exogenous availability related to nutrient deficiency (Ipsen et al. 2014). Ex-vivo longitudinal CNS OS detection demonstrated an increased superoxide anion (\({\text{O}}_{2}^{.-}\)) production in BDL rats, which was due to enhanced formation of ROS rather than a decrease of antioxidants activity (elevated levels of SOD1/2 and GPX-1 were observed) (Pierzchala et al. 2022).
As a more complex system to simple cell cultures, regular brain cell 3D cultures (i.e., mixed-cell cultures) prepared from the telencephalon of rat embryos have also been challenged with ammonia (5 mM) (Braissant et al. 2008; Braissant et al. 2002). In 3D brain cell culture, ammonia caused a decrease of endogenous synthesis of creatine and induced astrocyte swelling. Creatine co-treatment prevented this effect (Braissant et al. 2002).
The majority of the studies published till now focused on astrocytes (Table 2) and showed an ammonia concentration dependent (5–10 mM) swelling/edema. The organotypic slice model of ammonia-induced (10 mM) brain swelling additionally demonstrated an increase of neuronal soma diameter associated with large swollen nuclei (Back et al. 2011). It is important to highlight that the majority of ammonia levels used in these studies might have been too high and thus not physiologically relevant for both type A and C HE. Therefore, it remains to conclude if such high ammonia concentrations are required to induce swelling/edema or if the detection techniques used to date were not sensitive enough to detect edema/swelling at lower ammonia concentrations. Measuring brain tissue ammonia levels is difficult due to the multiple preanalytical steps (i.e., extraction of the brain, postmortem metabolic changes, the detection technique) with cerebrospinal fluid (CSF) and microdialysates measurements of ammonia being performed more often (DeMorrow et al. 2021). We encourage the development/validation of new techniques to measure brain ammonia in type A and C HE and afterwards establish if these levels of ammonia would lead to the same amount of swelling/edema. Moreover, an experimental correlation between brain, blood and CSF ammonia levels in the same animal model is highly warranted for the HE community and worth considering in the future. Additional works should also focus on characterizing other cell types and not only astrocytes.
What we learned from animal models of type A HE
Ammonia neurotoxic outcomes are based on glial and neuronal cell dysfunction (Braissant et al. 2019; Rangroo Thrane et al. 2013). In-vivo type A HE manifests with a progressive increase in the water content of cortical gray matter, astrocyte morphology alterations/swelling of an intracellular compartment (Blei et al. 1994; Master et al. 1999; Norenberg 1977, 1987; Norenberg and Lapham 1974; Rangroo Thrane et al. 2013; Swain et al. 1991, 1992; Traber et al. 1986, 1987, 1989; Zamora et al. 1973) as well as a strong link between astrocyte swelling and OS (Reinehr et al. 2007).
Overall, based on in-vitro studies, neurotoxicity may rise not just from the consequences of tissue/astrocyte swelling and elevated ICP, but also from acute ammonia toxicity to neurons, in particular when extremely high ammonia levels are persisted for a longer period (Back et al. 2011). Given the fact that GS in the brain is located mainly in astrocytes, its role is to protect neurons from excitotoxicity caused by excess of ammonia by converting it into Gln in the presence of Glu and ATP (Suarez et al. 2002; Traber et al. 1989). However, the potential to improve ammonia detoxification by GS is limited (Butterworth et al. 1988). Therefore, the protection of neurons by astrocytes may be compromised, and accumulation of water and cytotoxic compounds, among which are ROS and reactive nitrogen species (RNS) (Pierzchala et al. 2022), would cause deleterious effects on neurological function.
Similar to the results obtained from brain slices exposed to the toxic levels of ammonia (Benjamin et al. 1978), in-vivo studies confirmed that ammonia has an impact on astrocyte potassium buffering, leading to a rise in extracellular \({\left[{\text{K}}^{+}\right]}_{\text{o}}\) (~ 2 mM) with immediate and acute neurological deterioration (seizures in awake animals) (Rangroo Thrane et al. 2013). Excess of both \({\left[{\text{N}\text{H}}_{4}^{+}\right]}_{\text{o}}\) and \({\left[{\text{K}}^{+}\right]}_{\text{o}}\) promotes further overactivation of the Na+-K+-2Cl− cotransporter isoform 1 (NKCC1) in neurons and selectively impairs cortical inhibitory neurotransmission, while NKCC1 inhibition reduces the neurological symptoms of acute ammonia intoxication (Rangroo Thrane et al. 2013). Furthermore, inhibition of GS worsened the neurological outcome by increasing the \({\left[{\text{N}\text{H}}_{4}^{+}\right]}_{\text{o}}\) and \({\left[{\text{K}}^{+}\right]}_{\text{o}}\) load on neurons (Rangroo Thrane et al. 2013).
EM studies on the brains of type A HE animal models disclosed morphological changes of astrocytes. In the early phase represented by cellular hypertrophy, an increased number of cytoplasmic organelles including mitochondria, rough and smooth endoplasmic reticulum was observed (Norenberg 1977). This early reaction may occur to preserve cellular homeostasis and ammonia detoxification. During the late/coma phase, astrocyte cytoplasmic vacuolization and mitochondrial contraction (Norenberg 1977; Schliess et al. 2009) is a direct sign of inclined deterioration of their protective role, changes in synaptic transmission, and cell death (Master et al. 1999; Shubin et al. 2016).
Methionine-sulfoximine (MSO), an irreversible GS inhibitor, has been shown to avoid the negative effects of hyperammonemia on brain glucose consumption, improve the degree of cerebral edema and the rise in CBF, and prevent intracellular Gln buildup in a dose-dependent manner in the portocaval anastomosis (PCA) model with intravenous infusion of ammonia (Master et al. 1999). The decrease of intracellular Gln was accompanied by an increase in brain Glu levels, as well as plasma and CSF ammonia concentrations (Master et al. 1999).
Brain edema occurs rapidly in type A HE animal models. According to published data (Table 3), brain edema, predominantly attributable to astrocyte swelling, is the major neuropathological hallmark of ALF and plays a critical role in the development of high ICP (Traber et al. 1986) which is related to pathophysiological ammonia levels. Furthermore, there is a significant correlation between Gln levels in the blood, brain, and CSF and the level of ICP in ALF. Additionally, acute Aquaporin 4 (Aqp4) protein rise in the brain is unfavorable, indicating a higher risk of brain edema.
In addition to type A HE animal models, recent in-vivo hyperammonemia studies (two-photon microscopy) have shown no astrocyte swelling or brain edema in the acute phase of ammonia increase. Instead astrocyte shrinkage was observed lasting for 30 min after intraperitoneal (i.p.) ammonia injection (Rangroo Thrane et al. 2013). To induce brain edema and astrocyte swelling (50 mM in-situ), a lethal dose of ammonia (10 mmol/kg i.p.) was required (Rangroo Thrane et al. 2013).
What we learned from type C HE
In-vivo – animal models of type C HE
Several studies have investigated the changes in brain water content in animal models of type C HE, with the BDL model being the widest used, a model endorsed by ISHEN (DeMorrow et al. 2021). The results of these studies are summarized in Table 4. As indicated, the gravimetry technique seems to be the most used. Nonetheless, there are only a few published studies using these techniques, and the results appear to be controversial, with studies reporting changes in BDL rats while others not. Of note, the changes reported are small, e.g., 1–2% increase in water content, similar with what was measured in humans using absolute water measurements with MRI. Complementary methods to evaluate the hydration status of the brain might be useful. MRI and MRS offer the possibility to evaluated in-vivo and longitudinally changes in brain structure/microstructure, volumes, osmoregulation, metabolism and can be used complementary to any ex-vivo end point measurement as gravimetry or EM. The increase in brain Gln was a consistent finding in the majority of type C HE work published till now, in animal models but also in humans. In addition, a decrease of other brain osmolytes, as an osmotic counteraction of the increase in brain Gln, was also measured (Table 4). These findings were interpreted to reflect a volume regulatory response to compensate for Gln induced osmotic imbalance in astrocytes, as Gln is considered to be mainly located in astrocytes. It has been speculated that there is a brain regional difference in type C HE (Mosso et al. 2022b; Flatt et al. 2021), therefore it would be interesting to establish if brain metabolism/microstructure and other parameters are brain region dependent, together with possible changes in the morphology/structure of astrocytes, neurons, microglia and endothelial cells. These studies would be instrumental in identifying the ammonia fingerprints in the brain.
As mentioned before, HE is a multifactorial disease with different mechanisms acting together inducing neurological implications. As such, it has been shown in animal models that ammonia, OS, and inflammation all play a role in the pathogenesis of type C HE (Pierzchala et al. 2022; Wright et al. 2007a) (Table 4), as well as associated brain edema (cortex) (Bosoi et al. 2012). Treatment with allopurinol (an inhibitor of the oxidant enzyme xanthine oxidase (XO), an OS marker) reduced OS while normalizing water content in male rats (Bosoi et al. 2012), whilst treatment with probiotics reduced brain Gln concentrations (Rackayova et al. 2021). Furthermore, endotoxemia has been shown to impact brain edema in BDL rats following an increased inflammatory response (Wright et al. 2007a). Elevated ammonia, OS, and simultaneous inflammation induced ribonucleic acid (RNA) oxidation (8-Oxo-2’-deoxyguanosine (Oxo-8-dG) accumulation) in primary astrocytes cultures (Giorgio et al. 2020; Gorg et al. 2008; Korkmaz 2018; Kruczek et al. 2011; Schliess et al. 2009) and in the BDL rat model of type C HE, with dominating cytoplasmic localization indicating interplay of cytosolic and mitochondrial nucleic acids with hydroxyl radical (\({\text{HO}}^{\cdot}\))(Pierzchala et al. 2022). RNA oxidation alters translational machinery and gene expression, revealing a link between ammonia-induced OS (Pierzchala et al. 2022) and cognitive impairment via altered protein synthesis (Nunomura et al. 2017) and neurotransmission (Kumar et al. 2018). EM of BDL cortical samples revealed partially collapsed microvessels, as well as minimal water accumulation in the astrocytic endfeet and perivascular tissue (Wright et al. 2007a). Enlarged astrocytic endfeet, perivascular edema, and collapsed microvessels were also evident in BDL rats following an acute inflammatory response (Wright et al. 2007a). Several brain areas from BDL rats showed enlarged mitochondria with ruptured membranes, unfolded cristae, and a non-dense matrix (Dhanda et al. 2018). These morphological alterations have been shown to contribute to the reported impaired ATP synthesis and amplify mitochondrial OS (Dhanda et al. 2018).
Human type C HE
The main findings using in-vivo MRI and MRS techniques in patients with HE have been previously discussed (Cudalbu and Taylor-Robinson 2019). Volumetric MRI results have highlighted a decrease in brain volume in type C HE, mainly in gray matter , with functionally well-compensated patients with cirrhosis showing no brain volume changes. The changes in brain volume measured in chronic HE were mainly associated with brain atrophy (Bosoi and Rose 2013). A few absolute brain water content measurements have also been done in type C HE, showing a small increase in brain water content (around 1%) (Shah et al. 2008; Winterdahl et al. 2019) with other studies showing no significant increases in patients with covert HE (minimal HE or HE grade 1) (Oeltzschner et al. 2016). Magnetization transfer (MT) MR experiments highlighted a small decrease in magnetization transfer ratio in type C HE, which was consistent among studies and was interpreted as low-grade astrocytic/cerebral edema. This might also be linked to alterations in membrane permeability and cytoplasmic structure and to subsequent shifts in the distribution of macromolecules and intracellular water, with subtle alterations in intracellular and extracellular edema. Some studies where DW-MRI experiments were performed showed a mild increase in ADC in patients with cirrhosis, even with minimal HE . The overall agreement of all these measurements seems to be linked to an increase in water content, however additional studies are required to unravel the cellular origin which remains controversial. Future experiments using adapted biophysical models would potentially help elucidating these questions.
Based on the data published to date, the small change in water content in patients with type C HE are consistent and in line with the results obtained in animal studies. These results seem to highlight that in addition to brain edema other mechanisms are involved in type C HE, however further experiments are required.
1HMRS studies in patients with type C HE highlighted stronger changes in brain metabolites (Glx (Glu + Gln)/Cr, mIns/Cr, and tCho/Cr) in overt HE patients, while in minimal HE, the decrease in mIns/Cr was observed more often than an increase in Glx/Cr. In functionally well-compensated liver cirrhosis patients, no significant changes were measured (Cudalbu et al. 2023). Overall, these studies have been performed at lower magnetic fields, raising the questions as to whether metabolite changes occur in well-compensated liver disease patients, or if these changes are very small, and thus not detectable at lower magnetic fields.
New approaches to evaluate brain edema and/or extra- and intracellular water content and microstructural changes
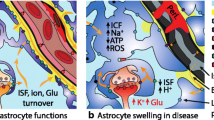
By virtue of the perivascular location of astrocytes and their high expression of water transporting membrane proteins, water is believed to preferentially enter via astrocyte membranes (Papadopoulos et al. 2004). This renders astrocytes more susceptible to swelling than neurons, especially when their adaptive capacity for volume regulation is exhausted (Bemeur et al. 2016; Bosoi and Rose 2013; Papadopoulos et al. 2004; Thrane et al. 2015). Morphological features of astrocytes, such as fine processes make up a large proportion of the cell volume and are in close contact with synapses and other components of brain parenchyma such as the vasculature (Wilhelmsson et al. 2006). Antibodies against GFAP, the most frequently used astrocyte marker (Eng et al. 2000), reveal the cytoskeletal structure but do not label all portions of the astrocyte. GFAP is entirely absent from the finely branching processes and is often not detectably present in the cell body. Consequently, GFAP immunohistochemistry can markedly underestimate the extent of astrocyte branching and territory, especially when performed in 2D. While other molecular markers have been used for immunohistochemical identification of astrocytes (e.g. GS (Norenberg 1979)), these molecules are also expressed by other cells (Sofroniew and Vinters 2010).
Astrocytes dynamically alter their volume to regulate physiological brain function, such as during sleep (O’Donnell et al. 2015) or extracellular potassium buffering (Florence et al. 2012). It is therefore essential to consider the importance of studying astrocytes in live tissue (in-vivo or ex-vivo) when investigating their role in brain edema. Two indirect parameters of astrocyte morphology have been established, which, while not fully resolving local geometry, provide statistical measures of astrocyte morphology (Minge et al. 2021). These are the fraction of tissue volume that astrocytes occupy and the density of resolvable astrocytic processes. They are relatively straightforward to obtain both in-vivo and in acute brain slices by impaling and iontophoretically injecting astrocytes with a fluorescent dye through a craniotomy (Wilhelmsson et al. 2004). Alexa Fluor 594 has often been chosen for this method as it provides 100% rapid staining of thin processes and even penetrates gap junctions (Savtchenko et al. 2019). Using two-photon excitation fluorescence microscopy, a widely available technique, z-stack of images can be collected containing the entire visible astrocyte structure, which is then analysed in a 3D format (Savtchenko et al. 2019). More powerful scanners that can rapidly scan many focal planes have also been combined with advanced genetic tools for monitoring Ca2+ gradients with high sensitivity, which allow for 3D Ca2+ imaging of a whole astrocyte evaluating its activity (Bindocci et al. 2017; Savtchouk et al. 2018). This method is highly complementary to morphological assessments but very challenging to analyse (Savtchouk et al. 2018). Furthermore, rodents can be habituated to be head-restraint so that they are awake during imaging, overcoming the need for general anaesthesia (Shih et al. 2012), but increasing further the complexity of data processing especially with Ca2+ imaging (Savtchouk et al. 2018). These type of imaging procedures require a chronic cranial window preparation, which is technically challenging, as well as imaging training. However, once mastered, they can be conducted without inducing astrogliosis (Hefendehl et al. 2011), and can also be applied to other cell types.
The definition of cytotoxic edema (isolated fluid shift from the interstitial to the intracellular compartment) might be controversial, as cytotoxic swelling would plausibly always be accompanied by some degree of net brain edema through other mechanisms, such as osmotic gradient across the BBB. Therefore, extracellular space volume measurements in addition to the morphological assessment of astrocytes are essential for the investigation of brain edema. There are several techniques available for the investigation of brain extracellular space volume (reviewed in (Nicholson and Hrabetova 2017; Soria et al. 2020; Sykova and Nicholson 2008)), which are mostly invasive (or at an experimental stage) and therefore mainly applied in rodent studies. Real-time iontophoresis (RTI) is the current gold-standard method for experimentally addressing extracellular space properties (volume and tortuosity), by measuring diffusional spread of a molecule, usually tetramethylammonium cation (TMA+) in ECS (Nicholson and Phillips 1981). TMA+ is applied iontophoretically via a glass capillary into the tissue, and time-dependent changes in concentration of TMA+ are detected within a known distance (typically 100–150 μm) by a TMA+ ion-selective microelectrode. Studies have reported adapted versions of the standard experimental procedure, where the use of fluorescent probes (e.g., Alexa488 and Alexan568) in both electrodes are included (Xie et al. 2013). This allows for their distance to be determined more accurately with fluorescence microscopy. A smaller extracellular space results in reduced TMA+ dilution, reflected by higher levels of detected TMA+ and vice versa.
The advantage of the RTI method is that it can be performed in brain slices, as well as in-vivo, and therefore be combined with the proposed cell morphological assessments. Due to the diffusion properties of TMA+, repeated time-lapse measurements (~ 5 min apart) can be obtained. This technique has also recently been used in awake animals (Xie et al. 2013), eliminating the effects of general anaesthesia, which have been shown to impair glymphatic system clearance and reduce extracellular space volume (Gakuba et al. 2018). On the other hand, the disadvantage associated with this method is that it does not visualize ECS structure and has very low spatial resolution relative to ECS structural geometries. Interpolation and averaging of tissue properties across the given distance are therefore required. Furthermore, due to the calibration being performed in aqueous agar, which represents an unhindered, homogeneous diffusion (Odackal et al. 2017) in regions with anisotropic diffusion, the ECS volume fraction has to be re-calculated in a different way (Vorisek and Sykova 1997). Combining this technique with complementary methods of microscopy, such as super-resolution shadow imaging (SUSHI), can eliminate many of the above limitations and provide more accurate and comprehensive analysis of the ECS volume. SUSHI is based on labelling of the interstitial fluid with a freely diffusible hydrophilic fluorophore that renders cellular structures visible as shadows. As the interstitial compartment is identical to the ECS in structural geometry, the ECS meshwork becomes directly visible allowing for its geometrical analysis (Soria et al. 2020; Tonnesen et al. 2018).
In addition to these techniques, DW-MRI and DW-MRS have been shown to be useful in HE due to their ability to characterize the microstructure of the brain in-vivo and non-invasively using water and metabolites complementary information. The estimated microstructure parameters resulting from these diffusion measures could extremely well complement the measurements described above (Fig. 2).
Conclusion
The role of brain edema as a central event in HE has been questioned in the past, especially in type C HE where changes in water content are smaller, variable and sometimes controversial. It is now highly accepted that type A and type C HE are biologically and clinically different, leading to different manifestations of brain edema and that HE is a multifactorial disease where brain edema might not play a sole central role. The present review describes the main published in-vitro, ex-vivo/in-vivo findings using cell cultures, animal models and humans with HE, highlighting the presence of brain edema in HE. The majority of the in-vitro studies published till now focused on astrocytes and astrocytic swelling while other cell types such as the endothelial cells, neurons, microglia and pericytes in the brain are involved in the pathogenesis of HE. Additional studies should therefore focus on all cell types and asses their morphological changes using physiological ammonia concentrations relevant for type A and C HE. The water content changes reported in animal models of type C HE are small, variable but similar with what was measured in humans using absolute water measurements with MRI. The increase in brain Gln and decrease of the main osmolytes was consistent between animal models and human studies with type C HE, highlighting the presence of a volume regulatory response. In this context, methods with increased specificity, resolution and precision are required to assess absolute brain water changes together with cellular changes (both the extracellular space and cell volume/morphology) in HE. The usage of a multimodal approach combining in-vivo with ex-vivo experiments and allowing to monitor the progression of the disease longitudinally, could be instrumental in elucidating the central role of brain edema as a cause or consequence of HE and its implication in the development of the neurological alterations linked to HE.
Data Availability
Not applicable.
Abbreviations
- ALF:
-
Acute liver failure
- ACLF:
-
Acute on chronic liver failure
- ADC:
-
Apparent diffusion coefficient
- BBB:
-
Blood brain barrier
- BDL:
-
Bile-duct ligation
- CBF:
-
Cerebral blood flow
- CLD:
-
Chronic liver disease
- CNS:
-
Central nervous system
- Cr:
-
Creatine
- CSF:
-
Cerebrospinal fluid
- DW-MRI:
-
Diffusion-weighted MRI
- DW-MRS:
-
Diffusion-weighted MRS
- ECS:
-
Extracellular space
- Gln:
-
Glutamine
- Glu:
-
Glutamate
- GM:
-
Gray matter
- GS:
-
Glutamine synthetase
- HE:
-
Hepatic Encephalopathy
- \({\text{HO}}^{\cdot}\) :
-
Hydroxyl radical
- ICP:
-
Intracranial pressure
- mIns:
-
Myo-inositol
- MRI:
-
Magnetic resonance imaging
- MRS:
-
Magnetic resonance spectroscopy
- MT:
-
Magnetization transfer
- NAA:
-
N-acetylaspartate
- NF-κB:
-
Nuclear factor-κ B
- IFN-γ:
-
Interferon-γ
- IL-1β:
-
Interleukin 1β
- IL-6 :
-
Interleukin 6
- OS:
-
Oxidative stress
- \({\text{O}}_{2}^{\cdot -}\) :
-
Superoxide anion
- Oxo-8-dG:
-
8-Oxo-2’-deoxyguanosine
- PCA:
-
Portocaval anastomosis
- Tau:
-
Taurine
- tCho:
-
Total choline
- TNF-α:
-
Tumor necrosis factor-α
- WM:
-
White matter
References
Agarwal AN, Mais DD (2019) Sensitivity and specificity of Alzheimer type II astrocytes in hepatic encephalopathy. Arch Pathol Lab Med 143(10):1256–1258
Angelova PR, Kerbert AJC, Habtesion A, Hall A, Abramov AY, Jalan R (2022) Hyperammonaemia induces mitochondrial dysfunction and neuronal cell death. JHEP Rep 4(8):100510
Arroyo V, Moreau R, Jalan R (2020) Acute-on-chronic liver failure. N Engl J Med 382(22):2137–2145
Back A, Tupper KY, Bai T, Chiranand P, Goldenberg FD, Frank JI, Brorson JR (2011) Ammonia-induced brain swelling and neurotoxicity in an organotypic slice model. Neurol Res 33(10):1100–1108
Bemeur C, Cudalbu C, Dam G, Thrane AS, Cooper AJ, Rose CF (2016) Brain edema: a valid endpoint for measuring hepatic encephalopathy? Metab Brain Dis 31(6):1249–1258
Benjamin AM, Okamoto K, Quastel JH (1978) Effects of ammonium ions on spontaneous action potentials and on contents of sodium, potassium, ammonium and chloride ions in brain in vitro. J Neurochem 30(1):131–143
Benveniste H, Hedlund LW, Johnson GA (1992) Mechanism of detection of acute cerebral ischemia in rats by diffusion-weighted magnetic resonance microscopy. Stroke 23(5):746–754
Bindocci E, Savtchouk I, Liaudet N, Becker D, Carriero G, Volterra A (2017) Three-dimensional ca2+ imaging advances understanding of astrocyte biology. Science 356(6339):eaai8185. https://doi.org/10.1126/science.aai8185
Blei AT, Olafsson S, Therrien G, Butterworth RF (1994) Ammonia-induced brain edema and intracranial hypertension in rats after portacaval anastomosis. Hepatol 19(6):1437–1444
Bobermin LD, Quincozes-Santos A, Guerra MC, Leite MC, Souza DO, Goncalves CA, Gottfried C (2012) Resveratrol prevents ammonia toxicity in astroglial cells. PLoS One 7(12):e52164
Bosoi CR, Rose CF (2013) Brain edema in acute liver failure and chronic liver disease: similarities and differences. Neurochem Int 62(4):446–457
Bosoi CR, Yang X, Huynh J, Parent-Robitaille C, Jiang W, Tremblay M, Rose CF (2012) Systemic oxidative stress is implicated in the pathogenesis of brain edema in rats with chronic liver failure. Free Radic Biol Med 52(7):1228–1235
Bosoi CR, Zwingmann C, Marin H, Parent-Robitaille C, Huynh J, Tremblay M, Rose CF (2014) Increased brain lactate is central to the development of brain edema in rats with chronic liver disease. J Hepatol 60(3):554–560
Braissant O, Cagnon L, Monnet-Tschudi F, Speer O, Wallimann T, Honegger P, Henry H (2008) Ammonium alters creatine transport and synthesis in a 3D culture of developing brain cells, resulting in secondary cerebral creatine deficiency. Eur J Neurosci 27(7):1673–1685
Braissant O, Henry H, Villard AM, Zurich MG, Loup M, Eilers B, Parlascino G, Matter E, Boulat O, Honegger P, Bachmann C (2002) Ammonium-induced impairment of axonal growth is prevented through glial creatine. J Neurosci 22(22):9810–9820
Braissant O, Rackayova V, Pierzchala K, Grosse J, McLin VA, Cudalbu C (2019) Longitudinal neurometabolic changes in the hippocampus of a rat model of chronic hepatic encephalopathy. J Hepatol 71(3):505–515
Budde MD, Frank JA (2010) Neurite beading is sufficient to decrease the apparent diffusion coefficient after ischemic stroke. Proc Natl Acad Sci U S A 107(32):14472–14477
Butterworth RF, Girard G, Giguere JF (1988) Regional differences in the capacity for ammonia removal by brain following portocaval anastomosis. J Neurochem 51(2):486–490
Callaghan PT, Jolley KW, Lelievre J (1979) Diffusion of water in the endosperm tissue of wheat grains as studied by pulsed field gradient nuclear magnetic resonance. Biophys J 28(1):133–141
Chavarria L, Alonso J, Garcia-Martinez R, Aymerich FX, Huerga E, Jacas C, Vargas V, Cordoba J, Rovira A (2011) Biexponential analysis of diffusion-tensor imaging of the brain in patients with cirrhosis before and after liver transplantation. AJNR Am J Neuroradiol 32(8):1510–1517
Chavarria L, Alonso J, Garcia-Martinez R, Simon-Talero M, Ventura-Cots M, Ramirez C, Torrens M, Vargas V, Rovira A, Cordoba J (2013a) Brain magnetic resonance spectroscopy in episodic hepatic encephalopathy. J Cereb Blood Flow Metab 33(2):272–277
Chavarria L, Oria M, Romero-Gimenez J, Alonso J, Lope-Piedrafita S, Cordoba J (2013b) Brain magnetic resonance in experimental acute-on-chronic liver failure. Liver Int 33(2):294–300
Chavarria L, Oria M, Romero-Gimenez J, Alonso J, Lope-Piedrafita S, Cordoba J (2010) Diffusion tensor imaging supports the cytotoxic origin of brain edema in a rat model of acute liver failure. Gastroenterology 138(4):1566–1573
Claria J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M, Amoros A, Titos E, Alcaraz-Quiles J, Oettl K, Morales-Ruiz M, Angeli P, Domenicali M, Alessandria C, Gerbes A, Wendon J, Nevens F, Trebicka J, Laleman W, Saliba F, Welzel TM, Albillos A, Gustot T, Benten D, Durand F, Gines P, Bernardi M, Arroyo V, Consortium, C. S. I. o. t. E.-C. & the European Foundation for the Study of Chronic Liver, F (2016) Systemic inflammation in decompensated cirrhosis: Characterization and role in acute-on-chronic liver failure. Hepatology 64(4):1249–1264
Cordoba J, Ventura-Cots M, Simon-Talero M, Amoros A, Pavesi M, Vilstrup H, Angeli P, Domenicali M, Gines P, Bernardi M, Arroyo V, Consortium CSI o. E.-C (2014) Characteristics, risk factors, and mortality of cirrhotic patients hospitalized for hepatic encephalopathy with and without acute-on-chronic liver failure (ACLF). J Hepatol 60(2):275–281
Cudalbu C, Taylor-Robinson SD (2019) Brain edema in chronic hepatic encephalopathy. J Clin Exp Hepatol 9(3):362–382
Cudalbu C, Xin L, Marechal B, Lachat S, Zangas-Gheri F, Valenza N, Hanquinet S, McLin VA (2023) High field brain proton magnetic resonance spectroscopy and volumetry in children with chronic, compensated liver disease - A pilot study. Anal Biochem 675:115212 https://doi.org/10.1016/j.ab.2023.115212
Davies NA, Wright G, Ytrebo LM, Stadlbauer V, Fuskevag OM, Zwingmann C, Davies DC, Habtesion A, Hodges SJ, Jalan R (2009) L-ornithine and phenylacetate synergistically produce sustained reduction in ammonia and brain water in cirrhotic rats. Hepatology 50(1):155–164
DeMorrow S, Cudalbu C, Davies N, Jayakumar AR, Rose CF (2021) 2021 ISHEN guidelines on animal models of hepatic encephalopathy. Liver Int 41(7):1474–1488. https://doi.org/10.1111/liv.14911
Dhanda S, Sunkaria A, Halder A, Sandhir R (2018) Mitochondrial dysfunctions contribute to energy deficits in rodent model of hepatic encephalopathy. Metab Brain Dis 33(1):209–223
Dixit V, Chang TM (1990) Brain edema and the blood brain barrier in galactosamine-induced fulminant hepatic failure rats. An animal model for evaluation of liver support systems. ASAIO Trans 36(1):21–27
Ebisu T, Naruse S, Horikawa Y, Ueda S, Tanaka C, Uto M, Umeda M, Higuchi T (1993) Discrimination between different types of white matter edema with diffusion-weighted MR imaging. J Magn Reson Imaging 3(6):863–868
Eefsen M, Jelnes P, Schmidt LE, Vainer B, Bisgaard HC, Larsen FS (2010) Brain expression of the water channels aquaporin-1 and – 4 in mice with acute liver injury, hyperammonemia and brain edema. Metab Brain Dis 25(3):315–323
Eng LF, Ghirnikar RS, Lee YL (2000) Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem Res 25(9–10):1439–1451
Flatt E, McLin VA, Braissant O, Pierzchala K, Mastromarino P, Mitrea SO, Sessa D, Gruetter R, Cudalbu C (2021) Probiotics combined with rifaximin influence the neurometabolic changes in a rat model of type C HE. Sci Rep 11(1):17988. https://doi.org/10.1038/s41598-021-97018-8
Florence CM, Baillie LD, Mulligan SJ (2012) Dynamic volume changes in astrocytes are an intrinsic phenomenon mediated by bicarbonate ion flux. PLoS One 7(11):e51124
Gakuba C, Gaberel T, Goursaud S, Bourges J, Di Palma C, Quenault A, de Martinez S, Vivien D, Gauberti M (2018) General anesthesia inhibits the activity of the “Glymphatic System. Theranostics 8(3):710–722
Ganz R, Swain M, Traber P, DalCanto M, Blei AT (1989) Ammonia-induced swelling of rat cerebral cortical slices: implications for the pathogenesis of Brain Edema in Acute hepatic failure. Metab Brain Dis 4:213–223
Garcia-Martinez R, Rovira A, Alonso J, Jacas C, Simon-Talero M, Chavarria L, Vargas V, Cordoba J (2011) Hepatic encephalopathy is associated with posttransplant cognitive function and brain volume. Liver Transpl 17(1):38–46
Giorgio M, Dellino GI, Gambino V, Roda N, Pelicci PG (2020) On the epigenetic role of guanosine oxidation. Redox Biol 29:101398
Goodman JA, Ackerman JJ, Neil JJ (2008) Cs + ADC in rat brain decreases markedly at death. Magn Reson Med 59(1):65–72
Gorg B, Qvartskhava N, Keitel V, Bidmon HJ, Selbach O, Schliess F, Haussinger D (2008) Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology 48(2):567–579
Gregorios JBM, Mozes LW, Norenberg L-OB, Norenberg MD (1985a) Morphologic effects of ammonia on primary astrocytes cultures. I. Light microscopic studies. J Neuropathol Exp Neurol 44(4):397–403
Gregorios JBM, Mozes LW, Norenberg MD (1985b) Morphologic effects of ammonia on primary astrocyte cultures. II. Electron microscopic studies. J Neuropathol Expt Neurol 44(4):404–414
Hadjihambi A, Cudalbu C, Pierzchala K, Simicic D, Donnelly C, Konstantinou C, Davies N, Habtesion A, Gourine AV, Jalan R, Hosford PS (2022) Abnormal brain oxygen homeostasis in an animal model of liver disease. JHEP Rep 4(8):100509
Haussinger D (2006) Low grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology 43(6):1187–1190
Haussinger D, Dhiman RK, Felipo V, Gorg B, Jalan R, Kircheis G, Merli M, Montagnese S, Romero-Gomez M, Schnitzler A, Taylor-Robinson SD, Vilstrup H (2022) Hepatic encephalopathy. Nat Rev Dis Primers 8(1):43
Haussinger D, Laubenberger J, vom Dahl S, Ernst T, Bayer S, Langer M, Gerok W, Hennig J (1994) Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 107(5):1475–1480
Häussinger D, Warskulat U, Schliess F (1997) Osmosignalling and osmolytes in liver and astrocytes. In: Felipo V, Grisolía S (eds) Advances in cirrhosis, hyperammonemia, and hepatic encephalopathy. Adv Expt Med Biol 420: 195–215
Hayazaki K, Matsuoka Y, Kan M, Hakuba A (1995) Variation in equation coefficients in the gravimetric method to determine brain water content. Neurol Med Chir (Tokyo) 35(2):69–74
Hefendehl JK, Wegenast-Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME, Kohsaka S, Eichner M, Jucker M (2011) Long-term in vivo imaging of beta-amyloid plaque appearance and growth in a mouse model of cerebral beta-amyloidosis. J Neurosci 31(2):624–629
Ipsen DH, Tveden-Nyborg P, Lykkesfeldt J (2014) Does vitamin C deficiency promote fatty liver disease development? Nutrients 6(12):5473–5499
Ito J, Marmarou A, Barzo P, Fatouros P, Corwin F (1996) Characterization of edema by diffusion-weighted imaging in experimental traumatic brain injury. J Neurosurg 84(1):97–103
Jaeger V, DeMorrow S, McMillin M (2019) The direct contribution of astrocytes and microglia to the pathogenesis of hepatic Encephalopathy. J Clin Transl Hepatol 7(4):352–361
Jalan R, Damink O, Hayes SW, Deutz PC, Lee A (2004) Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol 41(4):613–620
Jalan R, Rose CF (2022) Heretical thoughts into hepatic encephalopathy. J Hepatol 77(2):539–548
Jelescu IO, Ciobanu L, Geffroy F, Marquet P, Le Bihan D (2014) Effects of hypotonic stress and ouabain on the apparent diffusion coefficient of water at cellular and tissue levels in Aplysia. NMR Biomed 27(3):280–290
Jelescu IO, Palombo M, Bagnato F, Schilling KG (2020) Challenges for biophysical modeling of microstructure. J Neurosci Methods 344:108861
Joshi D, O’Grady J, Patel A, Shawcross D, Connor S, Deasy N, Willars C, Bernal W, Wendon J, Auzinger G (2014) Cerebral oedema is rare in acute-on-chronic liver failure patients presenting with high-grade hepatic encephalopathy. Liver Int 34(3):362–366
Kale RA, Gupta RK, Saraswat VA, Hasan KM, Trivedi R, Mishra AM, Ranjan P, Pandey CM, Narayana PA (2006) Demonstration of interstitial cerebral edema with diffusion tensor MR imaging in type C hepatic encephalopathy. Hepatology 43(4):698–706
Kasthuri N, Hayworth KJ, Berger DR, Schalek RL, Conchello JA, Knowles-Barley S, Lee D, Vazquez-Reina A, Kaynig V, Jones TR, Roberts M, Morgan JL, Tapia JC, Seung HS, Roncal WG, Vogelstein JT, Burns R, Sussman DL, Priebe CE, Pfister H, Lichtman JW (2015) Saturated reconstruction of a volume of neocortex. Cell 162(3):648–661
Kato M, Hughes RD, Keays RT, Williams R (1992) Electron microscopic study of brain capillaries in cerebral edema. From Fulminant Hepatic Failure 15(6):1060–1066
Korkmaz KSDB, Roggenbuck B (2018) Detection of 8-OHdG as a diagnostic biomarker. J Lab Precision Med 3:95–95
Korogod N, Petersen CC, Knott GW (2015) Ultrastructural analysis of adult mouse neocortex comparing aldehyde perfusion with cryo fixation. Elife 4:e05793. https://doi.org/10.7554/eLife.05793
Kreis R, Ross BD, Farrow NA, Ackerman Z (1992) Metabolic disorders of the brain in chronic hepatic encephalopathy detected with H-1 MR spectroscopy. 182(1). https://doi.org/10.1148/radiology.182.1.1345760
Kruczek C, Görg B, Keitel V, Bidmon HJ, Schliess F, Häussinger D (2011) Ammonia increases nitric oxide, free Zn2+ and metallothionein mRNA expression in cultured rat astrocytes. Biol Chem 392(12):1155–1165. https://doi.org/10.1515/BC.2011.199
Kumar A, Yegla B, Foster TC (2018) Redox signaling in neurotransmission and cognition during aging. Antioxid Redox Signal 28(18):1724–1745
Latour LL, Hasegawa Y, Formato JE, Fisher M, Sotak CH (1994) Spreading waves of decreased diffusion coefficient after cortical stimulation in the rat brain. Magn Reson Med 32(2):189–198
Linster CL, Van Schaftingen E (2007) Vitamin C. biosynthesis, recycling and degradation in mammals. FEBS J 274(1):1–22
Lodi R, Tonon C, Stracciari A, Weiger M, Camaggi V, Iotti S, Donati G, Guarino M, Bolondi L, Barbiroli B (2004) Diffusion MRI shows increased water apparent diffusion coefficient in the brains of cirrhotics. Neurology 62(5):762–766
Lu K, Zimmermann M, Gorg B, Bidmon HJ, Biermann B, Klocker N, Haussinger D, Reichert AS (2019) Hepatic encephalopathy is linked to alterations of autophagic flux in astrocytes. EBioMedicine 48:539–553
Mardini H, Smith FE, Record CO, Blamire AM (2011) Magnetic resonance quantification of water and metabolites in the brain of cirrhotics following induced hyperammonaemia. J Hepatol 54(6):1154–1160
Marmarou A, Poll W, Shulman K, Bhagavan H (1978) A simple gravimetric technique for measurement of cerebral edema. J Neurosurg 49(4):530–537
Martinez-Hernandez A, Bell KP, Norenberg MD (1977) Glutamine synthetase: glial localization in brain. Science 195(4284):1356–1358
Master S, Gottstein J, Blei AT (1999) Cerebral blood flow and the development of ammonia-induced brain edema in rats after portacaval anastomosis. Hepatol 30(4):876-80. https://doi.org/10.1002/hep.510300428
Minge D, Domingos C, Unichenko P, Behringer C, Pauletti A, Anders S, Herde MK, Delekate A, Gulakova P, Schoch S, Petzold GC, Henneberger C (2021) Heterogeneity and development of fine astrocyte morphology captured by Diffraction-Limited Microscopy. Front Cell Neurosci 15:669280
Moreau R, Gao B, Papp M, Banares R, Kamath PS (2021) Acute-on-chronic liver failure: a distinct clinical syndrome. J Hepatol 75(Suppl 1):S27–S35
Moseley ME, Cohen Y, Mintorovitch J, Chileuitt L, Shimizu H, Kucharczyk J, Wendland MF, Weinstein PR (1990) Early detection of regional cerebral ischemia in cats: comparison of diffusion- and T2-weighted MRI and spectroscopy. Magn Reson Med 14(2):330–346
Mosso J, Rey M, Simicic D, Pierzchala K, Jelescu I, Cudalbu C (2022a) Diffusion MRI and MRS probe cerebellar microstructure alterations in the rat developing brain during hepatic encephalopathy. Proc Intl Soc Magn Reson Med 30:2022. Access to a conference proceeding: https://inspire-medfiles.icometrix.com/0155_mosso_ismrm2022.pdf
Mosso J, Yin T, Poitry-Yamate C, Simicic D, Lepore M, McLin VA, Braissant O, Cudalbu C, Lanz B (2022b) PET CMR(glc) mapping and (1)H-MRS show altered glucose uptake and neurometabolic profiles in BDL rats. Anal Biochem 647:114606
Mosso J, Valette J, Katarzyna P, Simicic D, Jelescu I, Cudalbu C (2021) Diffusion-weighted magnetic resonance spectroscopy in the cerebellum of a rat model of hepatic encephalopathy at 14.1T. Proc Intl Soc Magn Reson Med 29. Access to a conference proceeding: https://cibm.ch/wpcontent/uploads/abstract_0342_combined-Jessie-Julie-Mosso.pdf
Murthy CR, Hertz L (1988) Pyruvate decarboxylation in astrocytes and in neurons in primary cultures in the presence and the absence of ammonia. Neurochem Res 13(1):57–61
Murthy CRK, Norenberg MD (2002) Suppression of ammonia-induced swelling by aspartate but not by ornithine in primary cultures of rat astrocytes. Neurochem Int 41:171–176
Nicholson C, Hrabetova S (2017) Brain extracellular space: the final frontier of neuroscience. Biophys J 113(10):2133–2142
Nicholson C, Phillips JM (1981) Ion diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J Physiol 321:225–257
Norenberg MD (1977) A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest 36(6):618–627
Norenberg MD (1979) Distribution of glutamine synthetase in the rat central nervous system. J Histochem Cytochem 27(3):756–762
Norenberg MD (1987) The role of astrocytes in hepatic encephalopathy. Neurochem Pathol 6(1–2):13–33
Norenberg MD, Baker L, Norenberg L-OB, Blicharska J, Bruce-Gregorios JH, Neary JT (1991) Ammonia-induced astrocyte swelling in primary culture. Neurochem Res 16(7):833–836
Norenberg MD, Lapham LW (1974) The astrocyte response in experimental portal-systemic encephalopathy: an electron microscopic study. J Neuropathol Exp Neurol 33(3):422–435
Nunomura A, Lee HG, Zhu X, Perry G (2017) Consequences of RNA oxidation on protein synthesis rate and fidelity: implications for the pathophysiology of neuropsychiatric disorders. Biochem Soc Trans 45(5):1053–1066
O’Donnell J, Ding F, Nedergaard M (2015) Distinct functional states of astrocytes during sleep and wakefulness: is norepinephrine the master regulator? Curr Sleep Med Rep 1(1):1–8
Ochoa-Sanchez R, Tamnanloo F, Rose CF (2021) Hepatic encephalopathy: from metabolic to neurodegenerative. Neurochem Res 46(10):2612–2625
Odackal J, Colbourn R, Odackal NJ, Tao L, Nicholson C, Hrabetova S (2017) Real-time iontophoresis with tetramethylammonium to quantify volume fraction and tortuosity of brain extracellular space. J Vis Exp (125):55755. https://doi.org/10.3791/55755
Oeltzschner G, Butz M, Wickrath F, Wittsack HJ, Schnitzler A (2016) Covert hepatic encephalopathy: elevated total glutathione and absence of brain water content changes. Metab Brain Dis 31(3):517–527
Papadopoulos MC, Manley GT, Krishna S, Verkman AS (2004) Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 18(11):1291–1293
Pelle J, Castelli FA, Rudler M, Alioua I, Colsch B, Fenaille F, Junot C, Thabut D, Weiss N (2022) Metabolomics in the understanding and management of hepatic encephalopathy. Anal Biochem 636:114477
Pierzchala K, Simicic D, Sienkiewicz A, Sessa D, Mitrea S, Braissant O, McLin VA, Gruetter R, Cudalbu C (2022) Central nervous system and systemic oxidative stress interplay with inflammation in a bile duct ligation rat model of type C hepatic encephalopathy. Free Radic Biol Med 178:295–307
Rackayova V, Braissant O, Mclin VA, Berset C, Lanz B, Cudalbu C (2016) H-1 and P-31 magnetic resonance spectroscopy in a rat model of chronic hepatic encephalopathy: in vivo longitudinal measurements of brain energy metabolism. Metab Brain Dis 31(6):1303–1314
Rackayova V, Braissant O, Rougemont AL, Cudalbu C, McLin VA (2020) Longitudinal osmotic and neurometabolic changes in young rats with chronic cholestatic liver disease. Sci Rep 10(1):7536
Rackayova V, Flatt E, Braissant O, Grosse J, Capobianco D, Mastromarino P, McMillin M, DeMorrow S, McLin VA, Cudalbu C (2021) Probiotics improve the neurometabolic profile of rats with chronic cholestatic liver disease. Sci Rep 11(1):2269. https://doi.org/10.1038/s41598-021-81871-8
Rama Rao KV, Reddy PV, Tong X, Norenberg MD (2010) Brain edema in acute liver failure: inhibition by L-histidine. Am J Pathol 176(3):1400–1408
Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M (2013) Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 19(12):1643–1648
Rao KV, Jayakumar AR, Tong X, Alvarez VM, Norenberg MD (2010) Marked potentiation of cell swelling by cytokines in ammonia-sensitized cultured astrocytes. J Neuroinflammation 7:66. https://doi.org/10.1186/1742-2094-7-66
Rao KV, Panickar KS, Jayakumar AR, Norenberg MD (2005) Astrocytes protect neurons from ammonia toxicity. Neurochem Res 30(10):1311–1318
Reinehr R, Gorg B, Becker S, Qvartskhava N, Bidmon HJ, Selbach O, Haas HL, Schliess F, Haussinger D (2007) Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices. Glia 55(7):758–771
Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R (2000) The systemic inflammatory response syndrome in acute liver failure. Hepatology 32(4 Pt 1):734–739
Rose CF (2010) Increase brain lactate in hepatic encephalopathy: cause or consequence? Neurochem Int 57(4):389–394
Rose CF, Amodio P, Bajaj JS, Dhiman RK, Montagnese S, Taylor-Robinson SD, Vilstrup H, Jalan R (2020) Hepatic encephalopathy: novel insights into classification, pathophysiology and therapy. J Hepatol 73(6):1526–1547
Savtchenko LP, Bard L, Jensen TP, Reynolds JP, Kraev I, Medvedev N, Stewart MG, Henneberger C, Rusakov DA (2019) Author correction: disentangling astroglial physiology with a realistic cell model in silico. Nat Commun 10(1):5062
Savtchouk I, Carriero G, Volterra A (2018) Studying Axon-Astrocyte Functional interactions by 3D two-photon ca(2+) imaging: a practical guide to experiments and “Big Data” analysis. Front Cell Neurosci 12:98
Sawhney R, Holland-Fischer P, Rosselli M, Mookerjee RP, Agarwal B, Jalan R (2016) Role of ammonia, inflammation, and cerebral oxygenation in brain dysfunction of acute-on-chronic liver failure patients. Liver Transpl 22(6):732–742
Schliess F, Görg B, Häussinger D (2009) RNA oxidation and zinc in hepatic encephalopathy and hyperammonemia. Metab Brain Dis 24(1):119–134
Schliess F, Sinning R, Fischer R, Schmalenbach C, Häussinger D (1996) Calcium-dependent activation of Erk-1 and Erk-2 after hypo-osmotic astrocyte swelling. Biochem J 320(Pt 1):167–171
Sen S, Rose C, Ytrebo LM, Davies NA, Nedredal GI, Drevland SS, Kjonno M, Prinzen FW, Hodges SJ, Deutz NE, Williams R, Butterworth RF, Revhaug A, Jalan R (2006) Effect of albumin dialysis on intracranial pressure increase in pigs with acute liver failure: a randomized study. Crit Care Med 34(1):158–164
Shah NJ, Neeb H, Kircheis G, Engels P, Haussinger D, Zilles K (2008) Quantitative cerebral water content mapping in hepatic encephalopathy. Neuroimage 41(3):706–717
Shih AY, Driscoll JD, Drew PJ, Nishimura N, Schaffer CB, Kleinfeld D (2012) Two-photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab 32(7):1277–1309
Shubin AV, Demidyuk IV, Komissarov AA, Rafieva LM, Kostrov SV (2016) Cytoplasmic vacuolization in cell death and survival. Oncotarget 7(34):55863–55889
Simicic D, Cudalbu C, Pierzchala K (2022) Overview of oxidative stress findings in hepatic encephalopathy: from cellular and ammonium-based animal models to human data. Anal Biochem 654:114795
Sinke AP, Jayakumar AR, Panickar KS, Moriyama M, Reddy PV, Norenberg MD (2008) NFkappaB in the mechanism of ammonia-induced astrocyte swelling in culture. J Neurochem 106(6):2302–2311
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119(1):7–35
Soria FN, Miguelez C, Penagarikano O, Tonnesen J (2020) Current techniques for investigating the brain extracellular space. Front Neurosci 14:570750
Studer D, Humbel BM, Chiquet M (2008) Electron microscopy of high pressure frozen samples: bridging the gap between cellular ultrastructure and atomic resolution. Histochem Cell Biol 130(5):877–889
Suarez I, Bodega G, Fernandez B (2002) Glutamine synthetase in brain: effect of ammonia. Neurochem Int 41(2–3):123–142
Swain MS, Blei AT, Butterworth RF, Kraig RP (1991) Intracellular pH rises and astrocytes swell after Portacaval Anastomosis in rats. Am J Physiol 261(6):R1491–R1496
Swain M, Butterworth RF, Blei AT (1992) Ammonia and related amino-acids in the pathogenesis of Brain Edema in Acute ischemic liver-failure in rats. Hepatology 15(3):449–453
Sykova E, Nicholson C (2008) Diffusion in brain extracellular space. Physiol Rev 88(4):1277–1340
Thrane AS, Thrane R, Plog V, Nedergaard M (2015) Filtering the muddied waters of brain edema. Trends Neurosci 38(6):333–335
Tonnesen J, Inavalli V, Nagerl UV (2018) Super-resolution imaging of the extracellular space in living brain tissue. Cell 172(5):1108–1121
Traber PG, Canto D, Ganger M (1987) Electron microscopic evaluation of Brain Edema in rabbits with Galactosamine-Induced fulminant hepatic failure: Ultrastructure and Integrity of the blood-brain barrier. Hepatology 7(6):1272–1277
Traber P, DalCanto M, Ganger D, Blei AT (1989) Effect of body temperature on brain edema and encephalopathy in the rat after hepatic devascularization. Gastroenterology 96(3):885–891
Traber PG, Ganger DR, Blei AT (1986) Brain edema in rabbits with galactommine-induced fulminant hepatitis: regional differences and effects on intracranial pressure. Gastroenterology 91:1347–1356
Vorisek I, Sykova E (1997) Evolution of anisotropic diffusion in the developing rat corpus callosum. J Neurophysiol 78(2):912–919
Wendon JA, Harrison PM, Keays R, Williams R (1994) Cerebral blood flow and metabolism in fulminant liver failure. Hepatology 19(6):1407–1413
Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, Ellisman MH, Pekny M (2006) Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci U S A 103(46):17513–17518
Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, Pekny M (2004) Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci 24(21):5016–5021
Winterdahl M, Abbas Z, Noer O, Thomsen KL, Gras V, Nahimi A, Vilstrup H, Shah NJ, Dam G (2019) Cerebral water content mapping in cirrhosis patients with and without manifest HE. Metab Brain Dis 34(4):1071–1076
Wright G, Davies NA, Shawcross DL, Hodges SJ, Zwingmann C, Brooks HF, Mani AR, Harry D, Stadlbauer V, Zou Z, Williams R, Davies C, Moore KP, Jalan R (2007a) Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology 45(6):1517–1526
Wright G, Shawcross D, Damink OSW, Jalan R (2007b) Brain cytokine flux in acute liver failure and its relationship with intracranial hypertension. Metab Brain Dis 22(3–4):375–388
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M (2013) Sleep drives metabolite clearance from the adult brain. Science 342(6156):373–377
Zamora AJ, Cavanagh JB, Kyu MH (1973) Ultrastructural Responses of the Astrocytes to Portocaval Anastomosis in the Rat. 18(1):25–45
Acknowledgements
CC, KP, JM were supported by the Center for Biomedical Imaging of the UNIL, UNIGE, HUG, CHUV, EPFL, the Leenaards and Jeantet Foundations.
Funding
Open access funding provided by EPFL Lausanne The Swiss National Foundation (SNSF) project no 310030_201218 (CC); European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No 813120 (INSPiRE-MED) (CC and JM); AH is funded by the Foundation for Liver Research (REGISTERED CHARITY No.268211/1134579).
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Figure
Figure 1 was generated using pictures with adaptation (additional markings) from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pierzchala, K., Hadjihambi, A., Mosso, J. et al. Lessons on brain edema in HE: from cellular to animal models and clinical studies. Metab Brain Dis 39, 403–437 (2024). https://doi.org/10.1007/s11011-023-01269-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-023-01269-5