
Abstract
The Co–Mg–Al mixed metal oxides were prepared by calcination of co-precipitated hydrotalcite-like precursors at various temperatures (600–800 °C), characterised with respect to chemical (AAS) and phase (XRD) composition, textural parameters (BET), form and aggregation of cobalt species (UV–vis-DRS) and their redox properties (H2-TPR, cyclic voltammetry). Moreover, the process of thermal decomposition of hydrotalcite-like materials to mixed metal oxide systems was studied by thermogravimetric method combined with the analysis of gaseous decomposition products by mass spectrometry. Calcined hydrotalcite-like materials were tested as catalysts for methanol incineration. Catalytic performance of the oxides depended on cobalt content, Mg/Al ratio and calcination temperature. The catalysts with lower cobalt content, higher Mg/Al ratio and calcined at lower temperatures (600 or 700 °C) were less effective in the process of methanol incineration. In a series of the studied catalysts, the best results, with respect to high catalytic activity and selectivity to CO2, were obtained for the mixed oxide with Co:Mg:Al molar ratio of 10:57:33 calcined at 800 °C. High activity of this catalyst was likely connected with the presence of a Co–Mg–Al spinel-type phases, containing easy reducible Co3+ cations, formed during high-temperature treatment of the hydrotalcite-like precursor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Emissions of volatile organic compounds (VOCs) to the atmosphere from various sources are one of the most serious environmental problems [1]. Majority of VOCs is toxic and may take part in photochemical reactions in the atmosphere with other pollutants (e.g. nitrogen oxides) resulting in very dangerous secondary pollutants (e.g. ozone, aldehydes, peroxyacyl nitrates). The best way to reduce the VOCs emission is to eliminate them prior to their discharge. Among various methods employed for VOCs removal including thermal combustion, absorption, adsorptive recovery and biofiltration [2], the low-temperature catalytic incineration seems to be the most reliable and effective post-treatment technology [1]. The catalysts for VOCs incineration should effectively operate at relatively low temperatures to avoid additional heating of flue gases as well as to limit the formation of NOx by thermal mechanism [3]. Noble metals, such as Pt [4–7], Pd [8–10], Rh [11] and Au [12, 13], belong to the most active and effective components of the catalysts for incineration of broad range of various VOCs. On the other hand, high cost of noble metals significantly limits commercial utilisation of such catalysts. Cobalt belongs to the most promising components of the non-noble metal-based catalytic systems for VOCs incineration. Solsona et al. [14] reported the supported and unsupported nanocrystalline cobalt oxides as very effective catalysts for propane incineration (total conversion of propane was achieved at about 250 °C). The catalytic activity was mainly dependent on the crystallite size of cobalt oxides, decreasing with an increase in the crystallite size. The Co3O4 oxide was identified as the most catalytically active phase in the studied process. Jirátová et al. [15] studied total oxidation of toluene and ethanol over the hydrotalcite-derived Co–Mn–Al mixed oxide catalyst (Co:Mn:Al molar ratio of 4:1:1) modified with various amounts of potassium (0–3 mass%). The Co–Mn–Al mixed oxide showed very good catalytic performance in the studied processes and addition of low amounts of potassium promoter additionally activated mixed metal oxide catalyst mainly by modification of the surface acid–base properties. Thus, it was shown that both the nanocrystalline structure of cobalt oxide phases as well as the surface acid–base properties of such catalytic systems are very important parameters that determine the catalytic performance in the processes of VOCs incineration. These observations were inspiration for the present study. Similarly to report of Jirátová et al. [15] mentioned above, the hydrotalcite-like materials were used as precursors of mixed metal oxide catalysts for VOCs incineration. Thermal treatment of such precursors results in nanocrystalline materials with homogenously distributed active components. By tailoring of chemical composition of hydrotalcite-like precursors and conditions of their thermal treatment, mixed metal oxide systems with various phase composition can be prepared [16–23]. The surface acid–base properties of hydrotalcite-derived mixed metal oxide catalysts can be controlled by changing of the Mg/Al ratio in the hydrotalcite-like precursors.
The aim of the studies has been determination of the influence of the Mg/Al ratio in the cobalt-containing hydrotalcite-like precursors as well as calcination conditions on their catalytic efficiency in the process of methanol incineration. Methanol was used as a model VOC molecule in the presented studies.
Experimental
Samples preparation
The hydrotalcite-like precursors with the intended Co/Mg/Al molar ratios of 5/77/18, 10/72/18, 5/62/33 and 10/57/33 were synthesised by co-precipitation. The metal nitrates of p.a. purity, namely Co(NO3)2·6H2O, Mg(NO3)2·6H2O and Al(NO3)3·9H2O (all supplied by Sigma–Aldrich), NaOH (POCh), Na2CO3 (POCh) and distilled water were used for preparation of solutions. An aqueous solution (450 mL) of Co, Mg and Al nitrates in appropriate molar ratio and total metal ions concentration of 1.0 mol L−1 was added with flow rate of 7.5 mL min−1 into 1000-mL batch reactor containing 200 mL of distilled water. The flow rate of simultaneously added alkaline solution of NaOH (3.0 mol L−1) and Na2CO3 (0.5 mol L−1) was controlled to maintain reaction pH of 10.0 ± 0.2. The co-precipitation was carried out at vigorous stirring at 60 °C. The resulting suspension was stirred at 60 °C for 60 min. The product was filtered off, washed thoroughly with distilled water and dried overnight at 60 °C in air. Finally, the precursors were calcined at various temperatures (600, 700 or 800 °C) for 9 h in air. The calcined samples were kept in a desiccator in order to avoid their contact with ambient atmosphere. The prepared mixed oxide samples were labelled by acronyms with molar ratios of cation constituents and calcination temperature (e.g. Co10Mg57Al33-800).
Samples characterisation
The cation composition of the prepared hydrotalcite-like precursors was determined by atomic absorption spectroscopy. The samples were dissolved in a hydrochloric acid solution (36%), and the cation concentration in solutions was determined using 280FS AA instrument (Agilent Technologies) at the following wavelengths: Co 240.7 nm, Mg 285.2 nm and Al 309.3 nm.
The phase composition of the hydrotalcite-like precursors and related mixed metal oxides obtained by their calcination was determined by powder X-ray diffraction (XRD). The powder XRD patterns were recorded with a D2 Phaser diffractometer (Bruker) using Co Kα1 radiation (λ = 0.179 nm) in the 2ϴ range from 8° to 80°, step size 0.02°. The qualitative analysis was performed with a HighScore Plus 4.0 software package (PANanalytical). The hydrotalcite lattice parameters a and c were determined from XRD data using the following formulas: a = 2d 110 and c = (3d 003 + 6d 006)/2. The mean coherence length L was calculated from the FWHM values (broadening at half of the maximum intensity) of the diffraction lines using Scherrer equation.
Thermal decomposition of the hydrotalcite-like precursors was studied by thermogravimetric method coupled with QMS analysis of evolved gases. The TG-DTG-QMS measurements were carried out using a Mettler Toledo 851e instrument operated under a flow of air (80 mL min−1) in the temperature range of 25–1000 °C with a heating rate of 10 °C min−1. The gases evolved during the thermal decomposition of the samples were continuously monitored by quadrupole mass spectrometer ThermoStar (Balzers) connected directly to microbalance for the chosen mass numbers m/z (18-H2O+, 30-NO+ and 44-CO +2 ).
The specific surface area of the calcined samples was determined by the BET method using a 3Flex (Micromeritics) automated gas adsorption system. Prior to the nitrogen adsorption at −196 °C all the samples were outgassed under vacuum at 350 °C for 24 h.
The UV–vis-DR spectra of the calcined samples were recorded using an Evolution 600 (Thermo) spectrophotometer. The measurements were performed in the range from 190 to 900 nm with a resolution of 2 nm.
Cyclic voltammograms of the calcined samples were recorded in a three-electrode cell using a graphite paste electrode as the working electrode, platinum coil as the auxiliary electrode and Ag|AgCl as the reference electrode. Composite paste was prepared by mixing synthetic graphite (100–150 mg) with Nujol (0.05 mL) and a small amount of the mixed oxide catalyst (0.005–0.010 g). The measurements were performed in acetate buffer (pH = 4.6) as electrolyte at a scan rate of 50 mV s−1. Before experiment, the solutions were pretreated with argon to keep oxygen-free atmosphere during the measurement.
The reducibility of the calcined samples was studied by temperature-programmed reduction method (H2-TPR). Experiments were carried out in a fixed-bed flow microreactor starting from room temperature to 950 °C, with a linear heating rate of 10 °C min−1. H2-TPR runs were carried out in a flow (10 mL min−1) of 5 vol.% H2 diluted in Ar (N5 quality, Messer). The evolution of hydrogen was detected by microvolume TCD (Valco).
Catalytic studies
The mixed oxide samples obtained from hydrotalcite-like precursors at 600, 700 and 800 °C were tested as catalysts for methanol incineration. Catalytic experiments were performed in a fixed-bed flow quartz microreactor system under atmospheric pressure in the temperature range from 125 to 425 °C with an isothermal steps every 25 °C. For each test, 100 mg of catalyst was outgassed in a flow of air at 500 °C for 30 min. After cooling down to 125 °C, the gas mixture containing 3.7 vol% of methanol diluted in air (total flow rate of 20 mL min−1) was supplied into the reactor by the isothermal saturator (0 °C). The reaction products were analysed using a gas chromatograph SRI 8610C equipped with Hayesep D column as well as methaniser-FID detection system.
Results and discussion
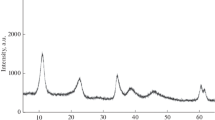
Formation of hydrotalcite-like materials after co-precipitation reaction was proven by powder XRD results. In the XRD patterns of the co-precipitated precursors (Fig. 1), only diffraction lines characteristic for hydrotalcite-like compounds were found, no other crystalline phase was detected. The hydrotalcite lattice parameters a and c, determined from XRD analysis, are compared in Table 1. Slightly smaller a parameter observed for the samples with increased aluminium content can be explained by smaller ionic radius of Al3+ (53 pm) in comparison with radius of Mg2+ (72 pm). The samples with increased aluminium loading in the hydrotalcite-like samples exhibited also slightly smaller d 003 basal spacing, which resulted in a decreased c parameter. Similar effect was observed by authors for the Cu–Mg–Al hydrotalcite-like materials [16]. This effect could be related to the increased Al3+ content in the brucite-like layers, which causes the higher net positive charge and tighter arrangement of the layers due to stronger electrostatic interactions between the layers and interlayer anions. The mean coherence length evaluated from XRD data is related to structure ordering; it was evaluated from broadening of hydrotalcite (hkl 003, 006, 110) diffraction line using Scherrer equation; the L values varied in the range of 9.1–12.8 nm and confirmed formation of well-crystalline product.

Powder XRD patterns of the co-precipitated hydrotalcite-like precursors
Cation composition of prepared hydrotalcite-like precursors was determined by AAS after samples dissolution in hydrochloric acid. The results presented in Table 2 show that molar ratios of cations calculated from the measured data are very close to the intended values, i.e. the molar ratios of cations adjusted in nitrate solutions used in co-precipitation reaction.
The process of thermal decomposition of the hydrotalcite-like samples into mixed oxides was studied by thermogravimetry coupled with analysis of gaseous products released during thermal treatment of the samples (TG-QMS). Results of thermal analysis are presented in Fig. 2. In general, thermal behaviour of hydrotalcite-like compounds may be characterised by two main transitions: (1) the loss of interlayer water without collapse of the layered hydrotalcite structure and (2) subsequent loss of hydroxyl groups accompanied by water release from the brucite-like layers and decomposition of volatile interlayer anions (e.g. carbonate or nitrate) at higher temperatures [24]. The temperature ranges of these two transitions depend on cationic composition of the hydroxide layers and interlayer anions. The first transition related to the loss of interlayer water is represented by DTG peaks and maxima of water evolution at temperatures below 250 °C. It should be noted that in the case of the samples with higher Al/Mg ratio (Co5Mg62Al33 and Co10Mg57Al33) these peaks were detected at temperatures of about 20–25 °C higher than in the samples with the lower Al/Mg ratio (Co5Mg77Al18 and Co10Mg72Al18). It could be explained by more effective stabilisation of interlayer water in the samples with the increased content of aluminium. Apart from interlayer water, very small amounts of carbon dioxide were also released at temperatures below 250 °C, likely as a result of CO2 desorption from the samples surface or thermal decomposition of unstable surface carbonates.

Results of thermogravimetry coupled with evolved gas analysis documenting thermal decomposition of the co-precipitated hydrotalcite-like precursors
The second stage of the hydrotalcite-like samples decomposition, including dehydroxylation of the brucite-like layers as well as decomposition of interlayer anions, was represented by DTG peaks and maxima of H2O, CO2 and NO evolution observed above 250 °C. Release of CO2 and H2O from the samples with the lower Al/Mg ratio (Co5Mg77Al18 and Co10Mg72Al18) was represented by single sharp maxima at about 355–375 °C (Fig. 2). For the samples with the higher Al/Mg ratio (Co5Mg62Al33 and Co10Mg57Al33), the profiles of H2O and CO2 evolution were more complex and consisted of at least two overlapping peaks of water release and at least three peaks of carbon dioxide evolution. The low-temperature peak of H2O release was located at about 310 °C, while the high-temperature peaks were located at 380 and 360 °C for the Co5Mg62Al33 and Co10Mg57Al33 samples, respectively. Comparing positions of these peaks with temperatures of pure Al(OH)3 and Mg(OH)2 dehydroxylation, which was determined to be 295 °C [25] and 377 °C [26], respectively, it could be suggested that the low-temperature peak was likely related to the release of OH− anions attached to Al3+, while the high-temperature peak to dehydroxylation of OH− attached to Mg2+ cations. Such splitting of the water evolution process was not observed for Co5Mg77Al18 and Co10Mg72Al18 precursors due to lower content of aluminium in these samples; however, the asymmetry and positions of the water release peaks may suggest that it is a superposition of the peaks related to dehydroxylation of OH− bounded to Mg2+ with the smaller contribution of OH− attached to Al3+. The profiles of CO2 evolution from Co5Mg62Al33 and Co10Mg57Al33 were also much more complex comparing to that observed for the Co5Mg77Al18 and Co10Mg72Al18 samples and consisted of at least four peaks. As it was mentioned above, small peaks located below 250 °C were possibly related to CO2 desorption from the samples surface or thermal decomposition of unstable surface carbonates. Peaks centred at about 310 and 395–400 °C were assigned to decomposition of interlayer carbonates and indicated the presence of CO3 2− anions stabilised in the interlayer with various strengths. It could be explained by different interaction and therefore also stabilisation of interlayer anions by Al3+ and Mg2+ cations in hydroxide layers. Moreover, CO2 release related to decomposition of very stable carbonates was detected at temperature of about 605–610 °C. Similar effect was reported in our previous studies for the Cu5Mg66Al29 and Mg71Al29 hydrotalcite-like samples, which released small amounts of CO2 at temperatures as high as 637 and 588 °C, respectively [27]. In the case of the samples with the lower Al/Mg ratio, only one peak related to the thermal decomposition of interlayer carbonates, centred at about 365 and 375 °C, was detected for Co5Mg77Al18 and Co10Mg72Al18, respectively. Release of small amounts of NO was observed only for the samples with the lower Al/Mg ratio and was represented by peaks located at about 355 and 470 °C in the NO evolution profile of Co5Mg77Al18 and maximum of NO evolution at about 500 °C for the Co10Mg72Al18 sample. These results may indicate the presence of a small amount of nitrate anions in the interlayer of the Co5Mg77Al18 and Co10Mg72Al18 samples, but the NO evolution was more likely connected with decomposition of residual nitrates, which were not removed from the samples surface after washing.
Based on the results of thermogravimetric analysis, temperatures of 600 °C and higher were chosen for calcination of the prepared hydrotalcite-like precursors. Such temperatures should ensure a complete decomposition of the hydrotalcite-like precursors and their transformation into mixed metal oxides. The variation of calcination temperatures should result in the preparation of the samples with various phase composition, crystallinity and textural properties.
Powder XRD patterns of the calcined samples are presented in Fig. 3. The diffraction lines characteristic for MgO (periclase) were found in the powder XRD patterns of the mixed oxides obtained at 600 and 700 °C (the lines at approximately 43°, 50° and 74° 2θ). Phases with spinel structure (represented by diffraction lines at about 22°, 36°, 52°, 70° and 77° 2θ) were identified, together with periclase-like oxides, in the powder XRD patterns of the samples with the higher Al/Mg ratio (Co5Mg62Al33 and Co10Mg57Al33) calcined at 800 °C. The spinel diffraction lines were not found in the samples with the lower Al/Mg ratio (Co5Mg77Al18 and Co10Mg72Al18). Comparing the powder XRD patterns of the samples with the higher Al/Mg ratio (Co5Mg62Al33 and Co10Mg57Al33) calcined at 600 and 800 °C, the positions of periclase diffraction lines were shifted from 50.8° to 50.2° and 74.4° to 73.7°, respectively. The corresponding slight increase in periclase lattice parameter from about 0.4211 to 0.4225 nm could be explained by incorporation of Al3+ cations into MgO lattice in the early stages of mixed oxide formation after thermal decomposition of the hydrotalcite-like precursors. Increasing calcination temperature enhanced crystallisation of the MgO-like phase and formation of Mg–Al spinel-type phase. The lattice parameter of MgO-like phase formed at 800° (0.4225 nm) is close to that reported for reference MgO (0.4223 nm, PDF No. 04-012-6481). Analogous changes in lattice parameters of NiO-like phase were observed during calcination of Ni–Al layered double hydroxides [28]. For the samples with the lower Al/Mg ratio (Co5Mg77Al18 and Co10Mg72Al18), only negligible shift (below 0.2°) in the positions of periclase diffraction lines was detected. Moreover, cobalt cations (both Co2+ and even Co3+) were very likely incorporated into the lattice of periclase-like and spinel-type oxides. Radius of cobalt cations is larger than that of Al3+ ones and not very different from the radius of Mg2+ cations; therefore, incorporation of small amounts of cobalt cations into the lattice of Mg–Al mixed oxides is practically impossible to detect by XRD.

Powder XRD patterns of the mixed oxides obtained at various temperatures from the hydrotalcite-like precursors
The measured BET surface areas of the mixed oxide samples obtained at various temperatures are compared in Table 3. The results lead to the following conclusions: (1) the increase in cobalt content in the samples decreased the BET surface area; (2) the samples with higher Al/Mg ratio showed larger surface area and (3) the increase in calcination temperatures resulted in gradual decrease in the surface area of the samples.
The cobalt species formed during calcination of hydrotalcite-like precursors were analysed by UV–vis-DR spectroscopy (Fig. 4). The spectra consisted of the bands in two regions characteristic of Co2+ cations below 330 nm and in the range 500–700 nm, as well as in the region characteristic of Co3+ cations in the range 330–480 nm [29]. The bands in the UV region below 330 nm were assigned to a low-energy charge transfer between the oxygen ligands and central Co2+ ion in tetrahedral symmetry [30]. The absorption bands, centred at 360 nm, were assigned to the 1A1g → 1T2g transition in Co3+ ions in octahedral [31, 32]. The bands mentioned above were present in the spectra of all the studied samples and indicated that major part of cobalt cations was present in the form of Co2+; a part of cobalt cations was oxidised during calcination in air to Co3+ species. Calcination of the samples at high temperature of 800 °C, especially in the case of the samples with the higher Al/Mg ratio (Co5Mg62Al33 and Co10Mg57Al33), resulted in the appearance of the well-defined triplet at 545, 590 and 635 nm (inserts in Fig. 4) corresponding to 4A2 → 4T1 (P) transition in Co2+ ions in tetrahedral environment, characteristic for CoAl2O4 spinel [33]. The formation of such spinel was reported to occur during thermal treatment of the samples containing small amounts of uniformly distributed cobalt species [34]. On the other hand, the band characteristic of Co3+ cations in the range 330–480 nm could indicate the presence of spinel phases containing trivalent cobalt, e.g. MgCo2O4, MgCoAlO4 and Co3O4. However, the absence of the band at about 690–700 nm corresponding to 1A1g → 1T1g transition in Co3+ ions in the octahedral coordination implied that Co3O4 spinel was not formed or was formed only in a very small amounts during calcination of the hydrotalcite-like precursors [34]. Moreover, it should be noted that the highest intensity of the band characteristic of Co3+ cations (330–480 nm) was observed for the samples with the higher cobalt content and lower Al/Mg ratio (Co5Mg77Al18 series). Therefore, it seems possible that this band represents mainly MgCo2O4 with the inverse spinel structure [35].

UV–vis-DR spectra of the mixed oxides obtained at various temperatures from the hydrotalcite-like precursors
The electrochemical properties of the mixed oxide samples prepared at various temperatures were examined by cyclic voltammetry. Figure 5 shows the cyclic voltammograms obtained for the Co10Mg57Al33 sample calcined at 600 and 800 °C. The oxidation wave for the sample calcined at 600 °C is visible at Epa = 301 mV and reduction wave can be observed at Epc = 8 mV. The shape of the voltammogram profiles shows that the redox process is irreversible and oxidation dominates over reduction in the mixed oxide catalyst. In the Co10Mg57Al33 sample calcined at 800 °C, the oxidation wave is present at Epa = 301 mV, so exactly at the same position as for the catalyst prepared at 600 °C. On the other hand, significant differences are observed in the reduction waves of the Co10Mg57Al33 sample calcined at 600 and 800 °C. In the catalyst calcined at higher temperature, the reduction wave was shifted to 21 mV and, moreover, additional low-intensive reduction wave appeared at about 121 mV. Such result may indicate that increase in calcination temperature from 600 to 800 °C generated more easily reducible cobalt species in the Co10Mg57Al33 sample.

Results of cyclic voltammetry measurements obtained for Co10Mg57Al33 mixed oxides obtained at 600 and 800 °C
Figure 6 presents the results of the multicycle voltammetry performed for the Co10Mg57Al33 sample calcined at 800 °C. In the initial cycles, splitting of the oxidation wave for two components was observed. This effect was less significant in the subsequent redox cycles, and finally an increase in the anodic current and the formation of one intensive oxidation wave occurred. As it was mentioned above, two reduction waves were observed in the profile of the Co10Mg57Al33 sample calcined at 800 °C. In the subsequent redox cycles, the intensity of these waves, especially that at higher positive potential, gradually increased. It could be explained by the formation of the more easily reducible cobalt species in the subsequent redox cycles. Such effects were not observed for the Co10Mg57Al33 sample calcined at 600 °C, for which no changes in the subsequent oxidation and reduction wave profiles were found.

Results of multicycle voltammetry measurements performed for the Co10Mg57Al33 mixed oxide sample obtained at 800 °C
Figure 7 presents the results of temperature-programmed reduction (H2-TPR) obtained for the Co10Mg57Al33 sample calcined at 600 and 800 °C. The reduction in cobalt species proceeded in two steps. In the first step, Co3+ was reduced to Co2+, while in the second step Co2+ to Co0 [36, 37]. It should be noted that in the case of the sample calcined at 800 °C the reduction of Co3+ to Co2+ took place at temperature lower by about 80 °C in comparison with the sample calcined at 600 °C. Thus, the results of these studies are fully consistent with the results of the cyclic voltammetry and support hypothesis that calcination of Co10Mg57Al33 at 800 °C generated cobalt species containing easy reducible Co3+.

Results of temperature-programmed reduction (H2-TPR) performed for the Co10Mg57Al33 mixed oxide sample obtained at 600 and 800 °C
Mixed oxides obtained from hydrotalcite-like precursors at various temperatures were tested as catalysts for incineration of methanol, which was used as a model VOC. It can be seen from Fig. 8 that catalytic performance of the studied samples depended on chemical composition as well as calcination temperature. In a series of the samples with the higher Al/Mg ratio (Co5Mg62Al33 and Co10Mg57Al33), an increase in calcination temperature resulted in catalytic activation of the samples. Moreover, for the catalysts calcined at elevated temperatures higher selectivity to CO2 was achieved (apart from CO2 only CO was detected as a by-product).

Results of catalytic tests of the obtained mixed oxide catalysts in the methanol incineration
For series of the catalysts with the lower Al/Mg ratio (Co5Mg77Al18 and Co10Mg72Al18), no simple correlation between their catalytic performance and calcination temperature was observed. In the Co5Mg77Al18 series, the sample calcined at 800 °C was significantly more active and selective to CO2 than the catalysts calcined at lower temperatures, which showed very similar catalytic activity in methanol incineration. The activity of the catalysts of Co10Mg72Al18 series was very similar and only slightly depended on the calcination temperature. Among the examined catalysts, the best results, with respect to activity and selectivity to CO2, were obtained for the Co10Mg57Al33 sample calcined at 800 °C. Over the presence of the latter catalyst, methanol was completely oxidised to CO2 from 325 °C. This sample belongs to the group of very effective catalysts for methanol incineration (excluding noble metal-based catalysts) reported in the literature [27, 38, 39]. Such significant differences in the catalytic performance of the studied samples can be explained by various phase composition and redox properties of cobalt species present in these phases. As it was shown by UV–vis-DRS and XRD measurements, cobalt is present in the form of divalent and trivalent cations in periclase-like and spinel-type mixed oxides (due to low content of cobalt and its high dispersion likely in the form of Mg1−xCoxO, Mg(1−x)CoxAl2O4 and MgCoxAl(2−x)O4). Content of the spinel phases increased with increasing calcination temperature. The samples calcined at higher temperatures showed also better catalytic activity in the methanol incineration, especially in the case of the catalysts with the higher Al/Mg ratio. Thus, it could be concluded that cobalt present in the spinel phases is catalytically more active in comparison with cobalt in periclase-like phase. It is well known that reduction of Co3+ to Co2+ in the spinel phases (e.g. Co3O4 or MgCoxAl(2−x)O4) occurs at temperatures significantly lower in comparison to the reduction of Co2+ to Co0 in CoAl2O3, CoxMg(1−x)Al2O4 or CoO [36, 37]. Thus, cobalt oxide phases with spinel structure containing easily reducible Co3+ species (Co3O4 or MgCoxAl(2−x)O4) could be expected to be active components of the effective catalysts for methanol incineration. The cyclic voltammetry and H2-TPR measurements showed the presence of easily reducible phase (Fig. 5, the reduction of Co3+ to Co2+) in the most active catalyst (Co10Mg57Al33 sample calcined at 800 °C). Moreover, this reduction process was intensified in the subsequent voltammetric cycles (Fig. 6). Such effects related to the presence of the easily reducible phase as well as intensification of the reduction process were not observed for other catalysts exhibiting lower catalytic activity. Therefore, it could be suggested that this easily reducible phases, formed during high-temperature treatment (800 °C) of hydrotalcite-like precursor, are responsible for high catalytic activity in the process of methanol incineration. Taking into account the fact that the most active catalyst (Co10Mg57Al33 sample calcined at 800 °C) contained more than three times higher aluminium content and nearly six times higher amount of magnesium in comparison with cobalt content, the formation of the MgCoxAl(2−x)O4 spinel phase is more favourable than formation of Co3O4. Moreover, the presence of Co3O4 was not confirmed by UV–vis-DR studies. Thus, it could be suggested that the MgCoxAl(2−x)O4 spinel-type mixed oxide is more catalytically active in comparison with other cobalt-containing phases present in the Co10Mg57Al33 catalysts (e.g. Mg1−xCoxO periclase-like oxide) and is responsible for the enhanced catalytic activity of the sample calcined at high temperature (800 °C).
Conclusions
The Co–Mg–Al mixed metal oxides were obtained from hydrotalcite-like precursors with various cobalt contents and different Mg/Al ratios by calcination at 600, 700 and 800 °C. The prepared mixed oxides were tested as catalysts in methanol incineration. It was shown that both cation composition of hydrotalcite-like precursors and calcination temperature strongly influenced the catalytic performance of the mixed metal oxides. Among the studied samples, the best catalytic results were obtained with the Co10Mg57Al33 sample having high cobalt content and lower Mg/Al ratio, calcined at 800 °C. High catalytic activity of the latter was related to the presence of Co3+-containing spinel-type phase, very likely the MgCoxAl(2−x)O4 mixed oxide; it was probably more catalytically active in methanol incineration than other cobalt-containing phases formed during calcination of the hydrotalcite-like precursor. High catalytic activity of the spinel phase was ascribed to the relatively easy reducibility of Co3+ to Co2+ cations identified by voltammetry and H2-TPR measurements.
References
Zhang Z, Jiang Z, Shangguan W. Low-temperature catalysis for VOCs removal in technology and application: a state-of-the-art review. Catal Today. 2016;264:270–8.
Chiang YC, Chiang PC, Huang CP. Effects of pore structure and temperature on VOC adsorption on activated carbon. Carbon. 2001;39:523–34.
Toof LJ. A model for the prediction of thermal, prompt, and fuel NOx emissions from combustion turbines. Eng Gas Turbines Power. 1986;108:340–7.
Morales-Torres S, Maldonado-Hódar FJ, Pérez-Cadenas AF, Carrasco-Marín F. Design of low-temperature Pt-carbon combustion catalysts for VOC’s treatments. J Hazard Mater. 2010;183:814–22.
Abdelouahab-Reddam Z, Mail ER, Coloma F, Sepúlveda-Escribano A. Platinum supported on highly-dispersed ceria on activated carbon for the total oxidation of VOCs. Appl Catal A Gen. 2015;494:87–94.
Usón L, Colmenares MG, Hueso JL, Sebastián V, Balas F, Arruebo M, Santamaría J. VOCs abatement using thick eggshell Pt/SBA-15 pellets with hierarchical porosity. Catal Today. 2014;227:179–86.
Mitu M, Razus D, Oancea D. Effect of CO2 dilution on propane–air isothermal catalytic combustion on Platinum. J Therm Anal Calorim. 2017. doi:10.1007/s10973-017-6167-x.
Kim SC, Shim WG. Properties and performance of Pd based catalysts for catalytic oxidation of volatile organic compounds. Appl Catal B Environ. 2009;92:429–36.
Jabłońska M, Król A, Kukulska-Zając E, Tarach K, Girman V, Chmielarz L, Góra-Marek K. Zeolites Y modified with palladium as effective catalysts for low-temperature methanol incineration. Appl Catal B Environ. 2015;166–167:353–65.
Huang H, Ye X, Huang H, Zhang L, Leung DYC. Mechanistic study on formaldehyde removal over Pd/TiO2 catalysts: oxygen transfer and role of water vapor. Chem Eng J. 2013;230:73–9.
Kucherov AV, Sinev IM, Ojala S, Keiski R, Kustov LM. Adsorptive-catalytic removal of CH3OH, CH3SH, and CH3SSCH3 from air over the bifunctional system noble metals/HZSM-5. Stud Surf Sci Catal. 2007;170:1129–36.
Chen BB, Zhu XB, Crocker M, Wang Y, Shi C. FeOx-supported gold catalysts for catalytic removal of formaldehyde at room temperature. Appl Catal B: Environ. 2014;154–155:73–81.
Delannoy L, Fajerwerg K, Lakshmanan P, Potvin C, Méthivier C, Louis C. Supported gold catalysts for the decomposition of VOC: total oxidation of propene in low concentration as model reaction. Appl Catal B: Environ. 2010;94:117–24.
Solsona B, Davies TE, Garcia T, Vazquez I, Dejoz A, Taylor SH. Total oxidation of propane using nanocrystalline cobalt oxide and supported cobalt oxide catalysts. Appl Catal B: Environ. 2008;84:176–84.
Jirátová K, Mikulová J, Klempa J, Grygar T, Bastl Z, Kovanda F. Modification of Co–Mn–Al mixed oxide with potassium and its effect on deep oxidation of VOC. Appl Catal A Gen. 2009;361:106–16.
Basąg S, Piwowarska Z, Kowalczyk A, Węgrzyn A, Baran R, Gil B, Michalik M, Chmielarz L. Cu-Mg-Al hydrotalcite-like materials as precursors of effective catalysts for selective oxidation of ammonia to dinitrogen—the influence of Mg/Al ratio and calcination temperature. Appl Clay Sci. 2016;129:122–30.
Jabłońska M, Chmielarz L, Wegrzyn A, Guzik K, Piwowarska Z, Witkowski S, Walton RI, Dunne PW, Kovanda F. Thermal transformations of Cu-Mg (Zn)-Al(Fe) hydrotalcite-like materials into metal oxide systems and their catalytic activity in selective oxidation of ammonia to dinitrogen. J Therm Anal Calorim. 2013;114:731–47.
Chmielarz L, Rutkowska M, Kuśtrowski P, Drozdek M, Piwowarska Z, Dudek B, Dziembaj R, Michalik M. An influence of thermal treatment conditions of hydrotalcite-like materials on their catalytic activity in the process of N2O decomposition. J Therm Anal Calorim. 2011;105(161):70.
Kuśtrowski P, Wȩgrzyn A, Chmielarz L, Bronkowska A, Rafalska-Łasocha A, Dziembaj R. Thermally induced transformations in polyoxometalate-pillared hydrotalcites. J Therm Anal Calorim. 2004;77(243):51.
Bankauskaite A, Baltakys K. Thermal, texture and reconstruction properties of hydrotalcites substituted with Na+ or K+ ions. J Therm Anal Calorim. 2015;121(227):33.
Smolakova L, Frolich K, Troppova I, Kutalek P, Kroft E, Capek L. Determination of basic sites in Mg–Al mixed oxides by combination of TPD-CO2 and CO2 adsorption calorimetry. When the same basic sites are reported from both techniques? J Therm Anal Calorim. 2017;127:1921–9.
Meloni D, Monaci R, Cutrufello MG, Rombi E, Ferino I. Adsorption microcalorimetry characterization of K-doped MgAl mixed oxide catalysts for soybean oil transesterification synthesized by impregnation and ball milling techniques. J Therm Anal Calorim. 2015;119:1023–36.
Coriolano ACF, Alves AA, Araujo RA, Delgado RCOB, Carvalho FR, Fernandes VJ Jr, Araujo AS. Thermogravimetry study of the ester interchange of sunflower oil using Mg/Al layered double hydroxides (LDH) impregnated with potassium. J Therm Anal Calorim. 2017;127:1863–7.
Cavani F, Trifiro F, Vaccari A. Hydrotalcite-type anionic clays: preparation, properties and applications. Catal Today. 1991;11:173–301.
Taguchi M, Nakane T, Hashi K, Ohki S, Shimizu T, Sakka Y, Matsushita A, Abe H, Funazukuri T, Naka T. Reaction temperature variations on the crystallographic state of spinel cobalt aluminate. Dalton Trans. 2013;42:7167–76.
Li Y, Lu G, Ma J. Highly active and stable nano NiO–MgO catalyst encapsulated by silica with a core–shell structure for CO2 methanation. RSC Adv. 2014;4:17420–8.
Jabłońska M, Chmielarz L, Węgrzyn A, Góra-Marek K, Piwowarska Z, Witkowski S, Bidzińska E, Kuśtrowski P, Wach A, Majda D. Hydrotalcite derived (Cu, Mn)-Mg-Al metal oxide systems doped with palladium as catalysts for low-temperature methanol incineration. App Clay Sci. 2015;114:273–82.
Kovanda F, Rojka T, Bezdička P, Jirátová K, Obalová L, Pacultová K, Bastl Z, Grygar T. Effect of hydrothermal treatment on properties of Ni–Al layered double hydroxides and related mixed oxides. J Solid State Chem. 2009;182:27–36.
Zhang Q, Chen C, Wang M, Cai J, Xu J, Xia C. Facile preparation of highly-dispersed cobalt-silicon mixed oxide nanosphere and its catalytic application in cyclohexane selective oxidation. Nanoscale Res Lett. 2011;6:1–7.
Morpurgo S, Lojacono M, Porta P. Pillared hydroxycarbonates and mixed oxides. Part 1. Copper-zinc-cobalt-aluminum system. J Mater Chem. 1994;4:197–204.
Zayat M, Levy D. Blue CoAl2O4 particles prepared by the sol-gel and citrate-gel methods. Chem Mater. 2000;12:2763–9.
Liotta LF, Pantaleo G, Macaluso A, Di Carlo D, Deganello G. CoOx catalysts supported on alumina and alumina-baria: influence of the support on the cobalt species and their activity in NO reduction by C3H6 in lean conditions. Appl Catal A Gen. 2003;245:167–77.
van de Water LGA, Leendert Bezemer G, Versluijs-Helder JABM, Weckhuysen BM, de Jong KP. Spatially resolved UV–vis microspectroscopy on the preparation of alumina-supported Co Fischer-Tropsch catalysts: linking activity to Co distribution and speciation. J Catal. 2006;242:287–98.
Vakros J, Kordulis C, Lycourghiotis A. Cobalt oxide supported γ-alumina catalyst with very high active surface area prepared by equilibrium deposition filtration. Langmuir. 2002;18:417–22.
Krezhov K, Konstantinov P. On the cationic distribution in MgxCo3−xO4 spinels. J Phys Condens Mater. 1992;4:L543–8.
Han J, Jia L, Hou B, Li D, Liu Y, Liu Y. Catalytic properties of CoAl2O4/Al2O3 supported cobalt catalysts for Fischer–Tropsch synthesis. J Fuel Chem Technol. 2015;43:846–51.
Yu C, Zhou X, Weng W, Hu J, Chen X, Wei L. Effects of alkaline-earth strontium on the performance of Co/Al2O3 catalyst for methane partial oxidation. J Fuel Chem Technol. 2012;40:1222–9.
Chmielarz L, Piwowarska Z, Rutkowska M, Wojciechowska M, Dudek B, Witkowski S, Michalik M. Total oxidation of selected mono-carbon VOCs over hydrotalcite originated metal oxide catalysts. Catal Commun. 2012;17:118–25.
Dziembaj R, Molenda M, Chmielarz L, Zaitz MM, Piwowarska Z, Rafalska-Łasocha A. Optimization of Cu doped ceria nanoparticles as catalysts for low-temperature methanol and ethylene total oxidation. Catal Today. 2011;69:112–7.
Acknowledgements
Part of the research was done with equipment purchased in the frame of European Regional Development Fund (Polish Innovation Economy Operational Program—Contract No. POIG.02.01.00-12-023/08).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Basąg, S., Kovanda, F., Piwowarska, Z. et al. Hydrotalcite-derived Co-containing mixed metal oxide catalysts for methanol incineration . J Therm Anal Calorim 129, 1301–1311 (2017). https://doi.org/10.1007/s10973-017-6348-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6348-7