
Abstract
Layered double hydroxides (LDHs) containing Mg2+, Cu2+ or Zn2+ cations in the MeII positions and Al3+ and Fe3+ in the MeIII positions were synthesized by co-precipitation method. Detailed studies of thermal transformation of obtained LDHs into metal oxide systems were performed using high temperature X-ray diffraction in oxidising and reducing atmosphere, thermogravimetry coupled with mass spectrometry and temperature-programmed reduction. The LDH samples calcined at 600 and 900 °C were tested in the role of catalysts for selective oxidation of ammonia into nitrogen and water vapour. It was shown that all copper congaing samples presented high catalytic activity and additionally, for the Cu–Mg–Al and Cu–Mg–Fe hydrotalcite samples calcined at 600 °C relatively high stability and selectivity to dinitrogen was obtained. An increase in calcination temperature to 900 °C resulted in a decrease of their catalytic activity, possibly due to formation of well-crystallised metal oxide phases which are less catalytically active in the process of selective oxidation of ammonia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrotalcite-like compounds belong to a group of layered double hydroxides (LDHs) and are represented by the general formula of \( \left[ {{{\text{M}}^{\text{II}}_{ 1- {\text{x}}}} {{\text{M}}}^{\text{III}}_{\text{x}} \left( {\text{OH}} \right)_{2} } \right]^{\text{x +} } [{{\text{A}}}^{\text{n -} }_{\text{x}/\text{n}}{\cdot} y{\text{H}}_{ 2} {\text{O}}]^{\text{x -} }\) where MII and MIII are divalent and trivalent metal ions and An− is an interlayer n-valent anion. Typically, the x value in hydrotalcites is in the range from 0.20 to 0.33 [1]. It is possible to produce hydrotalcite-like materials containing various di- (e.g. Mg, Cu, Co, Ni, Zn) and trivalent (e.g. Al, Ni, Co, Fe, Cr) cations as well as different interlayer anions (e.g. CO 2−3 , NO −3 , SO 2−4 , WO 2−4 , CrO 2−4 ). Thermal treatment of hydrotalcite-like materials results in the formation of mixed metal oxides. Phase composition of the calcination products is dependent on chemical composition of LDH precursor, calcination temperature and the atmosphere during heating (oxidising, reducing) [1]. Rock salt-type MIIO and spinel-like oxides are usually identified in powder X-ray diffraction (XRD) patterns of the LDH-related mixed oxides. Therefore, to a large extent, both chemical as well as phase composition of the metal oxide systems can be controlled by the selection of suitable chemical composition and calcination conditions of the hydrotalcite-like precursors. Thermal decomposition of Mg–Al hydrotalcites is well documented: the formation of the MgO phase starts at about 400 °C and an increase in calcination temperature results in formation of MgAl2O4 spinel, together with MgO, above 900 °C [e.g. 2, 3]. The crystallinity of these phases increases with increasing calcination temperature and depends also on the Mg/Al ratio in hydrotalcite precursors. There are also some reports describing thermal decomposition of Cu–Mg–Al hydrotalcite-like materials [e.g. 4–7]. Again, the formation of various phases depends on composition of hydrotalcite precursors as well as calcination temperature. Kovanda et al. [5] observed formation of the well-crystallised tenorite (CuO) phase from a Cu-containing hydrotalcite sample calcined at 400 °C. An increase in calcination temperature resulted in the appearance and then an increase of the crystallinity of the MgAl2O4 and CuAl2O4 spinel phases. At temperatures as high as 900–1000 °C reflections characteristic of CuO disappeared but gueggonite (2CuO·MgO) was detected. On the other hand, Chmielarz et al. [8] reported the formation of amorphous CuO for the Cu–Mg–Al hydrotalcite precursors calcined at 800 °C and copper containing spinel phases for the samples calcined at higher temperatures. Another interesting metal oxide system can be obtained from the Zn–Al hydrotalcite-like precursors [9]. The hydrotalcite structure of such samples decomposes at about 300 °C. An increase in calcination temperature to 600 °C resulted in formation of the well-crystallised zinc oxide phase, while ZnAl2O4 spinel was detected for the samples calcined at 700 °C. Ferreira et al. [10] studied thermal decomposition of Mg–Fe hydrotalcite-like materials using XRD, thermogravimetric analyses and Mössbauer spectroscopy. Authors reported destruction of layered structure at about 300 °C and formation of the MgO and MgFe2O4 spinel phases at temperature above 500 °C. This paper presents a study of the thermal transformations of hydrotalcite-like materials containing Mg–Cu–Fe, Cu–Zn–Al and Cu–Zn–Fe, which are significantly less studied than the above-mentioned Mg–Al, Cu–Mg–Al and Zn–Al systems.
Hydrotalcite-like materials, as-prepared or calcined, have been found to be active catalysts in various processes. The Mg–Al oxides systems were reported to be effective catalysts for phenol alkylation [11], condensation of nitroaldols to nitroalcohols [12] or epoxidation of various allylic alcohols [13]. Cu–Mg–Al hydrotalcite-like materials were used as catalysts for Bayer–Villiger oxidation of ketones [14]. Their calcined forms were used in the oxidative coupling of phenylethyne [15], adsorption and oxidation of ammonia [16], N2O decomposition [17] as well as in decomposition and reduction of nitrogen oxides and sulphur oxides [e.g. 6, 18–21]. The Zn–Al oxide systems were reported to be active catalysts of Friedel–Crafts alkylation of benzene [22] as well as UV-assisted methyl orange decomposition [23]. The catalysts based on Cu–Zn–Al hydrotalcites presented high performance in wet hydrogen peroxide oxidation of phenol [24]. Moreover, they are widely tested in the role of water–gas shift catalysts [e.g. 25, 26]. Our previous studies have shown that Cu–Mg–Fe hydrotalcites are excellent precursors of active catalysts for the selective oxidation of ammonia [27].
Hydrotalcites and the metal oxide systems obtained by their thermal treatment are clearly of great potential for catalysis; however, their catalytic performance depends on conditions of the hydrotalcite precursor synthesis as well as their thermal transformation into metal oxide systems. The present work is focussed on the mechanism of thermal decomposition of hydrotalcite-like materials containing magnesium, copper, zinc, iron and aluminium and optimisation of the calcination conditions leading to active and selective catalysts of ammonia oxidation to nitrogen and water vapour. This process is important to control emission of toxic ammonia into the atmosphere. Ammonia is used as a reactant or produced as a by-product in many chemical processes (e.g. nitric acid and nitrogen fertilizer production, urea manufacturing, hydrodenitrification process, DeNO x process). The selective catalytic oxidation of ammonia by oxygen to nitrogen and water vapour is considered as one of the most promising methods for the removal of NH3 from oxygen containing waste gases [e.g. 28–30].
Experimental
Sample preparation
The hydrotalcite-like samples with the intended Mg(Zn)/Cu/Al(Fe) molar ratios of 2.0/0.0/1.0 and 1.4/0.6/1.0 were synthesized by a co-precipitation method using aqueous solutions of the following metal nitrates: Mg(NO3)2·6H2O (Sigma), Al(NO3)3·9H2O (Fluka), Cu(NO3)2·H2O (Merck), Fe(NO3)3·9H2O (POCh) and Zn(NO3)2·6H2O (POCh). A solution of NaOH (POCh) was used as a precipitating agent. The solutions of nitrates and NaOH were simultaneously added to a vigorously stirred solution containing Na2CO3 (POCh). The pH was maintained constant at 10.0 ± 0.2 by dropwise addition of NaOH solution. The obtained slurry was stirred at 60 °C for a further 30 min, filtered, washed with distilled water and dried. The samples were calcined in an air atmosphere at 600 or 900 °C for 12 h and then were kept in a desiccator in order to avoid the reconstruction of the hydrotalcite structure.
Sample characterisation
The chemical composition of the samples was determined by the AAS method using Varian AA880 spectrometer. Prior to the measurement the sample (50 mg) was mineralised in 2 mL of 35 % hydrochloric acid and boiled for 5 min. The experimental error of this analysis was estimated to be about 3 %.
Specific surface area of the calcined hydrotalcite samples was determined by low-temperature nitrogen adsorption (BET) method using Quantasorb Junior sorptometer (Ankersmit). Prior to the nitrogen adsorption at −196 °C the samples were outgassed in nitrogen atmosphere at 250 °C for 2 h.
Thermal decomposition of the hydrotalcite samples was studied using thermogravimetric method coupled with MS analysis of evolved gases. The thermogravimetry coupled with mass spectrometry measurements were carried out using a Mettler Toledo 851e operated under a flow of synthetic air (80 mL min−1) in the temperature range of 25–1000 °C with a heating rate of 10 °C min−1. The gases evolved during the thermal decomposition process were continuously monitored by quadrupole mass spectrometer ThermoStar (Balzers) connected on-line to the microbalance.
In situ high temperature XRD (HT-XRD) measurements were carried out in the temperature range of 30–900 °C with steps of 50 °C in air or 5 vol% of H2 diluted in nitrogen using a Bruker D8 Advance X-ray diffractometer (Cu Kα1/2 radiation, λ = 1.5418 Å) equipped with a high temperature, reactive-gas chamber (Anton Paar XRK 900). Measurements were performed with a sequential temperature increase of 5 °C min−1 and with no temperature holding time before each analysis. Additionally, XRD patterns of the as-synthesized and calcined (600 and 900 °C) samples were recorded using a D2 Phaser diffractometer (Bruker) using Cu Kα1 radiation (λ = 1.54060 Å).
The reducibility of the samples was studied by temperature-programmed reduction method (H2-TPR). Experiments were carried out in a fixed-bed flow microreactor starting from room temperature to 1100 °C, with a linear heating rate of 5 °C min−1. H2-TPR runs were carried out in a flow (6 mL min−1) of 5 vol% H2 diluted in Ar (N5 quality, Messer). Water vapour was removed from effluent gas by means of a cold trap. The evolution of hydrogen was detected by micro volume TCD (Valco).
Catalytic studies
The hydrotalcite samples calcined at 600 and 900 °C were tested as catalysts for the selective oxidation of ammonia to nitrogen and water vapour. Catalytic tests were performed in a fixed-bed flow microreactor system. The analysis of the reaction products was performed using QMS detector (PREVAC). Prior to the activity tests the sample of catalyst (100 mg) was outgassed at 600 °C for 1 h in a flow of pure helium (20 mL min−1). Catalytic tests were performed in a flow of the reaction mixture containing: [NH3] = 0.5 vol%, [O2] = 2.5 vol%, [He] = 97 vol%. The total flow rate of the reaction mixture was 40 mL min−1. The studies were performed in the temperature range of 150–500 °C with a linear heating rate of 10 °C min−1. Additionally, for the selected, most active catalysts long-term stability tests in isothermal conditions were done. The procedure of the sample outgassing and the composition of the reaction mixture were the same as in the case of the polythermic catalytic tests.
Results and discussion
The surface area (S BET) of the samples calcined at 600 and 900 °C as well as the results of their chemical analysis are shown in Table 1. It should be noted that the determined chemical composition of the samples is very similar to the intended composition. Only small discrepancies were found in the content of aluminium and iron. Surface area of the samples depends on both chemical composition as well as calcination temperature. It was found that a series of the Mg-containing samples has higher surface area compared to the Zn-containing samples. Incorporation of copper into the samples reduced their surface area. The replacement of aluminium for iron decreased the surface area; however, in this case described effect was much more significant. An increase in calcination temperature from 600 to 900 °C dramatically diminished the surface area of the samples.
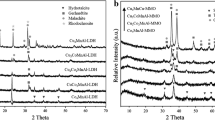
X-ray diffractograms of the dried samples, presented in Fig. 1 (bottom part), are characteristic of the hydrotalcite structure without any reflections of the other possible phases. The only exception is the Cu0.6Zn1.4Fe1.0 sample, which is very amorphous. The sharp diffraction lines of low intensity correspond to ZnO.

The XRD patterns of hydrotalcite-like materials: a Mg2.0Al1.0 sample: H hydrotalcite-like phase, P MgO (periclase), S MgAl2O4 (magnesium aluminate); b Cu0.6Mg1.4Al1.0 sample: H hydrotalcite-like phase, P MgO (periclase), T CuO (tenorite), S MgAl2O4 (magnesium aluminate) and/or CuAl2O4 (copper aluminate), G Cu2MgO3 (gueggenite); c Cu0.6Mg1.4Fe1.0 sample: H hydrotalcite-like phase, P MgO (periclase), T CuO (tenorite), M γ-Fe2O3 (maghemite), G Cu2MgO3 (gueggenite), S MgFe2O4 (magensioferrite) and/or (Cu,Mg)Fe2O4 (cuprospinel) and/or Fe3O4 (magnetite); d Zn2.0Al1.0 sample: H hydrotalcite-like phase, Z ZnO (zincite), S ZnAl2O4 (gahnite); E Cu0.6Zn1.4Al1.0 sample: H hydrotalcite-like phase, Z ZnO (zincite), T CuO (tenorite), S CuAl2O4 (copper aluminate) and/or ZnAl2O4 (gahnite); f Cu0.6Zn1.4Fe1.0 sample: Z ZnO (zincite), T CuO (tenorite), M γ-Fe2O3 (maghemite), S ZnFe2O4 (franklinite)
Diffractograms recorded for the Mg2.0Al1.0 sample calcined at 600 and 900 °C for 12 h are presented in Fig. 1a. In diffractograms of Mg–Al hydrotalcite calcined at 600 °C broad reflections characteristic of periclase (MgO) were identified. An increase in calcination temperature to 900 °C resulted in an appearance of new sharp reflections attributed to the presence of MgAl2O4 and an increase in the intensity of peaks characteristic of MgO. Figure 1b presents diffractograms of the calcined Cu0.6Mg1.4Al1.0 sample. For hydrotalcite calcined at 600 °C broad reflections characteristic of the MgO phase were identified. Calcination at 900 °C resulted in the formation of spinels (MgAl2O4 or/and CuAl2O4), CuO and gueggenite (Cu2MgO3). In the diffractogram of Cu–Mg–Fe hydrotalcite calcined at 600 °C reflections characteristic of the possible spinel phases (MgFe2O4, CuFe2O4, Fe3O4), MgO and CuO appeared (Fig. 1c). An increase in calcination temperature to 900 °C resulted in an increase in the intensity of peaks formed at lower temperatures and the appearance of new reflections characteristic of Cu2MgO3. Also the presence of Fe2O3 cannot be excluded because its characteristic reflections overlap with peaks of the other phases. Figure 1d shows diffractograms of the Zn2.0Al1.0 sample calcined at 600 and 900 °C. Calcination at lower temperature resulted in the formation of the ZnO phase, while for the sample calcined at higher temperature the crystallinity of these phases increased and the reflections characteristic of ZnAl2O4 spinel appeared. In the diffractogram of Cu0.6Zn1.4Al1.0 hydrotalcite calcined at 600 °C broad reflections characteristic of ZnO, CuO and the possible spinel phases (ZnAl2O4, CuAl2O4) were identified (Fig. 1e). Calcination of the sample at 900 °C resulted in an increase of the intensity of these reflections as well as the appearance of new reflection characteristic of the aforementioned phases. The diffraction pattern of the Cu0.6Zn1.4Fe1.0 sample calcined at 600 °C, presented in Fig. 1f, contains reflections characteristic of ZnO, CuO and the possible spinel phases (ZnFe2O4, CuFe2O4, Fe3O4). An increase in calcination temperature to 900 °C resulted in an increase of the intensity of these reflections.
Apart from XRD analysis of the samples calcined at 600 or 900 °C for 12 h, the changes in phase composition of the samples induced by thermal treatment were studied by in situ HT-XRD measurements in oxidising (air) and reducing (5 vol% H2 in N2) atmospheres. In this case temperature was increased with a linear rate of 5 °C min−1. The selected results of these studies are presented in Figs. 2 and 3 (all other diffractograms are attached into supplementary file). As it was mentioned above, all the as-prepared samples exhibited the characteristic hydrotalcite structure [2]. The only exception was Cu0.6Zn1.4Fe1.0, which consists of ZnO and other amorphous components.

In-situ HT-XRD diffraction patterns of the Cu–Mg–Al hydrotalcite-like material recorded in oxidising (a) and reducing (b) conditions. H hydrotalcite-like phase, P MgO (periclase), C Cu2O (cuprite), S MgAl2O4 (magnesium aluminate) and/or CuAl2O4 (copper aluminate), B CuAlO2

In-situ HT-XRD diffraction patterns of the Cu–Zn–Al hydrotalcite-like material recorded in oxidising (a) and reducing (b) conditions. H hydrotalcite-like phase, Z ZnO (zincite), S CuAl2O4 (copper aluminate) and/or ZnAl2O4 (gahnite), U and B CuAlO2
The hydrotalcite structure of Cu0.6Mg1.4Al1.0 (Fig. 2) hydrotalcite is stable up to about 150 °C. Detailed analysis of the positions of 00l reflections shows that the interlayer distance was slightly reduced for increased temperatures of thermal treatment. These results are consistent with TG-QMS analysis (Figs. 5, 6), which showed the evolution of water vapour in this temperature range. Therefore, it could be concluded that decrease in the interlayer distance is related to the release of interlayer water. The Cu0.6Mg1.4Al1.0 sample (Fig. 2) heated at temperatures 300–500 °C, both in oxidising and reducing atmospheres, exhibited an amorphous character. At higher temperatures broad reflections at 2θ values of 35, 43 and 64°, characteristic of MgO developed. At 800 °C, new peaks at 31, 36, 45, 65 and 74° that could be related to the MgAl2O4 and/or CuAl2O4 spinel phases appeared in diffractograms recorded in oxidising atmosphere. Additionally, reflections characteristic of cuprite (Cu2O) were found at 30, 36 (superposition of reflections characteristic of Cu2O and spinel), 42, 61 and 73°. An increase in temperature to 900 °C resulted in the appearance of peaks at 43 and 50° characteristic of CuAlO2 phase [e.g. 31]. Therefore, reduction Cu2+ to Cu1+ occurred at high temperatures. This effect, called auto-reduction or self-reduction, was reported formerly for processes performed in inert [e.g. 7, 31, 32] and oxidising atmospheres [33, 34]. Dalconi et al. [33], who studied oxidation state of copper species in the over-loaded ZSM-5 under heating treatment, reported formation of not only Cu1+ but also Cu0 in air atmosphere. Authors suggest that the formation and sintering of Cu0 is strictly related to the decomposition of copper hydroxide species and to the dehydration of zeolite through a process auto-catalytic reduction of copper ions. Liu and Robota [34], who studied oxidation state of copper by in situ XANES method, reported that there is a significant population of Cu1+ ions formed during the catalytic process of NO decomposition. Authors suggest that Cu1+ is formed by reductive desorption of oxygen from Cu2+ at elevated temperatures. Some mechanisms of Cu2+ auto-reduction was proposed [35–37]. An example could be mechanism proposed by Iwamoto et al. [35] and Jang et al. [36] for copper exchange zeolites, which involves (i) condensation and dehydration of Cu2+(OH−) species followed by (ii) formation of [–Cu2+–O2−–Cu2+–] dimers or oligomers and (iii) desorption of bridging extra-lattice oxygen atoms as oxygen molecules accompanied by reduction of Cu2+ to Cu+ ions.
The reflections characteristic of Cu2O and CuAlO2 were not present in diffractograms recorded for the Cu0.6Mg1.4Al1.0 sample in reducing atmosphere; however, new peaks related to the presence of metallic copper were found at 44 (superposition of reflections characteristic of MgO and Cu0), 51 and 73°.
Comparison of the XRD patterns obtained for the Cu0.6Mg1.4Al1.0 sample calcined at 600 °C for 12 h (Fig. 1b) and recorded at this temperature from in situ XRD measurements (Fig. 2) does not show any significant differences. In both cases only reflections characteristics of MgO were identified. The duration of the thermal treatment process at 900 °C significantly influenced the phase composition of the Cu0.6Mg1.4Al1.0 sample. The presence of gueggonite (Cu2MgO3) was observed only for the sample calcined for 12 h, while the HT-XRD diffractogram recorded at 900 °C contained no reflections characteristic of this phase. On the other hand, the Cu2O and CuAlO2 phases were identified only in the HT-XRD diffractogram. Thus, it seems that Cu1+ ions present in the above-mentioned phases are not stable and are re-oxidised to Cu2+. Therefore, the CuO and CuAl2O4 phases were identified in its diffractogram (Fig. 1b). It should also be noted that long calcination process significantly increased crystallinity of the identified phases.
The structure of Cu0.6Zn1.4Al1.0 hydrotalcite was stable to about 100–150 °C both in oxidising and reducing atmospheres (Fig. 3). At temperatures of about 200 °C the formation of poorly crystallised zincite (ZnO) started and the crystallinity of this phase increased with temperature. The reflections characteristic of ZnO were located at 2θ angles of 32, 34, 36, 47, 56, 63, 68, 69 and 77° [38] and possible spinel phases (CuAl2O4 and ZnAl2O4), represented by reflections at 31, 37, 45, 49, 55, 58, 65 and 74° [39, 40] were detected at higher temperatures. Additionally, the formation of the CuAlO2 phase, represented by characteristic reflections at 43, 50, 65 and 73° appeared for the Cu0.6Zn1.4Al1.0 sample treated at 600–650 °C in oxidising atmosphere. The formation of Cu2O cannot be also excluded; however, its characteristic reflections at 37° as well as 43 and 73° overlap with reflections of the spinel and CuAlO2 phases, respectively [e.g. 41]. Such thermal reduction of Cu2+ to Cu1+ was reported in literature [e.g. 7, 31, 32]; however, it should be noted that such reduction for the Cu0.6Zn1.4Al1.0 sample occured at temperature significantly lower than for Cu0.6Mg1.4Al1.0. Formation of ZnO started at about 600–650 °C in reducing atmosphere; however, at temperatures above 750 °C peaks related to this phase disappeared due to the reduction of ZnO to metallic zinc. On the other hand, zinc cations present in ZnAl2O4 are probably more resistant to reduction because this spinel phase was present in the high temperature range. The presence of the second possible spinel phase—CuAl2O4, which is characterised by reflections at the same positions as ZnAl2O4, is excluded in the high temperature range due to redox properties of copper [42]. Formation of metallic copper, which is a possible product of CuO and CuAl2O4 reduction was observed from temperature about 250 °C and is represented by reflections at 44, 51 and 73°.
The main difference in the phase composition of the Cu0.6Zn1.4Al1.0 sample treated in oxidising atmosphere for shorter (HT-XRD, Fig. 2d) and longer (12 h, Fig. 1e) times is related to the presence of Cu1+ containing phases (CuAlO2 and possibly also Cu2O) identified only in the former experiment and the presence of CuO which was found only in the latter measurements. It seems that re-oxidation of Cu1+ present in CuAlO2 and Cu2O occurred during cooling the sample calcined at 900 °C and, therefore, CuO and spinel phases were identified (Fig. 1e). It should be noted that an increase in thermal treatment duration increased the crystallinity of ZnO, CuO and spinel phases present in this sample.
Figure 4 summarises the changes in the phase composition of the hydrotalcite-like samples that occurred during their thermal treatment in oxidising and reducing atmosphere (determined from in situ HT-XRD). For each phase representative reflections were used to estimate crystallite sizes at a given temperature. Peak broadening was used to calculate crystallite size using Sherrer equation:
in which k is the crystallite size, λ is the wave length, β is the peak broadening, θ is the peak position. In Fig. 4 grey stripes represent phases and their width corresponds to crystallite size. For selected phases, if more that one peak was possible to use for calculations, discrepancies between calculations results were indicated as dotted lines. The original HT-XRD diffractograms were attached as supplementary file. The analysis of the phase composition of the samples was based on the following reports [38–45]. The main conclusions of the in situ HT-XRD studies are: (i) Mg-containing hydrotalcite-like materials are characterised by lower crystallinity than the Zn-containing samples; (ii) in general, thermal decomposition of the hydrotalcite structure occurs at temperatures below 250 °C, both in oxidising and reducing atmosphere; (iii) the formation of poorly crystallised MgO and ZnO was observed both in oxidising and reducing atmosphere at temperature above 250 °C and the crystallinity of these phases increased with an increase in temperature; (iv) depending on the hydrotalcite composition the formation of various spinel phases as well as copper and iron oxides was detected at elevated temperatures; (v) part of copper present in the samples was thermally reduced to Cu1+ (Cu2O and CuAlO2) in oxidising atmosphere at high temperatures; (vi) reduction of copper and iron containing phases into metallic copper and iron was observed only in reducing atmosphere; (vii) comparison of diffractograms recorded for the calcined samples and in situ HT-XRD for the as-prepared samples shows that duration of thermal treatment (calcination) influences phase composition and crystallinity of the indented phases.

Evolution of phase composition of the as-prepared samples during their thermal treatment in oxidising (air) and reducing (5 % H2 diluted in N2) atmosphere; H hydrotalcite-like phase, P MgO (periclase), C Cu2O (cuprite), U Cu (copper), T CuO (tenorite), M γ-Fe2O3 (maghemite), I Fe (iron), F (FeO)x·(MgO)1–x, Z ZnO (zincite), R corrundum (Al2O3), N zinc (Zn), A (Zn0.3Al0.7)Al1.7O4, S possible spinel phases: Cu0.6Mg1.4Al1.0 sample—MgAl2O4 (magnesium aluminate), CuAl2O4 (copper aluminate); Cu0.6Mg1.4Fe1.0 sample—MgFe2O4 (magensioferrite), (Cu,Mg)Fe2O4 (cuprospinel), FeIIFe III2 O4 (magnetite); Zn2.0Al1.0 sample—ZnAl2O4 (gahnite); Cu0.6Zn1.4Al1.0 sample—CuAl2O4 (copper aluminate), (gahnite); Cu0.6Zn1.4Fe1.0 sample—ZnFe2O4 (franklinite)
The TG-QMS method was another technique applied for study of thermal decomposition of the catalyst precursors. The thermal behaviour of hydrotalcites is generally characterised by two main transitions: (i) the loss of interlayer water without collapse of the hydrotalcite structure at low temperature and (ii) the loss of hydroxyl groups from the brucite-like layers and decomposition of interlayer anions at higher temperature [e.g. 2, 46, 47]. It must be underlined that these decomposition stages strongly depend on the cationic composition of the layers. Thermal decomposition of the Mg2.0Al1.0 sample proceeded in two stages (Fig. 5a). The first peak, centred at 198 °C, is related to removal of interlayer water, and the unresolved double DTG minima at 327 and 400 °C are caused by the evolution of water vapour produced by dehydroxylation of OH− anions from brucite-like layers. The evolution of CO2 and NO is related to thermal decomposition of interlayer anions (CO 2−3 and NO −3 ). It should be noted that a small amount of carbonates was stable at temperatures higher than 600 °C.

Results of thermogravimetric analysis of the Mg2.0Al1.0, Cu0.6Mg1.4Al1.0 and Cu0.6Mg1.4Fe1.0 samples. DTG (bottom) and m/z signals of H2O (18), CO2 (44) and NO (30) evolution (upper)
Partial substitution of Mg cations with Cu ions resulted in modification of the thermal decomposition pattern (Fig. 5b). The removal of interlayer water from Cu0.6Mg1.4Al1.0 was detected at 176 °C, so at a temperature 22 °C lower than Mg2.0Al1.0. Two DTG peaks centred at 279 and 366 °C are ascribed to dehydroxylation of the brucite-like layers and thermal decomposition of interlayer carbonate anions. Also, these stages of Cu0.6Mg1.4Al1.0 decomposition occurred at temperatures lower than that observed for Mg2.0Al1.0. Therefore, it could be concluded that incorporation of Cu cations into brucite-like sheets decreased the thermal stability of Mg–Al hydrotalcite. The presence of a high temperature peak (at 628 °C) related to the evolution of CO2 should also be noted. Therefore, it seems that thermally stable carbonates are present in this sample.
Substitution of iron for aluminium in Cu0.6Mg1.4Al1.0 hydrotalcite resulted in additional decrease in its decomposition temperature (Fig. 5c). The release of interlayer water from Cu0.6Mg1.4Fe1.0 occurred at 127 °C, while dehydroxylation of the brucite-like layers was observed in the range of 200–400 °C. The main peaks of CO2 and NO evolution, which are the products of thermal decomposition of interlayer CO 2−3 and NO −3 anions, were centred at about 370 °C. It should also be noted that in this case a small amount of carbonates was present in the sample at temperatures higher than 600 °C.
A maximum of interlayer water evolution from Zn2.0Al1.0 hydrotalcite was found at 174 °C, while the peak at 240 °C is associated to dehydroxylation of the brucite-like layers (Fig. 6a). Dehydroxylation temperature of Zn–Al hydrotalcite is lower than that measured for Mg–Al, which could be related to the different strengths of the Zn–OH and Mg–OH bonds. Such an effect was previously reported by Seftel et al. [48]. The evolution of CO2 and also a small amount of NO, which occurred in the range of 400–800 °C, shows the high stability of carbonate and nitrate anions present in this sample.

Results of thermogravimetric analysis of the Zn2.0Al1.0, Cu0.6Zn1.4Al1.0 and Cu0.6Zn1.4Fe1.0 samples. DTG (bottom) and m/z signals of H2O (18), CO2 (44) and NO (30) evolution (upper)
Similar to the previously described hydrotalcite samples, thermal decomposition of Cu0.6Zn1.4Al1.0 proceeded in two main steps (Fig. 6b). Removal of interlayer water took place at about 170 °C, while dehydroxylation at about 230 °C, so at temperatures very similar to those observed for decomposition of Zn2.0Al1.0 hydrotalcite. The evolution of CO2, which was formed by thermal decomposition of interlayer CO 2−3 anions, occurred in the range of 500–700 °C. Only small amounts of NO were produced from decomposition of NO −3 anions at temperature below 600 °C.
As was mentioned previously, XRD studies showed that Cu0.6Zn1.4Fe1.0, in contradiction to the described above samples, does not exhibit the hydrotalcite-like structure (Fig. 1f). Therefore, it is not surprising that results of TG-QMS obtained for Cu0.6Zn1.4Fe1.0 are different from those measured for the hydrotalcite-like samples (Fig. 6c). First of all, the mass loss is significantly lower (16 wt%) for Cu0.6Zn1.4Fe1.0 compared to the hydrotalcite-like materials (32–42 wt%). The evolution of water vapour was observed at temperatures below 500 °C and proceeded in three steps (72, 161, 272 °C). However, it should be noted that the amount of formed water vapour is significantly lower than for the hydrotalcite-like samples. Carbon dioxide was the main gaseous product of the Cu0.6Zn1.4Fe1.0 decomposition. A broad maximum of CO2 evolution was centred at about 350 °C. Also, the amount of NO emitted from the sample was significantly larger compared to the hydrotalcite-like samples. Thus, it seems that apart from the ZnO phase, which was identified by XRD, this sample contains amorphous carbonates and nitrates.
The reducibility of hydrotalcite-like materials calcined at 600 and 900 °C was studied by temperature-programmed reduction (H2-TPR). The results of these studies are presented in Fig. 7. The TPR pattern of the Cu0.6Mg1.4Al1.0 sample calcined at 600 °C (HT-600-Cu0.6Mg1.4Al1.0) contains a sharp peak centred at 223 °C with a small shoulder at 230 °C (Fig. 7a). According to our previous study [27], this low-temperature peak could be attributed to the reduction of highly dispersed copper oxide species, which include isolated copper ions and small 2D and 3D clusters. An increase in calcination temperature resulted in splitting of this reduction peak into two peaks located at 223 and 252 °C. The first peak, similar to the results obtained for the sample calcined at 600 °C, is attributed to the reduction of isolated and small copper oxide clusters, while the peak located at higher temperature (252 °C) is related to the reduction of copper in CuAl2O4 and bulky CuO clusters [8, 49]. Both these copper containing phases were identified by XRD analysis. The reduction of copper oxide species in Cu–Zn–Al hydrotalcite calcined at 600 and 900 °C occurred at higher temperatures compared to the calcined Cu0.6Mg1.4Al1.0 sample (Fig. 7c). This is not surprising, as it was shown by XRD studies (Fig. 1) that the crystallinity of the CuO phase detected in the Cu0.6Zn1.4Al1.0 sample, especially that calcined at 900 °C, is higher compared to Cu0.6Mg1.4Al1.0. The same effect was observed for spinel the phases, possibly also including CuZn2O4. Similar to the Cu0.6Mg1.4Al1.0 sample, an increase in calcination temperature from 600 to 900 °C resulted in a shift of the reduction peak recorded for Cu0.6Zn1.4Al1.0 to higher temperatures due to the formation of larger and better crystallised copper oxide phases, which were identified by XRD studies (Fig. 1). The broad reduction peak located at about 665 °C is related to the reduction of ZnO to metallic zinc [50], while the roughness of reduction plots observed above 800 °C is probably a result partial sublimation of metallic zinc [51]. The reduction profiles of the samples containing both copper and iron are much more complex (Fig. 7b, d). For the calcined Cu0.6Mg1.4Fe1.0 samples, peaks related to the reduction of the copper oxide phases appeared in the low temperature range (T < 350 °C), while at higher temperatures peaks related to the reduction of various iron oxide species were found (Fig. 7b). For the Cu0.6Mg1.4Fe1.0 sample calcined at 600 °C the reduction of copper present in the form of CuO, Cu2MgO3 and possibly also CuFe2O4 proceeded in the range of 190–280 °C. The peak centred at 205 °C is related to the reduction of highly dispersed copper oxide species, which include isolated copper ions and small 2D and 3D clusters [52–56], while the peak at 227 °C is associated with the reduction of more aggregated copper oxide species. An increase in calcination temperature to 900 °C resulted in a shift of these peaks to higher temperatures (260–380 °C). This effect could be explained by the formation of bulky copper oxide species at elevated calcination temperature, which are characterised by lower reducibility [52–56]. The peaks related to the reduction of iron species appeared at temperatures of 417, 525 and 740 °C. XRD studies have shown that iron could be present in the form of CuFe2O4, MgFe2O4 and Fe3O4. In the case of CuFe2O4, the reduction of copper should occur at lower temperature with the formation of metallic copper and Fe2O3. Basińska et al. [56] and Liang et al. [50] reported that Fe2O3 was reduced to Fe3O4 at about 390–450 °C. Therefore, the peaks at 417 and 435 °C, found for the Cu0.6Mg1.4Fe1.0 sample calcined at 600 and 900 °C, respectively, can be assigned to this process (Fig. 7c). The reduction of both Fe3O4 and MgFe2O4 spinel phases by hydrogen proceeds in two steps [55, 57]:
Therefore, the reduction peak at 525 °C for the sample calcined at 600 °C and peak located at 565 °C for the sample calcined at 900 °C are probably related to reduction of the spinel phases to FeO. Strobbe et al. [58] showed that due to the low stability of FeO, which can be easily reduced to metallic iron, the more stable solid solution (FeO)x·(MgO)1−x can be formed when a well-dispersed MgO phase is present. The presence of (FeO)x·(MgO)1−x phase was confirmed by XRD studies performed in reducing atmosphere (supplementary file). Taking into account that for the Cu0.6Mg1.4Fe1.0 sample calcined at 600 °C there is less crystallised MgO and iron oxide phases, the contact between these phases is better and, therefore, the stabilisation of FeO in the form of (FeO)x·(MgO)1−x solid solution is preferred compared to the sample calcined at 900 °C containing much better crystallised MgO and iron oxide phases. Of course, the reduction of more stable (FeO)x·(MgO)1−x solid solution occurred at temperatures higher than the less stable FeO and proceeded across a broader temperature range.

Results of H2-TPR studies for hydrotalcites calcined at 600 and 900 °C
The H2-TPR profiles obtained for the Cu0.6Zn1.4Fe1.0 sample calcined at 600 and 900 °C contain sharp low-temperature peaks related to the reduction of copper in oxide species (CuO and possibly also CuFe2O4) located at 275 and 330 °C, respectively (Fig. 7d). The shift of this peak to higher temperatures is a result of the thermally induced transformation of small copper oxide species into larger bulky clusters, which exhibit lower reducibility [52–56]. The reduction of iron oxide species occurred at higher temperatures. For the Cu0.6Zn1.4Fe1.0 sample calcined at 600 °C the reduction of iron oxide species (Fe2O3 and possibly ZnFe2O3 and Fe3O4) is represented by a broad reduction peak spread across the range of 330–800 °C, which contains overlapping reduction stages. The peak related to the reduction of Fe2O3 to Fe3O4, (Fe2O3 was identified by XRD (supplementary file)), could be expected in the range of 390–450 °C [e.g. 50]. It was also shown [50] that the reduction of ZnFe2O3 into ZnO and Fe3O4 as well as Fe2O3 to Fe3O4 proceeds in the same temperature range. Therefore, the small shoulder at about 430 °C could be related to the reduction of Fe2O3 and possibly also ZnFe2O3 into Fe3O4. The peaks at higher temperature are related to the reduction of Fe3O4 to metallic iron or, as it was suggested by some authors [e.g. 50], to unstable FeO which disproportionates to metallic iron and Fe3O4 [e.g. 59, 60]. The complex character of this part of the H2-TPR profile could be related to the varying reducibility of Fe3O4 aggregates of different size, or the influence of other components on Fe3O4 reducibility (e.g. ZnO, which is a typical semiconductor was reported to increase reducibility of iron oxide species [49]) as well as the possible stabilisation of FeO species. It should also be taken into account that the reduction of ZnO occurs in the same temperature range as reduction of Fe3O4. Thus, this part of the reduction profile represents a large number of possible transformations occurring in the sample. The H2-TPR profile of the Cu0.6Zn1.4Fe1.0 sample calcined at 900 °C includes the consecutive steps of Fe2O3 and ZnFe2O4 reduction to Fe3O4 (shoulder at 450 °C) and subsequent reduction of this iron oxide into metallic iron (possibly via FeO) represented by shoulder at 590 °C and maximum at 670 °C. Again the complexity of this part of the reduction pattern could be related to the varying reducibility of Fe3O4 aggregates of different size, the influence of other components on Fe3O4 reducibility or the possible stabilisation of FeO phase and reduction of ZnO, which occurs in similar temperature range.
The calcined samples were tested as catalysts for selective ammonia oxidation to dinitrogen and water vapour (4NH3 + 3O2 → 2N2 + 6H2O). Nitrogen oxides (NO, N2O, NO2) are possible side products of the ammonia oxidation process. Activity tests were performed for the catalyst samples calcined at 600 or 900 °C as well as without catalyst for comparison. The results of the study performed in the absence of catalyst are presented in Fig. 8. Ammonia oxidation started at about 375 °C and its conversion slowly increased to 13 % at 500 °C. Dinitrogen was the main reaction product; however, the formation of NO and N2O was also detected. NO2 was not detected. Figure 9 presents the results of catalytic tests performed for the Mg2.0Al1.0 and Zn2.0Al1.0 samples calcined at 600 or 900 °C. The reaction started at about 250–300 °C and the NH3 conversion slowly increased with an increase in reaction temperature. In the case of the samples calcined at 600 °C the best activity was found for HT-600-Zn2.0Al1.0, which was able to convert about 80 % of ammonia at 500 °C. The selectivity to dinitrogen obtained in the presence of this sample was much higher than for HT-600-Mg2.0Al1.0. An increase in calcination temperature of these catalysts to 900 °C resulted in their catalytic activation, especially in the case of HT-900-Mg2.0Al1.0, which was able to convert more than 95 % of ammonia, mainly to dinitrogen. Partial replacement of magnesium or zinc for copper significantly activated the catalysts in the ammonia oxidation process (Fig. 10). For the catalysts calcined at 600 °C (HT-600-Cu0.6Mg1.4Al1.0 and HT-600-Cu0.6Zn1.4Al1.0) oxidation of ammonia started at about 225 °C and its conversion gradually increased to 350 °C, where NH3 was completely oxidised in the reaction mixture. For both these catalysts a very similar activity was measured; however, they significantly differentiated in the selectivity of the reaction, especially at temperatures above 350 °C. Dinitrogen was the main product of ammonia oxidation; however, its contribution to the reaction products gradually decreased, while the content of NO increased, at elevated temperatures. The selectivity to N2 decreased at 500 °C to 72 and 52 % for the process performed in the presence of HT-600-Cu0.6Mg1.4Al1.0 and HT-600-Cu0.6Zn1.4Al1.0, respectively. An increase in calcination temperature to 900 °C decreased the activity of both the catalysts, which were able to completely oxidise ammonia in reaction mixture at temperature as high as 500 °C. This effect could be explained by decrease in surface area of the samples calcined at higher temperatures (Table 1) as well as the formation of more aggregated copper oxide species of lower reducibility. Another difference is related to significant changes in the selectivity to reaction products. For HT-900-Cu0.6Mg1.4Al1.0 the selectivity to dinitrogen decreased at higher temperature (43 % at 500 °C), while for HT-900-Cu0.6Zn1.4Al1.0 this effect was not so significant (74 % at 500 °C). Such significant differences in selectivity to dinitrogen could be explain by various redox properties of copper species present in both samples. Reduction of copper species in the HT-900-Cu0.6Zn1.4Al1.0 sample occurred at temperature significant higher than in the case of HT-900-Cu0.6Mg1.4Al1.0 (Fig. 7). Therefore, the oxidation of the ammonia to NO in the presence of the former catalysts is probably much slower and selectivity to dinitrogen is higher than that observed for HT-900-Cu0.6Mg1.4Al1.0. Figure 11 presents the results of catalytic tests for the Cu0.6Mg1.4Fe1.0 and Cu0.6Zn1.4Fe1.0 samples calcined at 600 or 900 °C. The HT-600-Cu0.6Mg1.4Fe1.0 sample was found to be more active in the low temperature range compared to the other catalysts. Oxidation of ammonia started at temperatures as low as 175 °C, while complete ammonia conversion in the reaction mixture was achieved at 375 °C. Dinitrogen was the main reaction product and its selectivity did not drop below 76 % in the studied temperature range. An increase in calcination temperature of the Cu0.6Mg1.4Fe1.0 sample to 900 °C decreased both the activity of the catalysts as well as its selectivity to dinitrogen. This effect could be explained by very significant decrease in surface area of the sample calcined at higher temperature (from 42 to 2 m2 g−1, Table 1) as well as the formation of more aggregated copper oxide species with lower reducibility (Fig. 7), which catalytically operate at higher temperatures. The HT-600-Cu0.6Zn1.4Fe1.0 catalyst was found to be less active than HT-600-Cu0.6Mg1.4Fe1.0, especially in the low temperature range. Additionally, this catalyst showed very poor selectivity to dinitrogen, which dropped to 17 % at 425 °C. At higher temperatures the selectivity to N2 increased probably due to a significant contribution of ammonia oxidation in the gas phase. An increase in calcination temperature resulted in a significant decrease in surface area (from 10 to 2 m2 g−1, Table 1); therefore, it is not surprising that the HT-900-Cu0.6Zn1.4Fe1.0 catalyst was found to be less active and more selective to dinitrogen than HT-600-Cu0.6Mg1.4Fe1.0. The differences in activity and selectivity observed for HT-900-Cu0.6Mg1.4Fe1.0 and HT-900-Cu0.6Zn1.4Fe1.0 samples could be related to significant differences in redox properties of these catalysts. The low-temperature peak of copper species reduction, present in the reduction profile of HT-600-Cu0.6Mg1.4Fe1.0, appeared at temperature lower by 60 °C than for HT-900-Cu0.6Zn1.4Fe1.0 (Fig. 7). Therefore, the rate of ammonia oxidation to NO is higher in the presence of the HT-600-Cu0.6Mg1.4Fe1.0 catalyst.

Results of activity tests for the selective ammonia oxidation in the absence of catalyst

Results of activity tests of the selective ammonia oxidation in the presence of the Mg2.0Al1.0 and Zn2.0Al1.0 samples calcined at 600 and 900 °C

Results of activity tests of the selective ammonia oxidation in the presence of the Cu0.6Mg1.4Al1.0 and Cu0.6Zn1.4Al1.0 samples calcined at 600 and 900 °C

Results of activity tests of the selective ammonia oxidation in the presence of the Cu0.6Mg1.4Fe1.0 and Cu0.6Zn1.4Fe1.0 samples calcined at 600 and 900 °C
For the most active catalysts, HT-600-Cu0.6Mg1.4Al1.0 and HT-600-Cu0.6Mg1.4Fe1.0, additional long-term stability tests were done (Fig. 12). As it can be seen, only small changes in ammonia conversion and selectivity to the N-containing reaction products were observed during 500 min of the catalytic test. Therefore, these catalysts presented high catalytic stability and could be promising candidates for the next stage of studied focussed on their commercialisation.

Results of long-term activity tests of the selective ammonia oxidation performed in the presence of the HT-600-Cu0.6Mg1.4Al1.0 (upper) and HT-600-Cu0.6Mg1.4Fe1.0 (bottom) catalysts
Analysis of the results of catalytic tests leads to the conclusion that the activity of the catalysts is determined mainly by the presence of copper oxide phases and their redox properties. It was found that for the samples containing easily reduced copper oxide phases higher catalytic activity in the low temperature range was observed. Iron introduced into the catalysts only slightly modified their catalytic activity, however, significantly decreased surface area, especially for the samples calcined at 900 °C. This effect is probably related to the formation of low surface area Fe2O3 and possible MgFe2O4, ZnFe2O4, Fe3O4 spinel phases. Such low surface area catalysts presented poor activity in the high temperature range. The analogous catalysts containing magnesium or zinc showed similar activity but different selectivity. For a series of Cu0.6Mg1.4Al1.0 and Cu0.6Mg1.4Fe1.0 calcined at 600 °C the selectivity to dinitrogen was significantly higher than for the analogous samples containing zinc. However, the opposite selectivity order was found for these samples calcined at 900 °C.
In our previous paper [27], the internal SCR mechanism (i-SCR) for ammonia oxidation over the hydrotalcite derived Cu–Mg–Fe oxide catalysts was proposed. According to this mechanism some ammonia is oxidised to NO, while in the second step NO is reduced by the remaining ammonia to N2 and H2O. N2O is a possible side product of the NO reduction process. Thus, the active and selective catalysts of ammonia oxidation should be able to catalyse ammonia oxidation to NO as well as reduction of NO with ammonia to N2 and H2O. Of course the relative rate of both these reactions is a key factor in determining the selectivity of the whole process. A higher rate of ammonia oxidation to NO results in lower selectivity to dinitrogen, while a higher rate of NO reduction by ammonia should not only lead to higher selectivity to dinitrogen but also lower catalytic activity. Copper was reported to be active in the low-temperature ammonia oxidation and SCR processes, while iron was found to be active in NO reduction by ammonia at higher temperatures [27]. The results of catalytic tests show that selectivity to dinitrogen strongly depends on calcination temperature, which determines the phase composition and crystallinity of the phases present in the samples. It seems that the most important are copper oxide phases, which, depending on their composition and aggregation, strongly influence the final selectivity of the process. Such an important role was not found for iron oxide species, which modified the catalytic performance of the catalysts only in the high temperature region.
Conclusions
Calcined Cu–Mg–Al, Cu–Mg–Fe and Cu–Zn–Al hydrotalcites were found to be active catalysts for the selective oxidation of ammonia. The activity of these catalysts depended on their chemical composition as well as calcination temperature. Calcination of Cu–Mg–Al hydrotalcite at 600 °C resulted in the formation of an amorphous structure in which MgO dominated, while for calcined Cu–Mg–Fe and Cu–Zn–Al hydrotalcites CuO, MgO, Fe2O3, ZnO and mixed oxides with spinel phases were identified. An increase in calcination temperature to 900 °C resulted in a significant increase in the crystallinity of the phases formed at lower temperatures. Additionally, the formation of Cu2MgO3 phase was observed for both Cu–Mg–Al and Cu–Mg–Fe. These catalysts presented high catalytic stability in the long-term reaction tests. An increase in calcination temperature significantly decreased the surface area of the samples, especially for the Fe-containing catalysts and additionally, shifted the process of the copper species reduction into higher temperatures. Hydrotalcites calcined at 900 °C were significantly less active in comparison to the samples calcined at 600 °C. Catalytic performance of the studied samples is determined mainly by the presence of copper oxide phases and their redox properties. The catalysts containing easily reduced copper oxide species presented not only catalytic activity at lower temperatures but also significant decrease in selectivity to nitrogen at higher temperatures.
References
Rives V. Study of layered double hydroxides by thermal methods. In: Rives V, editor. Layered double hydroxides: present and future. New York: Nova Science; 2001. p. 115–37.
Cavani F, Trifiró F, Vaccari A. Hydrotalcite-type anionic clays: preparation, properties and applications. Catal Today. 1991;11:173–301.
Miyata S. Physico-chemical properties of synthetic hydrotalcites in relation to composition. Clays Clay Miner. 1980;28:50–6.
Labajos FM, Rives V, Ulibarri MA. Effect of hydrothermal and thermal treatments on the physicochemical properties of Mg–Al hydrotalcite-like materials. J Mater Sci. 1992;27:1546–52.
Kovanda F, Jirátova K, Rymeš J, Koloušek D. Characterization of activated Cu/Mg/Al hydrotalcites and their catalytic activity in toluene combustion. Appl Clay Sci. 2001;18:71–80.
Chmielarz L, Kuśtrowski P, Rafalska-Łasocha A, Majda D, Dziembaj R. Catalytic activity of Co–Mg–Al, Cu–Mg–Al and Cu–Co–Mg–Al mixed oxides derived from hydrotalcites in SCR of NO with ammonia. Appl Catal B. 2002;35:195–210.
Chmielarz L, Kuśtrowski P, Rafalska-Łasocha A, Dziembaj R. Influence of Cu, Co and Ni cations incorporated in brucite-type layers on thermal behavior of hydrotalcites and reducibility of the derived mixed oxide systems. Thermochim Acta. 2003;395:225–36.
Chmielarz L, Piwowarska Z, Rutkowska M, Wojciechowska M, Dudek B, Witkowski S, Michalik M. Total oxidation of selected mono-carbon VOCs over hydrotalcite originated metal oxide catalysts. Catal Commun. 2012;17:118–25.
Theiss FL, Ayoko GA, Frost RL. Thermogravimetric analysis of selected layered double hydroxides. J Therm Anal Calorim. 2012. doi:10.1007/s10973-012-2584-z.
Ferreira OP, Alves OL, Gouveia DX, Souza Filho AG, de Paiva JAC, Mendes Filho J. Thermal decomposition and structural reconstruction effect on Mg–Fe–based hydrotalcite compounds. J Solid State Chem. 2004;177:3058–69.
Velu S, Swamy CS. Synthesis and physicochemical properties of a new copper–manganese–aluminium ternary hydrotalcite-like compound. J Mater Sci Lett. 1996;15:1647–77.
Bulbule VJ, Deshpande VH, Velu S, Sudalai A, Sivasankar S, Sathe VT. Heterogeneous Henry reaction of aldehydes: diastereoselective synthesis of nitroalcohol derivatives over Mg–Al hydrotalcites. Tetrahedron. 1999;55:9325–32.
Liu H, Xu W, Liu X, Guo Y, Lu G, Wang Y. Aldol condensation of furfural and acetone on layered double hydroxides. Kinet Catal. 2010;51:75–80.
Kaneda K, Ueno S, Imanaka T. Heterogeneous Baeyer–Villiger oxidation of ketones using an oxidant consisting of molecular oxygen and aldehydes in the presence of hydrotalcite catalysts. J Chem Soc Chem Commun. 1994;797–8.
Auer SM, Wandeler R, Göbel U, Baiker A. Heterogeneous coupling of phenylethyne over Cu–Mg–Al mixed oxides: influence of catalyst composition and calcination temperature on structural and catalytic properties. J Catal. 1997;169:1–12.
Trombetta M, Ramis G, Busca G, Montanari B, Vaccari A. Ammonia adsorption and oxidation on Cu/Mg/Al mixed oxide catalysts prepared via hydrotalcite-type precursors. Langmuir. 1997;13:4628–37.
Chmielarz L, Rutkowska M, Kuśtrowski P, Drozdek M, Piwowarska Z, Dudek B, Dziembaj R, Michalik M. An influence of thermal treatment conditions of hydrotalcite-like materials on their catalytic activity in the process of N2O decomposition. J Therm Anal Calorim. 2011;105:161–70.
Shannon IJ, Rey F, Sankar G, Thomas JM, Maschmeyer T, Waller AM, Palomares AE, Corma A, Dent AJ, Greaves GN. Hydrotalcite-derived mixed oxides containing copper: catalysts for the removal of nitric oxide. J Chem Soc Faraday Trans. 1996;92:4331–6.
Corma A, Palomares AE, Rey F, Màrquez F. Simultaneous catalytic removal of SO x and NO x with hydrotalcite-derived mixed oxides containing copper and their possibilities to be used in FCC units. J Catal. 1997;170:140–9.
Wen B, He M. Study of the Cu–Ce synergism for NO reduction with CO in the presence of O2, H2O and SO2 in FCC operation. Appl Catal B. 2002;37:75–82.
Salerno-Polato CM, Assumpçăo-Henriques C, Camacho-Rodrigues AC, Fontes-Monteiro JL. De–SO x additives based on mixed oxides derived from Mg, Al-hydrotalcite-like compounds containing Fe, Cu, Co or Cr. Catal Today. 2008;133–135:534–40.
Shimada H, Ogoshi T. Catalysis by calcined Zn2+/Al3+ layered double hydroxides in Friedel–Crafts alkylation of benzene with benzyl chloride. Bull Chem Soc Jpn. 2005;78:937–9.
Seftel EM, Popovici E, Mertens M, Stefaniak EA, Grieken R, Cool P, Vansant EF. SnIV-containing layered double hydroxides as precursors for nano-sized ZnO/SnO2 photocatalysts. Appl Catal B. 2008;84:699–705.
Zhang LH, Li F, Evans DG, Duan X. Cu–Zn–(Mn)–(Fe)–Al layered double hydroxides and their mixed metal oxides: physicochemical and catalytic properties in wet hydrogen peroxide oxidation of phenol. Ind Eng Chem Res. 2010;49:5959–68.
Ginés MJL, Marchi AJ, Apesteguía CR. Kinetic study of the reverse water–gas shift reaction over CuO/ZnO/Al2O3 catalysts. Appl Catal A. 1997;154:155–71.
Nishida K, Li D, Zhan Y, Shishido T, Oumi Y, Sano T, Takehira K. Effective MgO surface doping of Cu/Zn/Al oxides as water–gas shift catalysts. Appl Clay Sci. 2009;44:211–7.
Chmielarz L, Węgrzyn A, Wojciechowska W, Witkowski S, Michalik M. Selective catalytic oxidation (SCO) of ammonia to nitrogen over hydrotalcite originated Mg–Cu–Fe mixed metal oxides. Catal Lett. 2011;141:1345–54.
Brown DB, Mital R, Jaffri KA, Fuqua KB. US 7, 908, 845 B2, 2011.
Minami T. US 6, 823, 660 B2, 2004.
Funk W, Roberts P. US 7, 497, 076 B2, 2003.
Montanari B, Vaccari A, Gazzano M, Käßner P, Papp H, Makowski W, Łojewski T, Dziembaj R. Transition metal oxides supported on active carbons as low temperature catalysts for the selective catalytic reduction (SCR) of NO with NH3. Appl Catal B. 1996;16:205–13.
Kannan S, Rivers V, Knözinger H. High-temperature transformations of Cu-rich hydrotalcites. J Solid State Chem. 2004;177:319–31.
Dalconi MC, Cruciani G, Alberti A, Ciambelli P. Over-loaded Cu-ZSM-5 upon heating treatment: a time resolved X-ray diffraction study. Mesoporous Microporous Mater. 2006;94:139–47.
Liu DL, Robota HJ. In situ XANES characterization of the Cu oxidation state in Cu-ZSM-5 during NO decomposition catalysis. Catal Lett. 1993;21:291–301.
Iwamoto M, Yahiro H, Tanda K, Mizuno N, Mine Y, Kagawa S. Removal of nitrogen monoxide through a novel catalytic process: 1. decomposition on excessively copper ion exchanged ZSM-5 zeolites. J Phys Chem. 1991;95:3727–30.
Jang HJ, Hall WK, dItri JL. Redox behavior of CuZSM-5 catalysts: FTIR investigations of reactions of adsorbed NO and CO. J Phys Chem. 1996;100:9416–20.
Larsen SC, Aylor A, Bell AT, Reimer JA. Electron paramagnetic resonance studies of copper ion-exchanged ZSM-5. J Phys Chem. 1994;98:11533–40.
Valenzuela MA, Bosch P, Jiménez-Becerrill J, Quiroz O, Páez AI. Preparation, characterization and photocatalytic activity of ZnO, Fe2O3 and ZnFe2O4. J Photochem Photobiol A. 2002;148:177–82.
Maniecki TP, Mierczyński P, Maniukiewicz W, Gebauer D, Jóźwiak WK. The effect of spinel type support FeAlO3, ZnAl2O4, CrAl3O6 on physicochemical properties of Cu, Ag, Au, Ru supported catalysts for methanol synthesis. Kinet Catal. 2009;50:228–34.
Dhak D, Pramanik P. Particle size comparison of soft-chemically prepared transition metal (Co, Ni, Cu, Zn) aluminate spinels. J Am Ceram Soc. 2006;89:1014–21.
Zhao S, Li M, Liu X, Han G. Synthesis of CuAlO2 nanofibrous mats by electrospinning. Mater Chem Phys. 2009;116:615–8.
Morais Batista AH, Ramos FSO, Braga TP, Lima CL, Sousa FF, Barros EBD, Filho JM, Oliveira AS, Sousa JR, Valentini A, Oliveira AC. Mesoporous MAl2O4 (M = Cu, Ni, Fe or Mg) spinels: characterisation and application in the catalytic dehydrogenation of ethylbenzene in the presence of CO2. Appl Catal A. 2010;382:148–57.
Cammelli S, Degueldre C, Cervellino A, Abolhassani S, Kuri G, Bertsch J, Lützenkirchen-Hecht D, Frahm R. Cluster formation, evolution and size distribution in Fe–Cu alloy: analysis by XAFS, XRD and TEM. Nucl Instr Meth Phys Res B. 2010;268:632–7.
Fu L, Yang J, Bi Q, Liu W. Combustion synthesis immiscible nanostructured Fe–Cu alloy. J Alloys Compd. 2009;482:L22–4.
Ueda Y, Ikeda S, Mori Y, Zaman H. Magnetoresistance and magnetism in Fe–Cu alloys produced by electrodeposition and mechanical alloying methods. Mater Sci Eng A. 1996;217–218:371–5.
Palmer SJ, Grand LM, Frost RL. Thermal analysis of hydrotalcite synthesised from aluminate solutions. J Therm Anal Calorim. 2011;103:473–8.
Vulic T, Reitzmann A, Ranogajec J, Marinkovic-Neducin R. The influence of synthesis method and Mg–Al–Fe content on the thermal stability of layered double hydroxides. J Therm Anal Calorim. 2012;110:227–33.
Seftel EM, Popovici E, Pode V, Bandur G. Synthesis, characterization and thermal behavior of hydrotalcite-type Mg x Zn y Al. Rev Roum Chim. 2007;52:1033–7.
Vijayaraj M, Gopinath CS. On the “active spacer and stabilizer” role of Zn in Cu1−x Zn x Fe2O4 in the selective mono-N-methylation of aniline: XPS and catalysis study. J Catal. 2006;241:83–95.
Liang M, Kang W, Xie K. Comparison of reduction behavior of Fe2O3, ZnO and ZnFe2O4 by TPR technique. J Nat Gas Chem. 2009;18:110–3.
Khan A, Smirniotis PG. Relationship between temperature-programmed reduction profile and activity of modified ferrite-based catalysts for WGS reaction. J Mol Catal A. 2008;280:43–51.
Kim SK, Kim KH, Ihm SK. The characteristics of wet air oxidation of phenol over CuO x /Al2O3 catalysts: effect of copper loading. Chemosphere. 2007;68:287–92.
Chary KVR, Seela KK, Ramakanth DNP. Characterization and reductive amination of cyclohexanol and cyclohexanone over Cu/ZrO2 catalysts. Catal Commun. 2008;9:75–81.
Dow WP, Wang YP, Huang TJ. TPR and XRD studies of yttria-doped ceria/γ-alumina-supported copper oxide catalyst. Appl Catal A. 2000;190:25–34.
Balasamy RJ, Khurshid A, Al-Ali AAS, Atanda LA, Legata K, Asamoto M, Yahiro H, Nomura K, Sano T, Takehira K, Al-Khattaf SS. Ethylbenzene dehydrogenation over binary FeO x –MeO y /Mg(Al)O catalysts derived from hydrotalcites. Appl Catal A. 2010;390:225–39.
Basińska A, Jóźwiak WK, Góralski J, Domka F. The behavior of Ru/Fe2O3 catalysts and Fe2O3 supports in the TPR and TPO conditions. Appl Catal A. 2000;190:107–15.
Holgado MJ, Rivers V, Sanroman MS, Malet P. Hexacyanoferrate-interlayered hydrotalcite. Solid State Ion. 1996;92:273–83.
Strobbe DE, Buren FR, Strobbe-Kreemers AW, Dillen AJ, Geus JW. Iron oxide dehydrogenation catalysts supported on magnesium oxide. Part 1—preparation and characterization. J Chem Soc Faraday Trans. 1991;87:1623–9.
Pinna F, Fantinel T, Strukul G, Benedetti A, Pernicone N. TPR and XRD study of ammonia synthesis catalysts. Appl Catal A. 1997;149:341–51.
Pattek-Jańczyk A, Miczko B. Effect of preliminary heating on the activation of model iron catalyst for ammonia synthesis: I. Mössbauer studies of the catalyst reduction. Appl Catal A. 1995;124:253–65.
Acknowledgements
The research was partially carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (Contract No. POIG.02.01.00-12-023/08).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Jabłońska, M., Chmielarz, L., Węgrzyn, A. et al. Thermal transformations of Cu–Mg (Zn)–Al(Fe) hydrotalcite-like materials into metal oxide systems and their catalytic activity in selective oxidation of ammonia to dinitrogen. J Therm Anal Calorim 114, 731–747 (2013). https://doi.org/10.1007/s10973-012-2935-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-012-2935-9