
Abstract
The decomposition and thermal behavior of poly(ethylene terephthalate) (PET)/carbon nanotubes (CNTs) nanocomposites were studied using thermogravimetric (TG) analysis in air atmosphere. A series of PET/single-walled CNTs (SWCNTs) materials of varying nanoparticles concentration were prepared using the in situ polymerization technique. Transmission electron microscopy and scanning electron microscopy micrographs verified that the dispersion of the SWCNTs in the PET matrix was homogeneous, while some relatively small aggregates co-existed at higher filler concentration. Two-stage decomposition was observed in the experiments. During first stage, strong chemical bonds are broken, i.e., aliphatic bonds and benzyl ring containing molecules decompose into small molecules in the gaseous phase. During second stage, when temperature is higher, the remaining nanotubes along with the residues of the first stage are burned. Kissinger and Coats–Redfern (5, 10, 20, 50 K min−1) methods were applied to TG data to obtain kinetic parameters (activation energy, Arrhenius constant at 600 K and A factor) and Criado method to kinetics model analysis. In this kinetic model, energy activation is increasing with the increase of nanotubes concentration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Poly(ethylene terephthalate) (PET) is a semicrystalline polymer which has good mechanical properties, chemical resistance, thermal stability, and spinnability (ability to be spun, e.g., in the form of fibers). Depending on its processing and thermal history, PET may exist both as an amorphous (transparent) and as a semicrystalline material [1]. PET has been used in diverse fields such as food packaging, automotive, electrical, beverage containers, textile, and biomedical industries i.e., by Dacron [1, 2]. Introduction of nanofiller to the polymer matrix, depending on its kind, aims to give composites suitable/better mechanical, tribological, thermal, electrical, optical, and gas barrier properties [3–9]. Significant properties improvement of the polymer-matrix composites (PMCs) depends mainly on: the size and shape of nanofiller, surface area, degree of surface development, surface energy and the spatial distribution of the nanoparticles in the polymer matrix. The enhanced properties usually exhibited by such materials can significantly expand the potential applications of PET [10]. Apart from mechanical properties and processability, thermal properties and resistance to thermal degradation can be improved by the addition of appropriate nanoparticles [11–13]. However, in the case of PET nanocomposites, different results have been reported. Up to now, only a few researchers discovered that the organoclays can enhance the thermal stability of PET nanocomposites prepared by in situ polymerization or melt processing [14, 15]. Broza and Schulte [16] prepared PBT–PTMO/MWCNT nanocomposites by master batch process and concluded that mixing, extruding, granulating, and injection molding seems to be a good alternative to other production methods such as in situ polycondensation. However, in situ polymerization is a suitable method of obtaining well-dispersed nanofiller in the PET matrix [17, 18]. The intense dispersion process by ultrasonication and high speed stirring of CNTs in liquid monomer (1,4-butanediol) and subsequent in situ polycondensation enabled to receive PBT/SWCNT (single-walled CNT) nanocomposites in which the percolation threshold for conductivity was around 0.2 mass% of SWCNT [19]. Furthermore, significant enhancement in rheological properties and electrical conductivity (20 orders of magnitude to 1 S m−1 at 12 vol.%) for the PS/MWCNTs due to the formation of the interpenetrating phases was observed [20]. It is already known that PET/clay nanocomposites show onset decomposition temperature of 3–19 °C higher than that of pure PET [21]. Moreover, Yuan et al. [22] reported that during thermal decomposition in nitrogen the clay can slow down degradation of polymer by acting as a mass transport protective barrier, however, the catalytic effect of the metal derivatives in the clays could accelerate the decomposition behavior of PET. Vassiliou et al. also reported that the addition of MWCNTs during in situ polymerization of PET affected most of its properties. They claimed, that due to incorporation of up to 1 mass% MWCNTs into the PET matrix the decomposition temperature was shifted toward higher [23]. On the other hand, thermogravimetric (TG) analysis proved no influence of single wall nanotubes (SWNTs) on the thermal stability of PET [24].
This paper presents the research on how an introduction of CNTs to PET affects its thermal resistance. The above was tested by means of Coats–Redfern and Kissinger analytical methods which both showed one activation energy value. Obtained results allowed to use Criado and Selum methods to determine the kinetic model of the decomposition process.
Experimental
Materials
For the PET synthesis, the following chemicals were used: dimethyl terephthalate (DMT) (Sigma-Aldrich), ethan-1,2-diol (ED) (Sigma-Aldrich), zinc acetate Zn(CH3COO)2 (Sigma-Aldrich) as an ester exchange catalyst; antimony trioxide Sb2O3 (Sigma-Aldrich) as a polycondensation catalyst; Irganox 1010 (Ciba-Geigy, Switzerland) as a thermal stabilizer. The SWNTs (purity of 90 mass%) were purchased from Cheap Tubes Inc. (Brattleboro, USA). According to supplier’s data: the SWCNTs were produced in a high-yield catalytic process based on chemical vapor deposition with an outer diameter of 1–2 nm, inner diameter of 0.8–1.6 nm and about 5–30 μm of length. Before adding the SWCNTs to the reaction mixture, they were combined with ethanediol to split agglomerates and to improve further exfoliation.
Preparation of PET/SWCNT nanocomposites
Nanocomposites were synthesized by melt transesterification which was followed by polycondensation. Before polymerization, an appropriate amount of SWCNTs were dispersed in 250 mL of ED using ultrahigh speed stirrer (Ultra-Turrax® T25) and ultrasonic homogenizer (Sonopuls HD 2200, Bandelin with frequency of 20 kHz and 75 % of power 200 W). Complete dispersing time was 30 min. In addition, to improve the dispersion of SWCNT in ethanediol an ultrapower lower sonic bath (Bandelin, Sonorex Digitec, with frequency of 35 kHz and power 140 W) was applied for 8 h. In the first stage, the dispersion of SWCNTs in ED, DMT, and zinc acetate catalyst was charged into 1 dm3 steel reactor (Autoclave Engineers Inc., USA) equipped with a vacuum pump, condenser, and cold trap for collecting the by-products. The transesterification reaction was performed with a constant flow of nitrogen at a temperature range of 160–180 °C in the presence of catalyst for 1.5 h. During the reaction, methanol was distilled off. The course of the transesterification reaction was ascertained by monitoring the amount of effluent methanol. As the second stage commenced, the pressure was gradually decreased to about 0.1 h Pa, and the polycondensation was performed at a temperature of 275 °C and with continuous stirring (stirrer speed 40 min−1). The progress of the polymerization was monitored by measuring the changes of viscosity of the polymerization mixture, i.e., an increase in stirrer torque values during polycondensation. The reaction was considered complete when the viscosity of the system increased to 14 Pa s. Synthesis was finished when melt reached a defined value of melt viscosity corresponding to high molecular polymer mass. The obtained nanocomposite was extruded from the reactor under nitrogen flow, cooled to room temperature in a water bath and granulated. The neat PET polymer was synthesized following the same procedure without nanotubes. Subsequent to the granulation process, the granulate was used to obtain dumbbell-shaped samples (with a rectangular cross-section of 2 × 4 mm2) using Boy 15 (Dr BOY GmbH and Co., Germany) injection-molding machine. The injection-molding parameters were as follows: injection pressure 50 bar, melt temperature 265–279 °C, mold temperature 26 °C, hold time 10 s, and cool time 15 s.
Characterization of PET/SWCNT nanocomposites
The intrinsic viscosity [η] of the samples was determined at 30 °C in mixture phenol/1,1,2,2-tetrachloroethane (60/40 by mass). The polymer solution had a concentration of 0.5 g dL−1. The measurement was carried out on a capillary Ubbelohde viscometer (type Ic, K = 0.03294). In order to insure that the intrinsic viscosity will not be affected by present SWCNTs, the polymer nanocomposite solution was filtered through 0.2 μm pore size polytetrafluoromethylene filter (Whatman; membrane type TE 35). After filtration, the polymer was precipitated and re-dissolved. The Mark–Houwink equation [η] = 3.72 × 10−4·M 0.73 was used to calculate the viscosity average molecular mass of PET homopolymer [25].
Morphology and nanostructure
The structure of nanocomposites was observed by scanning electron microscopy (SEM) (JEOL JSM 6100 SEM). The samples were cryofractured in liquid nitrogen and then vacuum coated with a thin gold film prior to the test. Transmission electron microscopy (TEM, JEOL JEM-1200 EX Electron Microscope) micrograph was obtained using an acceleration voltage of 80 kV. For TEM measurement, ultrathin sections were prepared with a Reichert Ultracut R ultramicrotome using a diamond knife.
Theory and calculations
Activation energy
Kinetic studies start with the basic equation describing the relation of the rate of mass loss at a constant temperature to the fractional decomposition [26–28]:
where dα/dt is the rate of mass loss, α is the fractional decomposition at any time, and k is rate constant. The term f(α) is a function form of α; namely,
The rate constant, is dependent on the reaction temperature according to the Arrhenius expression:
where E d is the activation energy (in this case decomposition activation energy), A is a pre-exponential factor, R is the gas constant and T is the reaction temperature.
Combining Eqs. 1–3 an following expression is derived:
Due to that dα/dt = (dα/dT)(dT/dt) = β(dα/dT), the integrated form of Eq. 4 can be expressed as:
where β is the heating rate. The temperature dependent part of Eq. 5 cannot be solved analytically, therefore a number of approximation methods are proposed using TG and derivative TG (DTG) data.
Integration of Eq. 5 from an initial temperature, T 0, corresponding to a degree of conversion α 0, to the peak temperature, T p, where α = α p, gives:
If T 0 is low, it may be reasonably assumed that α 0 = 0, and considering that there is no reaction between 0 and T 0:
where g(α) is the integral function of conversion [29].
Coats and Redfern developed an integral method, which can be applied to TG data, assuming the order of reactions. The correct order is presumed to lead to the best linear plot, from which the activation energy is determined. The final form of the equation, which is used for the analysis, takes the following form [30, 31]:
Thus a plot of ln[1 − (1 − α)1 − n/(T 2(1 − n))] versus 1/T should result in a straight line of slope that equals −E/R for the correct value of reaction order, n.
On the other hand, activation energy could be determined by Kissinger method.
Kissinger [28, 30, 31] derived an equation that allows the calculation of activation energy using the temperature value, T m at the maxima of the first mass derivative curves at different heating rates, as shown in Fig. 3b. Because the maximum rate occurs when d2 α/dt 2 = 0, differentiation of Eq. 4 gives:

TEM micrograph of PET/SWCNTs nanocomposite containing 0.2 mass% SWCNTs
Kissinger assumes that the product \( n(1 - \alpha )_{\text m}^{{\text n} - {\text 1}} \) is independent of β. So, the following expression is derived:
Kinetic model of thermal degradation
In an attempt to develop a model for thermal behavior of polymeric materials in full scale systems, the main purpose is to describe the thermal degradation of polymers in terms of an intrinsic kinetics, in which heat and mass transfer limitations are not included. In general, kinetic models are proposed in the literature for plastics and thermosets. These models describe the process by means of a simplified reaction pathway. Each reaction phase is described by a number of complex degradation reactions [32].
The Coats–Redfern method [31] has been found to be the most versatile approach to calculate the kinetic parameters of thermal degradation processes. The Coats–Redfern method provides the following correlation:
g(α) is an integral function of conversion:
In order to investigate the solid-state degradation mechanism in our study, the Coats–Redfern algorithm was selected.
If the value of the activation energy is known, the kinetic model of the process can be found in the following way. Criado et al. [32–37] define the function:
where x is the reduced activation energy (E d/RT), β is the heating rate (K min−1), T is the absolute temperature (K) and π(x) is the expression of temperature integral. As was pointed out, π(x) can be described using a fourth order rational expression of Senum and Yang [38] as:
These equations are used to obtain the master curves as a function of the reaction degree corresponding to the different models listed in [32–37].
TG analysis
Described experiments were carried out by non-isothermal TG using a Q-5000 device (TA Instrument). Measurements were carried out in an oxidizing atmosphere i.e., dry, synthetic air (N2:O2 = 80:20 vol.%). The activation energies of the decomposition process were measured at heating rates 5, 10, 20, and 50 K min−1 in the temperature range of 313–1,173 K. Samples masses vary from about 8 to 12 mg. Air gas flowed into the furnace at 25 mL min−1.
Results and discussion
Characterization of the prepared materials
PET/SWCNTs nanocomposites were prepared in situ during polymerization of PET by the two-stage melt polycondensation method of DMT and ED in a 1 dm3 steel reactor in the presence of SWCNTs. The prepared nanocomposites of PET/SWCNT contained, respectively, 0.025, 0.05, 0.1, 0.2, and 0.4 mass% of nanotubes. In addition, for comparison purposes, unmodified PET was synthesized and characterized in the same manner as the nanocomposites. During the first stage of transesterification, oligomers were prepared, while methanol was removed as by-product. In order to prepare high molecular mass polyesters, the polycondensation of these oligomers ensued at higher temperature, with the simultaneous application of high vacuum. The intrinsic viscosity of PET that was prepared was 0.553 dL g−1. The presence of the SWCNTs in the polymerization mixture affected the reaction, leading to variations of the final intrinsic viscosities of the nanocomposites prepared. As shown in Table 1, the intrinsic viscosity increased at the SWCNTs concentration of 0.1 mass%, and, then, gradually decreased at higher concentrations. The similar effect was also observed in nanocomposites containing SiO2, which can also act as multifunctional additives, it was found that in small amounts SiO2 nanoparticles could act as chain extender, increasing the molecular mass of the polymer, while at higher contents, due to the extended degree of the aforementioned reaction, branched and crosslinked macromolecules are formed, leading to a reduce of molecular mass [39, 40]. This may explain the gradual decrease observed in molecular mass of the PET samples with concentrations of SWCNTs nanoparticles greater than 0.1 mass%.
From TEM micrograph (Fig. 1), it was observed that the dispersion of the SWCNTs into the PET matrix was homogeneous, while at higher concentrations of SWCNTs some small aggregates were also formed. This was due to the extremely high specific area of SWCNTs and the strong particle–matrix interactions that take place.
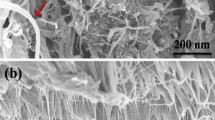
The dispersion of SWCNTs in ED produced quite good exfoliation as shown in the SEM image (Fig. 2) where some agglomerate particles have been seen and indicated by arrows, are clearly visualized embedded in the PET polymer matrix SEM images also indicate that CNTs were encapsulated by the PET matrix. This suggests a strong interaction between SWCNTs and PET matrix. Size of nanotubes in such agglomerate is increased by gold sputtering.

SEM micrograph of PET/SWCNTs nanocomposite containing 0.2 mass% SWCNTs
Decomposition process on TG
It seems clear that the introduction of the SWCNTs to PET should cause higher thermal resistance in comparison to the material obtained from the neat PET. Giraldo et al. [41] observed for PA6/MWCNTs nanocomposites the onset shift of thermal degradation to higher temperatures with increase of scratch hardness while avoiding the damage on the surface, without the presence of any debris in broad range of applied loads.
The composition process could be divided into two stages: stage 1 from 600 to 750 K, which described mainly the decomposition process; stage 2 from 750 to 900 K, in which remains from burning of black carbon along with remaining nanotubes were observed (Fig. 3).

Exemplary TG (a) and DTG (b) curves for PET with different nanotubes content (5 K min−1 heating rate)
For instance, Coats–Redfern plots for PET materials are presented in Fig. 4 for stage 1 (α from 0.05 to 0.65).

Exemplary Coats–Redfern plot of PET for stage 1 (5 K min−1 heating rate)
In the present study, 50 different reaction orders were selected in a range between 0.5 and 3.0 for which correlation coefficients greater than 0.9 were obtained.
The best approximations for first stage of PET materials with different nanotubes content and the following reaction orders are obtained: 2.5 (PET), 1.6 (PET with 0.05 mass%), 1.1 (PET with 0.10 mass%), 1.6 (PET with 0.20 mass%), 2.0 (PET with 0.40 mass%), respectively. It is known that values of reaction order strongly affect the activation energy value; therefore, in order to compare the influence of nanotubes content on this parameter average value n = 1.75 was chosen.
Calculated kinetic parameters were presented in Table 2 for first stage (for the obtained and selected reaction orders).
It can be seen that values of activation energy (average values) vary from 297 (PET with 0.10 nanotubes content) to 377 kJ mol−1 (PET without nanofiller). In general, for activation energy values calculated for different reaction orders, it can be observed that these parameters are reduced with a decrease in nanotube content, those values are lower in comparison to those obtained for PET without nanotubes. A similar tendency is observed in case of n = 1.75 and heating rate of 5 K min−1, the highest activation energy is calculated for materials without SWCNTs and values are smaller and similar for PET with nanotubes. The same tendencies are observed in case of pre-exponential A and rate constant k. The values of A vary from 4.43 × 1026 to 4.23 × 1029 min−1 and rate constant from 2.28 × 1059 to 5.85 × 1064 min−1. Comparison of activation energies obtained for first stage leads to the conclusion that introducing nanotubes causes a slight decrease in thermal resistance.
Activation energy could also be calculated from Kissinger method (with several heating rates)—Fig. 5.

Kissinger plot of PET for the first stage
The activation energy could be calculated from the slope of a plot of ln(β/\( T_{\text{m}}^{2} \)) as a function of 1/T (Tables 3, 4).
Surprisingly, the obtained trend value is quite different from the activation energy calculated by Coats–Redfern method. An increase in nanotubes content causes an increase of the activation energy value from about 189 (PET without SWCNTs) to 300 kJ mol−1 (PET with the highest nanotubes content). Obtained results allow to conclude that the introduction of nanotubes improved thermal resistance of nanocomposites.
The values of the activation energy for the second stage are similar in the range from 129 to 132 kJ mol−1, with the exception of the value obtained for the material with the highest SWCNTs content.
Kinetics model of degradation
Example z(α) plots, calculated on the basis of equations and derived from experimental data, are shown in Fig. 4 (for activation energy calculated by Coats–Redfern method for 5 K min−1 heating rate) and 5 (Kissinger). Comparing them with the master curves leads to an easy and precise determination of the degradation mechanism. It can clearly be seen that the experimental data of z(α) for PET are in accordance with the F1 master curve (Figs. 6, 7).

Theoretical z(α) master plots for the reaction models from Table 1 and experimental data for PET nanocomposites a obtained with activation energies calculated by different reaction order b with n = 1.75

Theoretical z(α) master plots for the reaction models from Table 1 and experimental data for PET nanocomposites obtained with activation energies calculated by Kissinger method
PET nanocomposites exhibit thermal degradation mechanism which is similar the most to a sigmoidal curve (Fn type mechanism), a random nucleation with one nucleus on the individual particle—F1. The curves shape is similar regardless to the nanotube content. The most similar to the mechanism F1 are curves obtained for PET with 0.40 and 0.20 nanotubes content. In case of results obtained for n = 1.75, curves similar to ones for reaction orders ranging between 0.05 and 0.30 (mechanism F1 or R3) are observed and with an increase in α value a change in the decomposition mechanism to D2 and D3 (two- and three-dimensional diffusion) is seen.
The degradation mechanism of the PET nanocomposites can be compared to the Fn type mechanism (deceleration curve), to be more precise F1—the random nucleation with one nucleus on the individual particle or R3—describing phase boundary controlled reaction in contracting volume. An increase of order causes decomposition mechanism changes from F1 or R3 to D2 and D3 decomposition mechanism (two- and three-dimensional diffusion).
Conclusions
By applying an intensive dispersion process consisting of the ultrasonication and subsequent ultrahigh-speed stirring of SWCNTs in monomer (ED) followed by in situ polymerization, it is possible to prepare PET/SWCNTs nanocomposites with good distribution of carbon nanoparticles in polymer matrix; however, occurrence of agglomerates is clearly seen in the SEM image. Introducing nanotubes (SWCNTs) improves thermal stability of PET nanocomposites. With an increase in nanotube content, an increase in activation energy values can be seen, particularly in the case of the values calculated by the Kissinger method. This method seems to be more appropriate to determine the activation energy rather than the Coats–Redfern method; however, the value calculated from both methods helped to determine the degradation kinetic model of described nanocomposites. The degradation kinetic process followed a deceleration curve and obeyed the random nucleation with one nucleus on the individual particle (F1 mechanism) and deceleration curve describing phase boundary controlled reaction in contracting volume (R3 mechanism). Increasing of reaction rate causes changes in degradation process from F1 or R3 to D2 and D3 (two- and three-dimensional diffusion). In this kinetic model, activation energy is increasing with the increase of nanotubes concentration.
References
Van Krevelen DW. Properties of polymers. 4th ed. Amsterdam: Elsevier; 2009.
Cowie JMG. Polymers: chemistry and physics of modern materials. 3rd ed. Boca Raton: Taylor and Francis Group; 2008.
Huang JC. Carbon black filled conducting polymers and polymer blends. Adv Polym Technol. 2002;21:299–313.
Monitruzzaman M, Winey KI. Polymer nanocomposites containing carbon nanotubes. Macromolecules. 2006;39:5194–205.
Okamoto M. Polymer/clay nanocomposites. Encyclopedia of nanoscience and nanotechnology, vol. 8. Stevenson Ranch: American Scientific Publishers; 2004.
Li Z, Luo G, Wei F, Huang Y. Microstructure of carbon nanotubes/PET conductive composites fibers and their properties. Compos Sci Technol. 2006;66:1022–9.
Zhu Z, Wang R, Dong Z, Huang X, Zhang D. Morphology, crystallization, and mechanical properties of poly(ethylene terephthalate)/multiwalled carbon nanotubes composites. J Appl Polym Sci. 2011;120:3460–8.
Roslaniec Z, Broza G, Schulte K. Nanocomposites based on multiblock polyester elastomers (PEE) and carbon nanotubes (CNTs). Compos Interface. 2003;10:95–102.
Brostow W, Broza G, Datashvili T, Hagg Lobland HE. Poly(butyl terephthalate)/oxytetramethylene + oxidized carbon nanotubes hybrids: mechanical and tribological behavior. J Mater Res. 2012;27(14):1815–23.
Frounchi M, Dourbash A. Oxygen barrier properties of poly(ethylene terephthalate) nanocomposites films. Macromol Mater Eng. 2009;294(1):68–74.
Mansour SA. Study of thermal stabilization for polystyrene/carbon nanocomposites via TG/DSC techniques. J Therm Anal Calorim. 2013;112:579–83.
Cheng HKF, Sahoo NG, Lu X, Li L. Thermal kinetics of montmorillonite nanoclay/maleic anhydride-modified polypropylene nanocomposites. J Therm Anal Calorim. 2012;109:17–25.
Li Y, Chenxia K, Huang C, Cheng Y. Effect of MnC2O4 nanoparticles on the thermal decomposition of TEGDN/NC propellant. J Therm Anal Calorim. 2012;109:171–6.
Lee HJ, Oh SJ, Choi JY, Kim JW, Han J, Tan LS, et al. In situ synthesis of poly(ethylene terephthalate) (PET) in ethylene glycol containing terephthalic acid and functionalized multiwalled carbon nanotubes (MWNTs) as an approach to MWNT/PET nanocomposites. Chem Mater. 2005;17:5057–64.
Guan G, Li C, Zhang D. Spinning and properties of poly(ethylene terephthalate)/organomontmorillonite nanocomposite fibers. J Appl Polym Sci. 2005;95:1443–7.
Broza G, Schulte K. Melt processing and filler/matrix interphase in carbon nanotube reinforced poly(ether–ester) thermoplastic elastomer. Polym Eng Sci. 2008;48:2033–8.
Hernández JJ, García-Gutiérrez MC, Nogales A, Rueda DR, Kwiatkowska M, Szymczyk A, Roslaniec Z, Concheso A, Guinea I, Ezquerra TA. Influence of preparation procedure on the conductivity and transparency of SWCNT–polymer nanocomposites. Compos Sci Technol. 2009;69:1867–72.
Paszkiewicz S, Szymczyk A, Spitalsky Z, Mosnacek J, Rosłaniec Z. Electrical conductivity of poly(ethylene terephthalate)/expanded graphite nanocomposites prepared by in situ polymerization. J Polym Sci B. 2012;50:1645–52.
Nogales A, Broza G, Roslaniec Z, Schulte K, Sics I, Hsiao BS, Sanz A, Garcia Gutierrez MC, Rueda DR, Domingo C, Ezquerra TA. Low percolation threshold in nanocomposites based on oxidized single wall carbon nanotubes and poly(butylene terephthalate). Macromolecules. 2004;37:7669–72.
Kota AK, Cipriano BH, Powell D, Raghavan SR, Bruck HA. Quantitative characterization of the formation of an interpenetrating phase composite in polystyrene from the percolation of multiwalled carbon nanotubes. Nanotechnology. 2007;18(50):505705–11.
Ke Y, Long C, Qi Z. Crystallization, properties, and crystal and nanoscale morphology of PET–clay nanocomposites. J Appl Polym Sci. 1999;71:1139–46.
Yuan X, Chuncheng L, Guan G, Xiao Y, Zhang D. Thermal degradation investigation of poly(ethylene terephthalate)/fibrous silicate nanocomposites. Polym Degrad Stab. 2008;93:466–75.
Vassiliou AA, Chrissafis ĆK, Bikiaris ĆDN. Thermal degradation kinetics of in situ prepared PET nanocomposites with acid-treated multi-walled carbon nanotubes. J Therm Anal Calorim. 2010;100:1063–71.
Anand KA, Agarwal US, Joseph R. Carbon nanotubes-reinforced PET nanocomposite by melt-compounding. J Appl Polym Sci. 2007;104:3090–5.
Samperi F, Puglisi C, Alicata R, Montaudo G. Thermal degradation of poly(ethylene terephthalate) at the processing temperature. Polym Degrad Stab. 2004;83:11–7.
Ozawa TT. A new method of analyzing thermogravimetric data. Bull Chem Soc Jpn. 1965;38:1881–6.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J Polym Sci C. 1964;6:183–95.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;21:1702–6.
Wang H, Yang J, Long S, Wang X, Yang Z, Li G. Studies on thermal degradation of poly(phenylene sulphide sulfone). Polym Degrad Stab. 2004;83(2):229–35.
Regnier N, Guibe C. Methodology for multistage degradation of polyimide polymer. Polym Degrad Stab. 1997;77:165–79.
Day M, Cooney JD, Wiles DM. The thermal stability of poly(aryl–ether–ether–ketone) as assessed by thermogravimetry. J Appl Polym Sci. 1989;38:323–37.
Criado JM, Malek J, Ortega A. Applicability of the master plots in kinetic analysis of non-isothermal data. Thermochim Acta. 1989;147:377–85.
Malek J. The kinetic analysis of non-isothermal data. Thermochim Acta. 1992;200:257–69.
Tonbul Y, Yurdakoc K. Thermogravimetric investigation of the dehydration kinetics of KSF, K10 and Turkish bentonite. Turk J Chem. 2001;25(3):333–9.
Coats AW, Redfern JP. Kinetic parameters from thermogravimetric data. Nature. 1964;201:68–9.
Vyazovkin S, Burnham AK, Criado JM, Perez-Maquedac LA, Popescu C, Sbirrazzuoli N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta. 2011;520:1–19.
Sbirrazzuoli N, Girault Y, Elegant L. The Malek method in the kinetic study of polymerization by differential scanning calorimetry. Thermochim Acta. 1995;249:179–86.
Senum GI, Yang RT. Rational approximations of the integral of the Arrhenius function. J Therm Anal Calorim. 1977;11:445–7.
Achilias DS, Bikiaris DN, Karavelidis V, Karayannidis GP. Effect of silica nanoparticles on solid state polymerization of poly(ethylene terephthalate). Eur Polym J. 2008;44(10):3096–107.
Bikiaris D, Karavelidis V, Karayannidis G. A new approach to prepare poly(ethylene terephthalate)/silica nanocomposites with increased molecular weight and fully adjustable branching or crosslinking by SSP. Macromol Rapid Commun. 2006;27:1199–205.
Giraldo LF, Brostow W, Devaux E, Lopez BL, Perez LD. Scratch and wear resistance of Polyamide 6 reinforced with multiwall carbon nanotubes. J Nanosci Nanotechnol. 2008;8(5):1–8.
Acknowledgments
The authors thank the financial support from National Science Centre Poland for sponsored the study (Project No NCN N N507 218340). Sandra Paszkiewicz also thanks for financial support from West Pomeranian University of Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Pilawka, R., Paszkiewicz, S. & Rosłaniec, Z. Thermal degradation kinetics of PET/SWCNTs nanocomposites prepared by the in situ polymerization. J Therm Anal Calorim 115, 451–460 (2014). https://doi.org/10.1007/s10973-013-3239-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-013-3239-4