
Abstract
Three competitive inhibitors of urease: boric acid, 2-mercaptoethanol and phosphate buffer pH 6.45, were subjected to the studies of temperature effects on their binding to the enzyme. The first of their kind for these systems, the studies were carried out at temperatures between 15 and 35 °C in HEPES buffers. The Michaelis K M and inhibition K i constants determined for the uninhibited and inhibited urease reactions, respectively, were found to increase with an increase in temperature, importantly, the inhibition remaining competitive irrespective of temperature. The inverse of the constants were further analyzed as equilibrium constants of substrate and inhibitor binding reactions resulting in the formation of ES and EI complexes. The reactions were thermodynamically analyzed with use of the van’t Hoff equation. Found pH-independent in the pH range 6.45–7.0, the changes in the standard enthalpy (ΔH° = −14 kJ mol−1) and the Gibbs free energy (\( \Delta G_{298}^{^\circ } \) = −14 kJ mol−1) for the substrate binding were negative, proving favorable for an exothermic and spontaneous reaction. With the standard entropy change ΔS° close to zero, the reaction is enthalpy driven. The thermodynamic functions of the inhibitor binding by contrast, were found strongly correlated with the inhibitory strengths of the inhibitors in the order: boric acid > 2-mercaptoethanol > phosphate buffer pH 6.45. Accordingly, boric acid demonstrated the changes in the thermodynamic functions considerably bigger (ΔH° = −42 kJ mol−1, \( \Delta G_{298}^{^\circ } \) = −23 kJ mol−1) than the substrate and the other two inhibitors. Those for 2-mercaptoethanol were ΔH° = −20 kJ mol−1 and \( \Delta G_{298}^{^\circ } \) = −17 kJ mol−1, while for phosphate buffer pH 6.45, ΔH° = −17 kJ mol−1, and \( \Delta G_{298}^{^\circ } \) = −13 kJ mol−1, and although slightly bigger, they were found practically comparable in value with those of the substrate, features that apparently result from the comparable values of K M and K is. Remarkably, the inhibitors showed negative values of ΔS° (to various degrees) proving that their binding to the enzyme, like that of the substrate, is driven by enthalpy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Urease (urea amidohydrolase, EC 3.5.1.5) is an enzyme whose catalytic function is to hydrolyze urea [1–3]. The products of the catalytic reaction are ammonia and carbamic acid, but on account of the spontaneous hydrolysis of the latter the observed products are ammonia and carbonic acid:

In view of the wide distribution of urease and wide availability of urea in nature, the catalytic reaction [Eq. (1)] makes urease an enzyme that plays a prominent role in the global N-cycle.
Urease is found in numerous plants, fungi, bacteria, algae, some invertebrates, and in soils as a soil enzyme [1–4]. Depending on the source, the structural organization of urease varies; typically, in plants and fungi urease is a hexamer of identical subunits (ca. 90 kDa each), whereas in bacteria (except Helicobacter pylori urease) it consists typically of three different subunits that join together to form trimers. The bacterial trimer aligns with the single subunit of plant/fungal enzyme exhibiting 50–60 % homology of amino acid sequences. Most importantly, irrespective of the source and composition, the salient feature of ureases is a two-Ni(II) ions-containing active site [5] with a characteristic structure found conserved among the enzymes (Fig. 1a) [6–10]. This fact allows assuming that all ureases employ the same catalytic mechanism.

Urease inhibition studies
Further to theoretical knowledge on urease biochemistry, studies of urease inhibitions are carried out to assure compounds that could control enzyme activity in an effective, dependable and safe manner. The need for such control arises from different medical, environmental and engineering circumstances as discussed below.
Of great significance, the hydrolysis of urea [Eq. (1)] has two consequential features: it produces NH3 and alkalinizes its environment, both capable of causing deleterious complications, notably in medical and agricultural contexts [1, 3]. In the former, bacterial ureases may act as a virulence factor giving rise to pathological conditions [11–13], such as urinary stone formation and pyelonephritis resulting from the infection of the urinary tracts (chiefly with Proteus mirabilis and Ureaplasma urealyticum), and peptic ulcer disease, gastric cancer and hepatic coma resulting from the infection of the gastrointestinal tracts (primarily with Helicobacter pylori). Due to the growing resistance of the pathogens to antibiotics, the use of urease inhibitors has been considered as a therapeutic alternative. In the agricultural context by contrast, urease in soils plays a crucial role in the N-uptake by plants in the form of NH3 produced from the hydrolysis of fertilizer urea [14–16]. However, if too rapid, the hydrolysis may lead to overproduction of ammonia resulting in detrimental effects, such as losses of nitrogen by ammonia volatilization, and ammonia- and alkaline-induced plant damage. Given the fact that urea now makes up over 50 % of the total N-fertilizer worldwide, the problem may have serious environmental and economic consequences. One way to counteract them is to suppress ureolytic activity of soils with use of urease inhibitors.
Another context, where urease activity participates, is calcium carbonate biomineralization. The process is achieved owing to an increase in pH and production of carbonate ions in the urea hydrolysis [Eq. (1)], which in the presence of Ca2+ ions are precipitated in the form of CaCO3 [17]. In addition to the significance in nature, this biocatalytic calcification offers potential in innovative engineering applications, such as preparation of advanced carbonate materials [17], cleaning waste- and groundwaters of Ca2+ and 90Sr2+ [18], and most excitingly, sealing cracks and fissures in buildings [19] and other stone historic monuments of cultural heritage for their restoration [20].
Interestingly, urease inhibition can also be advantageously exploited as an analytical technique in urease-based biosensors for the analysis of substances that act as inhibitors of the enzyme [21]. The measurements are based on the amount of inhibition provoked by the inhibitors, and they make use of enzyme sensitivity to sometimes infinitesimal concentrations of some inhibitors. As yet suffering from the lack of stability and selectivity, such biosensors offer enormous potential for measuring trace levels of compounds in environmental monitoring, food control and biomedical analysis.
Several classes of compounds are known to inhibit urease [1, 22–24]. These include amides and esters of phosphoric acid [7, 25–27], thiols [26–30], hydroxamic acids [26, 31–33], phosphinic and thiophosphinic acids [34–36], boric and boronic acids [37–43], heavy metal ions and their complexes [43–48], bismuth compounds [49], quinones [50–52], phosphate [26, 41, 53, 54], fluoride [27, 32, 55, 56] and to a lesser extent other compounds [1, 22–24, 57, 58]. Despite the ongoing research in this area, the choice of inhibitors qualifying for effective and dependable application in controlling urease activity is still very limited.
Of the inhibitors listed above, three competitive inhibitors of different inhibitory strengths, representing different classes of compounds, were chosen for this study: boric acid, 2-mercaptoethanol and phosphate buffer pH 6.45. Their inhibition constants reported in the literature are assembled in Table 1 and the schematic structures of their complexes with urease in Fig. 1. The following are the main features of the inhibitors.
Boric acid was shown to act as a urease inhibitor in the molecular form B(OH)3 [37, 41, 59]. The crystal structure of the boric acid–urease complex (Fig. 1b) [40] revealed that B(OH)3 replaces W1–W3 water molecules in the active site, leaving in place the bridging water WB. The inhibitor binds to the Ni ions with its two O-atoms, while the third one points toward the opening of the site. By contrast, 2-mercaptoethanol was shown to inhibit urease in its thiolate form OH–CH2–CH2–S− [26]. The crystal structure of the inhibitor–enzyme complex (Fig. 1c) [29] demonstrated that 2-mercaptoethanol binds to urease active site by displacing all four water molecules. The inhibitor S-atom bridges the Ni ions, while its OH group binds to Ni(1). Phosphate buffer, on the other hand, had its inhibitory strength shown pH-dependent; the strength decreases with an increase in pH to cease at pH 7.0–7.5 [26, 53]. The inhibitory action of the buffer was ascribed to H2PO −4 ion [26, 27, 53], a point verified by the crystal structure of the urease–phosphate complex at pH 6.3 [54]. In the structure (Fig. 1d), H2PO −4 ion replaces all four water molecules. One OH group of H2PO −4 bridges the Ni ions, while the other two O-atoms bind to the Ni ions and the fourth one points toward the opening of the site.
Competitive inhibition
In competitive inhibition, an inhibitor I binds to the free enzyme E in a manner that prevents substrate S binding [60]. The equilibria describing competitive inhibition are as follows:

where K D = E·S/ES is the equilibrium constant for the ES dissociation, K i = E·I/EI is the equilibrium constant for the EI dissociation, and k 2 is the rate constant for the breakdown of ES to E + P.
The Michaelis–Menten equation describing the uninhibited reaction is:
where K M is the Michaelis constant and v max is the maximum reaction rate, in competitive inhibition changes into the following rate equation:
where K i is the inhibition constant and the expression K M (1 + I/K i) denotes the apparent Michaelis constant K appM . As shown, competitive inhibition increases K M, but does not affect v max.
To diagnose the type of inhibition, most commonly the Lineweaver–Burk double reciprocal plot of the rate equation is used, which for the competitive inhibition has the form:
In competitive inhibition, the reciprocal plots, 1/v 0 versus 1/S, intersect at y-axis. Further, the replot of the slope = K appM /v max of the reciprocal plots [Eq. (5)] against inhibitor concentration I provides the inhibition constant K i = −intercept on the I-axis.
Alternatively, for diagnosing the inhibition the Dixon plot may be used. The plot is a linear transformation of Eq. (4) where the inhibitor concentration I is a variable:
In competitive inhibition, the plots 1/v 0 versus I recorded at unsaturating S concentrations intersect above the I-axis at I = −K i.
As argued in the preceding paragraphs, there is a compelling need to investigate urease inhibition in order to ascertain new inhibitors and even more importantly, to better develop strategies for knowledge-based urease activity control. Several approaches can be applied in such investigations, i.e., crystallographic studies of enzyme–inhibitor complexes [7, 29, 33, 40, 54, 56], structure-based molecular designs [34–36, 61] in addition to indispensable kinetic studies. It is in the latter context that in this work we offer a thorough temperature-dependent kinetic investigation of the inhibition of urease by three competitive inhibitors representing different inhibitory classes: boric acid, 2-mercaptoethanol and phosphate buffer pH 6.45. This investigation is the first of its kind performed for these systems. Earlier, only prefatory results on the thermodynamics of urease inhibition by heavy metal ions were reported in this journal [68, 69]. In this present work, the thermodynamics of the binding of the chosen inhibitors to the enzyme is comprehensively studied, interpreted in terms of their affinity to the enzyme and compared with the binding of the substrate. Further, the driving forces of the binding of the substrate and inhibitors are discussed.
Materials and methods
Materials
Urease (from jack beans, type III, with nominal activity 27.2 U mg−1 solid), urea (for Molecular Biology), HEPES buffer (SigmaUltra) and 2-mercaptoethanol (purity ≥ 99.0 %) were from Sigma-Aldrich. Boric acid and phosphoric acid were from POCh, Poland. 200 mM stock solutions of HEPES and phosphate buffers were prepared by neutralizing HEPES and phosphoric acid with NaOH. The HEPES stock solutions were further diluted to obtain the concentrations and pHs required for each individual inhibitor: 20 mM pH 7.0 for boric acid, 20 mM pH 6.45 for phosphate buffer pH 6.45, and 5 mM pH 6.84 for 2-mercaptoethanol. All the enzyme mixtures contained 1 mM EDTA. Ultrapure water (resistivity 18.2 MΩ cm) obtained from a Simplicity 185 Millipore system was used throughout.
Urease–inhibitors reactions in the absence of substrate
Urease–inhibitor incubation mixtures were prepared in the respective HEPES buffers. The mixtures contained 1 mg mL−1 urease and an inhibitor, either boric acid at concentrations from the range 15.625–250 mM, or phosphate buffer pH 6.45 at concentrations from the range 1–3.75 M. The mixtures were incubated with occasional stirring. During the incubations, periodically 0.4 mL aliquots were transferred into the assay mixtures (20 mL) containing 50 mM urea in 20 mM phosphate buffer pH 7.2, for enzyme residual activity (RA) determinations, where at 5 min reaction, samples of the reaction mixtures were withdrawn and the ammonia released was converted into indophenol, which was quantified by measuring the absorbance at 630 nm [62]. RA was expressed as percent of the activity of uninhibited urease. The measurements were carried out at room temperature. Interfering with the indophenol method [63], 2-mercaptoethanol was not included in the measurements.
Determination of urease initial reaction rates v 0 in the presence of substrate and inhibitors
For the urease reactions in the presence of boric acid and phosphate buffer pH 6.45, the initial reaction rates v 0 were determined by measuring the amount of ammonia released from the enzymatic reaction. The reactions were performed in mixtures (20 mL) containing 20 mM HEPES buffer of the respective pHs, urea and the inhibitor, the latter two at chosen concentrations. The reactions were initiated by the addition of 0.2 mL of urease solution (2 mg mL−1 freshly prepared in the respective buffers) and carried out for 5 min, when the samples were taken out for ammonia determination by the indophenol method [62].
Due to the interference with the indophenol method [63], the v 0 values of the urease reactions in the presence of 2-mercaptoethanol were assayed by a spectrophotometric method with use of phenol red [64]. The assays were performed in a stopped-flow unit (SX20, Applied Photophysics Ltd, Leatherhead, UK). The reaction mixtures in the unit were formed by mixing urease solution (0.04 mg mL−1) with the solution containing urea and the inhibitor, each at a chosen concentration, and phenol red (final concentration 6.675 × 10−3 mg mL−1) in 5 mM HEPES buffer pH 6.84. The reactions were monitored by the increase in the absorbance at 560 nm for 200 s [64].
The inhibitions were studied at five temperatures between 15 and 35 °C. At each temperature, the solutions to be mixed for the v 0 measurement were conditioned for ca. 20 min, whereas those in the stopped-flow unit, for ca. 5 min.
Determination of the inhibition constants K i of the studied inhibitors
The inhibition constants K i of boric acid and phosphate buffer pH 6.45 were derived from the v 0 rates measured in the presence of five inhibitor concentrations in the range of 0–0.45 mM for boric acid and 0–20 mM for phosphate buffer pH 6.45, at each inhibitor concentration for seven urea concentrations S from the range 2–50 mM in the respective buffers. The saturation curves, v 0 versus S, were constructed for each inhibitor concentration [Eq. (4)], from which the apparent Michaelis constants K appM and the maximum reaction rates v max were obtained by nonlinear regression. The inhibition constants K i were determined from the plots of the ratio of the apparent K appM to v max versus inhibitor concentration I.
The inhibition constants K i of 2-mercaptoethanol by contrast were obtained from the Dixon plots, 1/v 0 versus I [Eq. (6)]. For that, the v 0 rates were measured at two urea concentrations (15 and 25 mM urea), at each of them for four inhibitor concentrations I from the range 0.25–2.0 mM.
To establish the temperature dependence of the inhibition constants K i, each set of the measurements was repeated for each inhibitor at five temperatures (15, 20, 25, 30 and 35 °C).
The experimental uncertainties of the final values of K M, K i and the thermodynamic parameters were estimated from the regression analysis.
Results and discussion
Urease–inhibitor reactions in the absence of substrate: determination of IC50
Prior to measuring the urease v 0 rates in the urease–urea–inhibitor mixtures for K i determinations, it was checked whether the inhibitors equilibrated rapidly with urease to form urease–inhibitor complexes in a manner that is not time dependent. For that, the kinetic curves of urease–inhibitor reactions, RA versus time, were recorded for a range of the inhibitors’ concentrations (Fig. 2). The curves reveal that both boric acid and phosphate buffer equilibrate rapidly with urease within the time shorter than 30 s. Simultaneously, the IC50 values, i.e., inhibitor concentrations bringing about a 50 % inactivation of the enzyme, were estimated (insets to Fig. 2) at 46 mM for boric acid (in agreement with IC50 = 50 mM reported in [65]) and at ca. 4.7 M for phosphate buffer pH 6.45.

Kinetic curves of the reaction between urease and the inhibitors: boric acid and phosphate buffer pH 6.45 for different inhibitors’ concentrations, residual activity (RA) versus incubation time. Insets plots of RA as a function of inhibitors’ concentration in the incubation mixtures
Urease reactions in the presence of substrate and inhibitors: determination of K i
Having established that the inhibitors are rapidly binding to urease, the v 0 rates of the urease reactions were measured at the varied inhibitors’ and urea concentrations at each temperature.
For boric acid and phosphate buffer pH 6.45, the results were assembled in the form of saturation curves, v 0 versus S, as presented for three representative temperatures, 15°, 25° and 35° in Figs. 3a and 4a. The figures reveal that the urease reactions, whether in the presence or absence of the inhibitors, followed Michaelis–Menten kinetics [Eq. (3), (4)] at each temperature, and also that, as typically observed for enzymatic reactions, increasing the temperature increased the reaction rates. The kinetic parameters, K appM and v max, were derived from the curves, and the K M values for the uninhibited reactions are listed in Table 2.

Inhibition of urease by boric acid at 15, 25 and 35 °C: a saturation curves at different boric acid concentrations; b Lineweaver–Burk plots; c plots of the ratio of the apparent K appM /v max versus boric acid concentration

Inhibition of urease by phosphate buffer pH 6.45 at 15, 25 and 35 °C: a saturation curves at different phosphate buffer concentrations; b Lineweaver–Burk plots; c plots of the ratio of the apparent K appM /v max versus phosphate buffer concentration
To ascertain whether temperature over 15–35 °C affected the type of inhibition exerted by the studied inhibitors, the double reciprocal plots [Eq. (5)], 1/v 0 versus 1/S, were constructed. As shown in Fig. 3b and 4b, in all the cases the linear plots intersect on the y-axis, proving that the inhibition is competitive irrespective of temperature in the range of 15–35 °C. Further, the plots of the ratio of the apparent K appM to v max (values taken from the saturation curves) versus I were drawn (Fig. 3c, 4c), from which the inhibition constants K i of boric acid and phosphate buffer pH 6.45 were obtained. The K i values are assembled in Table 3.
The results concerning 2-mercaptoethanol are presented in Fig. 5 in the form of the Dixon plots, 1/v 0 versus 2-mercaptoethanol concentration. The plots form straight lines that intersect at each temperature above negative section of x-axis, proving that the inhibition is competitive. The K i values were read out as the negative values of the concentration of 2-mercaptoethanol corresponding to the intersection point. The values are listed in Table 3.

Inhibition of urease by 2-mercaptoethanol at 15, 25 and 35 °C: Dixon plots obtained for two non-saturating urea concentrations
Thermodynamic characterization of the uninhibited and inhibited urease reactions
Typically in the Michaelis–Menten mechanism [Eq. (2)], the binding step in enzyme reactions consists of the formation of the enzyme–substrate complex ES in a reaction: E + S ⇄ ES. The reaction is governed by the equilibrium constant equal to 1/K D, where K D is the equilibrium constant for the ES dissociation: ES ⇄ E + S [60].
By contrast, in competitive inhibition of enzymes [Eq. (2)], parallel to the substrate binding there occurs the inhibitor binding to the enzyme with the formation of the enzyme–inhibitor complex EI in a reaction: E + I ⇄ EI. The equilibrium of the reaction is expressed by the equilibrium constant equal to 1/K i, where K i is the equilibrium constant for the EI dissociation: EI ⇄ E + I [60].
Measuring the constants K D and K i at different temperatures enables the determination of the thermodynamic parameters for both the substrate and inhibitor binding to the enzyme, ΔH°, ΔS° and ΔG° (the standard enthalpy, entropy and the Gibbs free energy change of the reaction). The standard enthalpy ΔH° and the entropy ΔS° change can be assessed from the van’t Hoff plot:
where K eq is the equilibrium constant of a chemical reaction, T is the absolute temperature, and R stands for the gas constant. From this plot, ΔH°/R is the slope and −ΔS°/R is the intercept of the linear fit. By contrast, the Gibbs free energy change ΔG° for a given temperature can be obtained from the definition:
Effect of temperature on K M
To be able to analyze the thermodynamic parameters of the inhibitor binding relative to the substrate binding, first the uninhibited reaction will be thermodynamically characterized.
Listed in Table 2, the values of K M determined in HEPES buffer at pH 7.0, 6.84 and 6.45, at temperatures between 15 and 35 °C, agree with those reported in the literature [1]. A close inspection of the values reveals effects worthy of attention. One is that the K M constants obtained at pH 7.0 and 6.84 [64] are practically identical, whereas those obtained at pH 6.45 are lower. This observation is in agreement with the effect of pH on K M of urease reported in our previous works [41, 53]. Therein we showed that the dependence of pK M on pH takes up a form of a complex wave, where going from neutral toward lower pHs, K M decreases to assume the lowest values at pH around 6. Although this decrease is rather small, it indicates that at this pH urease has the highest affinity for the substrate, which must be a result of favorable ionization states of the residues of the active site involved in the substrate binding.
Another effect revealed by the data in Table 2 is that contrary to the previously reported independence of temperature [66], the K M values showed a slight increase by about 1.3 mM between 15 and 35 °C at each pH studied (ca. 1.4-fold increase). This indicates a small reduction in the enzyme affinity for the substrate at higher temperatures, which apparently can be regarded as resulting from the distortion of the active site structure.
Further, the thermodynamic parameters for the substrate–urease binding were examined. Importantly, in this examination, we followed the suggestion in the literature [67], based on insignificant variability of K M with pH [41, 53, 66], that urease features K M = K D. Correspondingly, the plots of the inverse of the urease Michaelis constant K M were constructed according to Eq. (7), ln K M versus 1/T, for the pHs studied (Fig. 6a), and the resulting parameters ΔH°, ΔS° and \( \Delta G_{298}^{^\circ } \) [Eq. (8)] were compiled in Table 4. As shown, the thermodynamic parameters for the formation of the urease–urea complex are, within the limits of the experimental error, pH-independent in the studied pH range. The standard enthalpy ΔH° and the Gibbs free energy \( \Delta G_{298}^{^\circ } \) changes are negative, proving favorable for an exothermic and spontaneous reaction. The standard entropy change ΔS°, on the other hand, was found to be close to zero. This indicates that the extent of disorder of the reactants before and after the reaction of substrate binding is comparable; however, the interpretation of this entropy change may be difficult, as the change may be a composite of many different contributions that partially cancel each other. Importantly, the values of ΔH° and ΔS° indicate that the substrate binding in the urease reaction is driven by enthalpy.

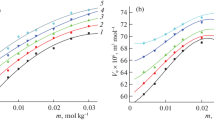
Effect of temperature in the range 15–35 °C on a the Michaelis constant K M at pH 7.0 and 6.45, b the inhibition constants K i of boric acid, 2-mercaptoethanol and phosphate buffer pH 6.45
Effect of temperature on K i
The inhibition constants K i of the studied inhibitors measured between 15 and 35 °C (Table 3) are consistent in magnitude with those presented in the literature for the ambient temperature (Table 1), thus confirming that the inhibitory strength of the inhibitors follows the order: boric acid > 2-mercaptoethanol > phosphate buffer pH 6.45.
The data in Table 3 reveal that for each studied inhibitor the inhibition constant K i grew with temperature in the range of 15–35 °C, nearly three times for boric acid, and less than two times for 2-mercaptoethanol and phosphate buffer pH 6.45. This growth of K i implies that temperature diminishes the affinity of the inhibitors to urease, an effect being more pronounced for boric acid than for 2-mercaptoethanol and phosphate buffer pH 6.45. To try to weigh whether these are temperature-provoked changes in the urease active site or in the inhibitors molecules that are responsible for the reduction of inhibitor-enzyme affinity, the Michaelis K M and inhibition K i constants were compared. As shown in Tables 2 and 3, while the K M value of urease increased ca. 1.4-fold between 15 and 35 °C independent of the pH studied, the K i value of boric acid increased ca. 2.8 times in the same temperature range. This comparison effectively implies that for the decrease in the affinity of boric acid to urease responsible are the changes in the boric acid molecules rather than in the urease active site. By the same token, the fact that K i of 2-mercaptoethanol and phosphate buffer pH 6.45 grew ca. 1.7 times between 15 and 35 °C means that the responsibility for the reduction of the affinity of these inhibitors to urease lies with both the active site and the inhibitors.
In order to carry out a thermodynamic analysis of the inhibition of urease by the three inhibitors studied, the equilibrium constants of the binding reactions [Eq. (2)] equal to the inverse of K i were plotted in the form of the van’t Hoff plots, ln K i versus 1/T [Eq. (7)]. The plots are presented in Fig. 6b and the resulting changes in the thermodynamic functions, in Table 5. Clearly, the location of the plots in Fig. 6b and the values of the thermodynamic functions are directly consequent on the inhibitory strength of the inhibitors; the stronger is the inhibitor (the lower is its K i value), the lower position its van’t Hoff plot occupies. In the studied group of inhibitors, boric acid is the strongest inhibitor showing its K i values that are, depending on temperature, two to one order of magnitude smaller than the K M values of urease, while the K i values of the other two inhibitors are comparable with K M. Consequently, boric acid demonstrated the changes in the thermodynamic functions in the binding reaction with urease considerably bigger than those of the substrate (Table 4) and those of the other two inhibitors (Table 5), a feature that clearly makes it distinct among the studied inhibitors. The change in the enthalpy of the reaction ΔH° = −42 kJ mol−1 proves that the reaction of boric acid with urease is markedly exothermic, more exothermic than the reaction of urease with the substrate (−14 kJ mol−1). Also, \( \Delta G_{298}^{^\circ } \) value of this reaction (−23 kJ mol−1) shows a similar feature, which proves that the reaction is more spontaneous than the other binding reactions under examination. Interestingly, compared to the reaction of substrate binding, boric acid binding shows a large negative value of ΔS° = −63 J K−1 mol−1, clearly indicating that the system after the reaction is more orderly than before the reaction.
By contrast, correlated with their inhibitory strengths, the other two inhibitors studied, 2-mercaptoethanol and phosphate buffer pH 6.45, showed the changes in the thermodynamic functions in their reactions with urease smaller than boric acid; the reactions are less exothermic (ΔH° = −20 and −17 kJ mol−1, respectively) and less spontaneous (\( \Delta G_{298}^{^\circ } \) = −17 and −13 kJ mol−1, respectively). Importantly, although these changes are slightly bigger than those for the substrate binding (Table 4), they are practically comparable in value, features that apparently result from the comparable values of K M and K is. The binding of the two inhibitors to the enzyme is also similar to the substrate binding in respect of entropy change ΔS°. The estimated values of ΔS° were found negative but quite small, and given the high experimental uncertainties, they can be viewed as close to zero.
Cumulatively, with respect to the driving force underlying the inhibitors’ binding, on the basis of the above data it is possible to conclude that like that of the substrate, the binding reactions of all the three inhibitors studied are driven by enthalpy.
In this connection, it would be interesting to compare the data collected in this work with other thermodynamic urease inhibition data. However, as mentioned earlier the reports on the thermodynamics of urease inhibition are scarce in the literature. The available data are those on heavy metal ions inhibition, Hg2+, Ag+, Cu2+, Zn2+ and Cd2+ ions, measured with use of isothermal titration calorimetry [68–70]. Importantly, unlike the competitive inhibitors in this work, heavy metal ions are comparatively strong inhibitors of urease, strongest being Hg2+, Ag+ and Cu2+ [44–46], that act on urease in a slow-binding mode [45]. The inhibitory action of the ions was proven to be primarily through the reaction with the enzyme thiol groups [46]. Intriguingly, the reported thermodynamic parameters for the binding of the ions to urease [68–70] do not vary among the ions, maintaining practically the same values irrespective of the ions’ inhibitory strength. The reported values are: ΔH° ≈ −14 kJ mol−1 (between −16 and −13 kJ mol−1), ΔS° ≈ 19 J K−1 mol−1 (between 17 and 20 J K−1 mol−1), \( \Delta G_{300}^{^\circ } \) ≈ −20 kJ mol−1 (between −21 and −20 kJ mol−1). Thus, like in our inhibitions, these bindings are exothermic and spontaneous, but the most striking difference is that the entropy of the reaction system increased upon metal ions binding, whereas it decreased upon binding of the inhibitors studied in the present work. Consequently, unlike our inhibitions, which are solely enthalpy driven, the inhibition by the heavy metal ions is driven by both enthalpy and entropy. Obviously needing further studies to account for, these distinct differences in the thermodynamic behavior of the two groups of the inhibitors might at least in part now be explained by different mechanisms of the reaction with urease, different inhibitory strengths of the inhibitors and inevitably, by different experimental techniques applied.
Conclusions
Temperature-dependent study of the inhibition of urease by three competitive inhibitors: boric acid, 2-mercaptoethanol and phosphate buffer pH 6.45, was performed in HEPES buffer in the temperature range 15–35 °C. The Michaelis K M and inhibition K i constants determined for the uninhibited and inhibited urease reactions, respectively, were found to increase with an increase in temperature with the inhibition remaining competitive irrespective of temperature. To thermodynamically characterize the reactions of substrate and inhibitor binding to the enzyme resulting in the formation of the enzyme–substrate ES and enzyme–inhibitor EI complexes, the inverse of the K M and K i constants were analyzed with use of the van’t Hoff equation. For the substrate binding, the changes in the standard enthalpy (ΔH° = −14 kJ mol−1) and the free energy (\( \Delta G_{298}^{^\circ } \) = −14 kJ mol−1) were found independent of pH in the range 6.45–7.0 and negative in value, proving favorable for an exothermic and spontaneous reaction. With the standard entropy change ΔS° close to zero, the reaction is enthalpy driven. The thermodynamic functions of the inhibitors’ binding reactions were found correlated with the inhibitory strengths of the inhibitors in the order: boric acid > 2-mercaptoethanol > phosphate buffer pH 6.45. Accordingly, the thermodynamic functions for boric acid binding were considerably bigger (ΔH° = −42 kJ mol−1, \( \Delta G_{298}^{^\circ } \) = −23 kJ mol−1) than those of the substrate and the other two inhibitors, with ΔS° assuming a large negative value of −63 J K−1 mol−1. 2-Mercaptoethanol and phosphate buffer pH 6.45, by contrast, showed the changes in the thermodynamic functions smaller than those of boric acid (ΔH° = −20 and −17 kJ mol−1, and \( \Delta G_{298}^{^\circ } \) = −17 and −13 kJ mol−1, respectively). Although slightly bigger, they were found practically comparable in value with those of the substrate, including the change in ΔS° found close to zero, features that apparently result from the comparable values of K M and K is. Importantly, all the three inhibitors showed negative values of ΔS° (to various degrees), proving that their binding to the enzyme, like that of the substrate, is driven by enthalpy.
References
Krajewska B. Ureases. I. Functional, kinetic and catalytic properties: a review. J Mol Catal B Enzym. 2009;59:9–21.
Maroney MJ, Ciurli S. Nonredox nickel enzymes. Chem Rev. 2014;114:4206–28.
Follmer C. Insights into the role and structure of plant ureases. Phytochemistry. 2008;69:18–28.
Polacco JC, Mazzafera P, Tezotto T. Opinion–Nickel and urease in plants: still many knowledge gaps. Plant Sci. 2013;199–200:79–90.
Dixon NE, Gazzola C, Blakeley RL, Zerner B. Jack bean urease (EC 3.5.1.5). A metalloenzyme. A simple biological role for nickel. J Am Chem Soc. 1975;97:4131–3.
Jabri E, Carr MB, Hausinger RP, Karplus PA. The crystal structure of urease from Klebsiella aerogenes. Science. 1995;268:998–1004.
Benini C, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. A new proposal for urease mechanism based on the crystal structures of the native and inhibited enzyme from Bacillus pasteurii: why urea hydrolysis costs two nickels. Structure. 1999;7:205–16.
Ha N-C, Oh S-T, Sung JY, Cha KA, Lee MH, Oh B-H. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat Struct Biol. 2001;8:505–9.
Balasubramanian A, Ponnuraj K. Crystal structure of the first plant urease from jack bean: 83 years of journey from its first crystal to molecular structure. J Mol Biol. 2010;400:274–83.
Balasubramanian A, Durairajpandian V, Elumalai S, Mathivanan N, Munirajan AK, Ponnuraj K. Structural and functional studies on urease from pigeon pea (Cajanus cajan). Int J Biol Macromol. 2013;58:301–9.
Follmer C. Ureases as a target for the treatment of gastric and urinary infections. J Clin Pathol. 2010;63:424–30.
Rutherford JC. The emerging role of urease as a general microbial virulence factor. PLoS Pathog. 2014;10:e1004062.
Burne RA, Chen Y-YM. Bacterial ureases in infectious diseases. Microb Infect. 2000;2:533–42.
Hasan HAH. Ureolytic organisms and soil fertility: a review. Commun Soil Sci Plant Anal. 2000;31:2565–89.
Mulvaney RL, Bremner JM. Control of urea transformation in soils. Soil Biochem. 1981;5:153–96.
Soares JR, Cantarella H, Menegale MLD. Ammonia volatilization losses from surface-applied urea with urease and nitrification inhibitors. Soil Biol Biochem. 2012;52:82–9.
Sondi I, Salopek-Sondi B. Influence of the primary structure of enzymes on the formation of CaCO3 polymorphs: a comparison of plant (Canavalia ensiformis) and bacterial (Bacillus pasteurii) ureases. Langmuir. 2005;21:8876–82.
Hammes F, Seka A, de Knijf S, Verstaete W. A novel approach to calcium removal from calcium-rich industrial wastewater. Water Res. 2003;37:699–704.
Sarayu K, Iyer NR, Murthy AR. Exploration on the biotechnological aspect of the ureolytic bacteria for the production of the cementitious materials: a review. Appl Biochem Biotechnol. 2014;172:2308–23.
Dhami NK, Reddy MS, Mukherjee A. Application of calcifying bacteria for remediation of stones and cultural heritage. Front Microbiol. 2014;5:304.
Krajewska B. Ureases. II. Properties and their customizing by enzyme immobilizations: a review. J Mol Catal B Enzym. 2009;59:22–40.
Upadhyay LSB. Urease inhibitors: a review. Indian J Biotechnol. 2012;11:381–8.
Kosikowska P, Berlicki Ł. Urease inhibitors as potential drugs for gastric and urinary tract infections: a patent review. Expert Opin Ther Pat. 2011;21:945–57.
Amtul Z, Rahman AU, Siddiqui RA, Choudhary MI. Chemistry and mechanism of urease inhibition. Curr Med Chem. 2002;9:1323–48.
McCarty GW, Bremner JM, Lee SJ. Inhibition of plant and microbial ureases by phosphoroamides. Plant Soil. 1990;127:269–83.
Todd MJ, Hausinger RP. Competitive inhibitors of Klebsiella aerogenes urease. Mechanisms of interaction with nickel active site. J Biol Chem. 1989;264:15835–42.
Dixon NE, Blakeley RL, Zerner B. Jack bean urease (EC 3.5.1.5). III. The involvement of active-site nickel ion in inhibition by β-mercaptoethanol, phosphoroamidate and fluoride. Can J Biochem. 1980;58:481–8.
Todd MJ, Gomez J. Enzyme kinetics determined using calorimetry: a general assay for enzyme activity? Anal Biochem. 2001;296:179–87.
Benini S, Rypniewski WR, Wilson KS, Ciurli S, Mangani S. The complex of Bacillus pasteurii with β-mercaptoethanol from X-ray data at 1.65 Å resolution. J Biol Inorg Chem. 1998;3:268–73.
Juszkiewicz A, Kot M, Zaborska W. Calorimetric study of inhibition of urease by 2-mercaptoethanol: procedures based upon integrated rate equations. Thermochim Acta. 1998;320:45–52.
Dixon NE, Hinds JA, Fihelly AK, Gazzola C, Winzor DJ, Blakeley RL, Zerner B. Jack bean urease (EC 3.5.1.5). IV. The molecular size and the mechanism of inhibition by hydroxamic acids. Spectroscopic titration of enzymes with reversible inhibitors. Can J Biochem. 1980;58:1323–34.
Krajewska B, Zaborska W, Leszko M. Inhibition of chitosan-immobilized urease by slow binding inhibitors: Ni2+, F− and acetohydroxamic acid. J Mol Catal B Enzym. 2001;14:101–9.
Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. The complex of Bacillus pasteurii with acetohydroxamate anion from X-ray data at 1.55 Å resolution. J Biol Inorg Chem. 2000;5:110–8.
Vassiliou S, Grabowiecka A, Kosikowska P, Yiotakis A, Kafarski P, Berlicki Ł. Design, synthesis, and evaluation of novel organophosphorus inhibitors of bacterial ureases. J Med Chem. 2008;51:5736–44.
Vassiliou S, Kosikowska P, Grabowiecka A, Yiotakis A, Kafarski P, Berlicki Ł. Computer-aided optimization of phosphinic inhibitors of bacterial ureases. J Med Chem. 2010;53:5597–606.
Berlicki Ł, Bochno M, Grabowiecka A, Białas A, Kosikowska P, Kafarski P. N-substituted aminomethanephosphonic and aminomethane-P-methylphosphinic acids as inhibitors of ureases. Amino Acids. 2012;42:1937–45.
Breitenbach JM, Hausinger RP. Proteus mirabilis urease. Partial purification and inhibition by boric acid and boronic acids. Biochem J. 1988;250:917–20.
Todd MJ, Hausinger RP. Reactivity of the essential thiol of Klebsiella aerogenes urease. Effect of pH and ligands on thiol modification. J Biol Chem. 1991;266:10260–7.
Krajewska B, Zaborska W, Leszko M, Brzózka Z. Inhibition of jack bean urease by a mixture of boric acid and phosphate buffer pH 6.96. Pol J Chem. 1999;73:359–66.
Benini S, Rypniewski WR, Wilson KS, Mangani S, Ciurli S. Molecular details of urease inhibition by boric acid: insights into the catalytic mechanism. J Am Chem Soc. 2004;126:3714–5.
Krajewska B, Ciurli S. Jack bean (Canavalia ensiformis) urease. Probing acid-base groups of the active site by pH-variation. Plant Physiol Biochem. 2005;43:651–8.
Reddy KRC, Kayastha AM. Boric acid and boronic acids inhibition of pigeonpea urease. J Enzyme Inhib Med Chem. 2006;21:467–70.
Kumar S, Kayastha AM. Inhibition studies of soybean (Glycine max) urease with heavy metals, sodium salts of mineral acids, boric acid, and boronic acids. J Enzyme Inhib Med Chem. 2010;25:646–52.
Krajewska B. Urease immobilized on chitosan membrane. Inactivation by heavy metal ions. J Chem Tech Biotechnol. 1991;52:157–62.
Zaborska W, Krajewska B, Olech Z. Heavy metal ions inhibition of jack bean urease: potential for rapid contaminant probing. J Enzyme Inhib Med Chem. 2004;19:65–9.
Krajewska B. Mono- (Ag, Hg) and di- (Cu, Hg) valent metal ions effects on the activity of jack bean urease. Probing the modes of metal binding to the enzyme. J Enzyme Inhib Med Chem. 2008;23:535–42.
Shi D-H, You Z-L, Xu C, Zhang Q, Zhu H-L. Synthesis, crystal structure and urease inhibitory activities of Schiff base metal complexes. Inorg Chem Commun. 2007;10:404–6.
Li Y-G, Shi D-H, Zhu H-L, Yan H, Ng SW. Transition metal complexes (M=Cu, Ni and Mn) of Schiff-base ligands: syntheses, crystal structure, and inhibitory bioactivities against urease and xanthine oxidase. Inorg Chim Acta. 2007;360:2881–9.
Zhang L, Mulrooney SB, Leung AFK, Zeng Y, Ko BBC, Hausinger RP, Sun H. Inhibition of urease by bismuth(III): implications for the mechanism of action of bismuth drugs. Biometals. 2006;19:503–11.
Zaborska W, Krajewska B, Kot M, Karcz W. Quinone-induced inhibition of urease. Elucidation of its mechanisms by probing thiol groups of the enzyme. Bioorg Chem. 2007;35:233–42.
Krajewska B, Zaborska W. Double mode of inhibition-inducing interactions of 1,4-naphthoquinone with urease. Arylation vs oxidation of enzyme thiols. Bioorg Med Chem. 2007;15:4144–51.
Zaborska W, Kot M, Superata K. Inhibition of jack bean urease by 1,4-benzoquinone and 2,5-dimethyl-1,4-benzoquinone. Evaluation of the inhibition mechanism. J Enzyme Inhib Med Chem. 2002;17:247–53.
Krajewska B, Zaborska W. The effect of phosphate buffer in the range of pH 5.80–8.07 on jack bean urease activity. J Mol Catal B Enzym. 1999;6:75–81.
Benini S, Rypniewski WR, Wilson KS, Ciurli S, Mangani S. Structure-based rationalization of urease inhibition by phosphate: novel insights into the enzyme mechanism. J Biol Inorg Chem. 2007;6:778–90.
Todd MJ, Hausinger RP. Fluoride inhibition of Klebsiella aerogenes urease: mechanistic implications of a pseudo-uncompetitive, slow-binding inhibitor. Biochemistry. 2000;39:5389–96.
Benini S, Cianci M, Mazzei L, Ciurli S. Fluoride inhibition of Sporosarcina pasteurii urease: structure and thermodynamics. J Biol Inorg Chem. 2014;19:1243–61.
Krajewska B. Hydrogen peroxide-induced inactivation of urease. Mechanism, kinetics and inhibitory potency. J Mol Catal B Enzym. 2011;68:262–9.
Krajewska B, Brindell M. Urease activity and l-ascorbic acid. J Enzyme Inhib Med Chem. 2011;26:309–18.
Krajewska B, Zaborska W, Leszko M. Inhibition of chitosan-immobilized urease by boric acid as determined by integration methods. J Mol Catal B Enzym. 1997;3:231–8.
Segel IH. Enzyme kinetics. New Jersey: Wiley; 1975.
Font M, Domínguez M-J, Sanmartín C, Palop JA, San-Francisco S, Urrutia O, Houdusse F, García-Mina JM. Structural characteristics of phosphoramide derivatives as urease inhibitors. Requirements for activity. J Agric Food Chem. 2008;56:8451–60.
Weatherburn MW. Phenol-hypochlorite reaction for determination of ammonia. Anal Chem. 1967;39:971–4.
Ngo TT, Phan APH, Yam CF, Lenhoff HM. Interference in determination of ammonia with the hypochlorite–alkaline phenol method of Berthelot. Anal Chem. 1982;54:46–9.
Krajewska B, van Eldik R, Brindell M. Temperature- and pressure-dependent stopped-flow kinetic studies of jack-bean urease. Implications for the catalytic mechanism. J Biol Inorg Chem. 2012;17:1123–34.
Krajewska B, Zaborska W. Jack bean urease. The effect of active-site binding inhibitors on the reactivity of enzyme thiol groups. Bioorg Chem. 2007;35:355–65.
Dixon NE, Riddles PW, Gazzola C, Blakeley RL, Zerner B. Jack bean urease (EC 3.5.1.5). V. On the mechanism of action of urease on urea, formamide, acetamide, N-methylurea, and related compounds. Can J Biochem. 1980;58:1335–44.
Lopreore C, Byers LD. The urease-catalyzed hydrolysis of thiourea and thioacetamide. Arch Biochem Biophys. 1998;349:299–303.
Behbehani GR, Saboury AA, Poorakbar E, Barzegar L. Application of the extended solvation model for thermodynamic study of copper ion binding to Jack bean urease. J Therm Anal Calorim. 2010;102:1141–6.
Behbehani GR, Saboury AA, Taherkhani A, Barzegar L, Mollaagazade A. A thermodynamic study on the binding of mercury and silver ions to urease. J Therm Anal Calorim. 2011;105:1081–6.
Behbehani GR, Saboury AA, Sabbaghy F. A calorimetric study on the interaction of zinc and cadmium ions with jack bean urease. Chin J Chem. 2011;29:446–50.
Acknowledgements
This work was supported by DS WCh/43 from Faculty of Chemistry of the Jagiellonian University, Kraków, Poland. The stopped-flow measurements were carried out with the equipment financed by the European Regional Development Fund within the framework of the Polish Innovation Economy Operational Program (Contract No. POIG.0 2.01.00-12-0 23/08).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Krajewska, B., Brindell, M. Thermodynamic study of competitive inhibitors’ binding to urease. J Therm Anal Calorim 123, 2427–2439 (2016). https://doi.org/10.1007/s10973-015-5145-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-5145-4