
Abstract
A need for method validation in radiochemical analyses of decommissioning waste is a challenging task due to lack of commercial reference materials. Participation in an intercomparison exercise is one way for a laboratory to assess their performance and validate their analysis results. A three-year project within the Nordic Nuclear Safety Research (NKS) community was initiated in order to carry out intercomparison exercises on difficult to measure (DTM) radionuclides in real decommissioning waste. Both Nordic and Non-Nordic laboratories participated. This paper reports the results from the final year of the project focusing on beta- and gamma emitter (i.e., easy to measure, ETM) analysis in spent ion exchange resin. The assigned values were derived from the participants’ results according to ISO 13528 standard and the performances were assessed using z scores. The results showed generally good performances for both DTMs and ETMs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Three years of intercomparison exercises within the Nordic Nuclear Safety Research (NKS) community on radiochemical analysis of difficult to measure (DTM) radionuclides in decommissioning waste finished in 2021 [1,2,3]. The need for the DTM intercomparison exercises was initiated by lack of DTM reference materials, with which laboratories could validate their radioanalytical methods and to gain valuable data for analytical service portfolio and accreditation purposes. The general yearly schedule was first implemented that the studied samples were homogenous. The homogeneity studies by the coordinator were carried out with gamma spectrometric analysis of an easy to measure (ETM) radionuclide e.g., 60Co, 137Cs or 152Eu. Secondly, the samples were distributed to the partners, who had several months to carry out the analyses. The analysis results corrected to a reference date were collected by the coordinator and discussed in a preliminary meeting, after which each partner had about a month to re-check their results and carry out new analyses if necessary or feasible. In cases when a reported result deviated significantly from the general trend, the participant was individually contacted by the coordinator and the participant was requested to re-evaluate the results and to find a possible calculation error. The project reports record the corrections [1,2,3]. At the end of each year, the results were statistically analysed according to the ISO 13528 standard [4] and discussed in a final meeting. In addition to the yearly reports in NKS report series [1,2,3], the results were further discussed and compared with activation calculation results in peer-reviewed articles [5, 6].
During the first year of the project, the intercomparison exercise was carried out on DTM analyses in activated steel [1, 5]. The participating laboratories were three from Finland, one from Sweden, one from Denmark, two from Norway and one from France [1]. The studied activated steel was a reactor pressure vessel (RPV) steel bar, which had been irradiated with neutrons in a nuclear power reactor in 1980s [5]. The RPV steel bar was cut to thin slices using electric discharge machine and each partner received 2–3 approximately 0.2 g samples [1, 5]. 14C, 55Fe and 63Ni were the main DTMs of interest, while also the gamma-emitter 60Co was analysed [1, 5]. Even though no major difficulties were reported in the 55Fe analysis, it was concluded that as 55Fe decays with electron capture and releases Auger electrons and low energy X-rays, it is especially sensitive to quenching in liquid scintillation counting (LSC) [5]. Therefore, careful preparation of liquid scintillation sample was needed as both sample solution and cocktail affect quenching [5]. 60Co spectral interference was concluded to be one of the major challenges in 63Ni analysis in steel samples, because Co and Ni have similar chemical behaviour and some of 60Co can be carried over into the 63Ni fraction causing increased signal in LSC spectrum [5]. Reliable 14C measurement was concluded to require further work as quantitative release of 14C as CO2 from the solid matrix was required and efficient capture of the released 14C into an absorption solution [5]. 60Co analysis was concluded to be relatively easy, even though careful analysis of the data was required [5].
The second year of the project focused on DTM analyses in activated concrete [2, 6]. The participating laboratories were the same as in the first year [1, 2]. The studied activated concrete originated from FiR1 research reactor biological shield [2, 6]. Each partner received approximately 20 g finely powdered sample [2, 6], which had been extracted from the activated concrete core using a dedicated drilling equipment [7]. 3H, 14C, 55Fe and 63Ni were the main DTMs of interest while 36Cl, 41Ca, 152Eu and 60Co were optionally analysed [2, 6]. Major difficulties were reported due to low activity and solubility of the studied material [2, 6]. Additionally, some laboratories did not carry out chemical composition analyses causing significant variations in 55Fe analysis, because the material contained large amounts of stable iron, which interfered both ion exchange separations and yield calculations [2, 6]. 63Ni, 14C and 36Cl results suffered mainly from low activities or low number of data entries whereas no major difficulties were reported for 3H [2, 6]. 41Ca analysis was reported to suffer from low activity, spectral interference and quenching [2, 6]. No major difficulties were reported in the ETM analysis [2, 6].
In the third and final year of the project, the intercomparison exercise was carried out on DTM and gamma-emitter analyses in a spent ion exchange resin [3]. The participating laboratories were three from Finland, one from Denmark, three from Norway, one from France and one from Taiwan [3]. Contrary to the first two years, the analysis of gamma-emitters was also a focus area. A wide range of gamma-emitters was expected to be present in the spent ion exchange resin, as an initial screening of the material revealed the presence of 60Co, 137Cs, and 54Mn. These three radionuclides indicated that the sample was fresh (presence of relatively short-lived corrosion product 54Mn), contained fission products (137Cs originating from a leaking spent fuel element), and corrosion products (60Co originating from corrosion of steel piping etc.). The main DTMs were selected to be corrosion products 55Fe and 63Ni and fission product 90Sr [3]. Additionally, some partners analysed 3H, 14C, and 99Tc [3]. This paper presents the final results obtained in the third year of the intercomparison exercises, further analyses, and discusses the results.
Sample history, homogeneity and stability
The studied spent ion exchange resin (FINEX C/A 850 H mixed exchanger in powder form, nuclear grade) originated from a nuclear power plant primary circuit. The resin material was well mixed, and approximately 0.8 g was weighed into nine glass liquid scintillation vials. Surface dose rate of each sample was approximately 30 μSv h−1. The guidelines for the initial review of the studied material was to establish sample homogeneity and stability [4]. Therefore, the homogeneity measurements were carried out the same way as in the first two years of the project [1, 2]. The homogeneity measurand was 137Cs activity concentration. The gamma measurements were carried out in 8096 channels using a p-type HPGe semiconductor detector with 18% relative efficiency and 1.8 keV FWHM at 1.33 MeV (ISOCS-characterized detector supplied by Mirion Technologies, connected to a DSA-LX multi-channel analyser and Genie 2000 software). The Geometry Composer v.4.4 software was utilised for efficiency calibrations. Each sample was carefully positioned at 20 cm distance on top the endcap in order to obtain a constant measurement geometry, low dead time, and low true coincidence summing (TCS) effects. The measurement time was 900 s. The 137Cs activity concentration of every sample was measured twice. Given that the relative standard deviation (RSD) of the measured specific activities was 1.1%, the samples were considered to be initially homogenous. In the end of the project, the homogeneity was verified using Eq. (1) as presented in the ISO 13528 standard [4].
where ss = between-sample standard deviation σpt = robust standard deviation of participant results.
The ss of the Eq. (1) was calculated from sample averages, between-test-portion ranges, general average, standard deviation of sample averages, within-sample deviation and between-sample standard deviation (equations presented in Annex B of the ISO 13528 standard) [4]. At the end of the project, when the σpt was calculated from the submitted results, Eq. (1) was verifed to hold true and therefore the samples were homogenous also according to the ISO 13528 standard.
As the stability of the sample is the other important parameter in the intercomparison exercise [4], stabilities of the ion exchange resin samples were linked with mass evolution, as evaporation could occur in moist resin samples. Therefore, each partner was committed to record the mass evolution of their sample and the storage conditions e.g. refrigerator, desiccator etc. The results showed that the mass of the sample did not alter significantly and therefore the samples did not suffer from instability [3].
Origin of the DTMs and ETMs in spent ion exchange resin
Radionuclides in the primary circuit of a nuclear reactor are divided into fission products, coolant activation products, and corrosion products [8]. Cleaning loops with ion exchange resins continuously remove radionuclides and other unwanted elements from a nuclear power plant primary circuit coolant. The main beta- and gamma-emitters of interest in this study are presented in Tables 1 (for DTMs of interest) and 2 (for ETMs of interest), respectively. The fission products originate from the fission reactions in the fuel. When a fuel leak occurs, actinides as well as fission products, such as 137Cs and 90Sr, are released into the coolant. Coolant activation products are radionuclides originating from water activation, activation of impurities contained in water and chemicals injected into the primary circuit [8]. The primary production mechanism (fuel excluded) of coolant activation product 14C is activation of 17O, rather than 14N production mechanism since the coolant is typically degassed and has a low dissolved 14N content [8]. 3H is produced from activation of 10B and 6Li in the coolant to control reactivity and pH, respectively [8]. 3H is also produced in activation and release from secondary start-up sources, namely Sb–Be source to produce neutrons for reactor start-up [8]. The activated corrosion products originate from corrosion of circuit surfaces releasing iron, nickel, cobalt and chromium into the coolant, which transfers the non-radioactive metallic elements as dissolved, colloidal or particulate species [8]. The metallic species may deposit on fuel rod surfaces and activate by absorbing a fast or thermal neutron [8]. The activated corrosion products can be released back into the coolant and distribute into the circuit [8].
Methodology for statistical analysis
Statistical analyses of the submitted results were performed using the ISO 13528 standard on proficiency testing by interlaboratory comparison [4]. As the studied spent ion exchange resin was not a reference material, the assigned values were calculated from the reported results of the participants. Robust means and robust standard deviations were calculated using Algorithm A, which is robust for outliers up to 20% [4]. The iterations of the robust mean and standard deviations should be carried out until there is no change in the third significant figure [4]. In this study, the iterations were continued until there was no change in 1 Bq g−1 digit for results below 1 kBq g−1 whereas for results above 1 kBq g−1, the iterations were continued until there was no change in 0.1 kBq g−1 digit.
Performance assessment was carried out using z score of Eq. (2), which is a recommended method in cases where the assigned value is calculated from the submitted results [4]. The submitted results (noted xi) were assessed against the assigned values derived from the participants’ results [4]. The z score results were acceptable when |z|≤ 2.0, a warning signal was given for results with 2.0 <|z|< 3.0, and |z|≥ 3.0 results were unacceptable [4].
where xi = the value given by a participant i xpt = the assigned value σpt = standard deviation for the proficiency assessment.
Overview of the DTM analyses
The radiochemical methods utilised in the DTM analysis of the spent ion exchange resin have been summarised by Leskinen et al. [3]. The procedures were mainly based on published references [10, 13–30]. The main focus was given for 90Sr, 55Fe and 63Ni whereas some analysed also 3H, 14C, and 99Tc. In general, the applied methods were divided between the volatile (i.e. 3H and 14C) and non-volatile (i.e. 90Sr, 55Fe and 63Ni) DTMs. Even though 99Tc is easily volatilised, in this report it is presented with the non-volatile DTMs. More specific information on the radiochemical analyses is presented in the project report [3] whereas a general overview is given in this section.
All partners carried out destruction of the solid matrix using acid digestion [3]. One partner reported a small solid residue after acid digestion using acid mixture of conc. HNO3, HCl and H2O2. The residue was filtered and measured using gamma spectrometry. The results showed that less than 1% of the total gamma activity had remained in the residue. Another partner reported that 99Tc analysis included acid leaching using 8M HNO3. Radioactive tracers (i.e., 63Ni, 99mTc, 85Sr) or stable carriers (i.e., Sr, Fe, Ni, Re as Tc surrogate) were added by all the partners whereas hold-back carriers (e.g. Cr, Mn, Cs, Sb, Nb, Zr, Mo, Cd, Eu, Y, Co) were added by some partners [3].
The radiochemical methods for separation of 55Fe, 63Ni, 90Sr and 99Tc included chloride, hydroxide and carbonate precipitations, ion exchange resin treatments and chromatographic resin treatments [3]. 90Sr was measured with LSC either directly after 90Sr fraction purification or with Cerenkov counting after ingrowth of 90Y. Additionally, one partner analysed 90Sr using mass spectrometry and another using proportional counter [4]. Yields were mainly determined by elemental analysis of stable Sr, but also with gamma spectrometric measurement of radioactive tracer 85Sr and EDTA titration [3]. 55Fe and 63Ni were measured using LSC and yields were determined by analysing stable Fe and Ni [3]. However, in one case Fe yield was estimated to be 90% and Ni yield was determined using standard addition [3]. 99Tc was measured using LSC and yields by determination of stable Re or 99mTc using gamma spectrometry [3].
The volatile 3H was analysed using thermal oxidation or alkaline distillation [3]. In thermal oxidation systems, the volatile DTMs are released from the solid matrix with increasing temperature and then trapped in trapping solutions. The volatile 14C was analysed using thermal oxidation or wet oxidation acid stripping [3]. The 3H and 14C were analysed using LSC and yields were determined either with spiked resin or analysis of a representative sample [3].
Overview of the ETM analyses
The geometries of the solid sample measurements were glass or plastic vials [3]. All the laboratories carried out gamma-emitter analysis in solid form, either in the original glass liquid scintillation vials provided by the organising laboratory or after transfer to other vials. Some laboratories carried out gamma analysis of dissolved samples, but the results suffered from poor statistics due to small sample sizes. The samples were placed on top of high purity Germanium (HPGe) detector endcaps at specified distances ranging from 0 to 20 cm. Dead times were in all cases below 4.3%. Acquisition times varied from a few hours to a few days.
True-coincidence-summing corrections were carried out by most, but not all participants, either on grounds of large source-detector distance, or of direct comparisons against the same radionuclides used during the efficiency calibrations (i.e., in a similar geometric setup).
Photo-peak efficiency calibrations were conducted in a number of ways: by using calibration solutions at a standard geometry; based on mathematical modelling (such as ISOCS/LabSOCS); and using multiple radionuclide calibration sources, followed by efficiency transfer corrections carried out with Monte Carlo codes. One of the participants compared activity estimates obtained through ISOCS models with those obtained from a calibration based on point-wise calibration sources, followed by Monte Carlo corrections. Results obtained by either approach were found to be in good agreement, as discussed in the next subsection.
Comparison of empirical and mathematical efficiency modelling approaches
For ETMs, several parameters play a crucial role in the quality of activity estimates. Absolute photo-peak detection efficiency is one of such parameters. In intercomparison exercises, nuclide activity concentrations are mainly considered, and thus valuable, intermediate information (such as efficiency) is not quite complete for direct comparison, given that laboratories carry out their measurements using different equipment, calibration, analysis protocols, data and tools. In view of this, it is highly valuable that participants strive to scrutinise and quantify the effect of different, widely accepted modelling practices on certain parameters and intermediate results.
One of the participants carried out efficiency calibrations using mathematical modelling (with ISOCS) as well as by resorting to a “traditional” calibration protocol, where point-like, certified calibration sources were measured, and subsequent Monte Carlo calculations were performed to cater for efficiency changes arising from geometrical differences and photon attenuation between the calibration and actual sample measurement setup. The latter procedure embodies the so-called efficiency transfer method. The aim of the efficiency comparison is not to judge on the quality of the ISOCS modelling approach, but rather to gain a better understanding about what are the main corrections to be applied to the standard calibration with a view to improving its quality. Also, this comparison provides an important verification of the efficiencies used in further analyses, since essentially independent methods are involved.
Absolute efficiency calibrations were carried out for a source-detector distance of 20 cm. Single-nuclide calibration sources containing 133Ba, 137Cs, 60Co, 241Am, 152Eu, 57Co, and 54Mn were employed. Dead times were kept below 1.4%. The resulting calibration data was valid in the approximate 70–1400 keV energy range. For the same energies of the main gamma lines considered for calibration, two calculation models were setup with the Monte Carlo code Serpent (version 2.1.31) [31]: one for the calibration source setup; and another one for the actual resin sample. Since Serpent does not model electron transport, calculations were performed in photon transport mode [32], and correction factors were computed as ratios of total photon attenuation inside the detector crystal as a function of the incoming photon energy. It is important to point out that the Monte Carlo methodology described here is not enough for TCS corrections. In this case, such corrections were not required, given the large source-detector distance.
Figure 1 presents efficiency comparisons (taking ISOCS as a reference) in the form of energy-dependent ratios for cases where Monte Carlo corrections were present or not. The combined uncertainties (at 1-sigma level) are mainly due to the calibration standards; photo-peak areas; Monte Carlo statistics; and ISOCS model uncertainties. The latter are by far the largest contributors to the combined uncertainty.

Absolute efficiency ratios between ISOCS and standard calibration sources. Relative uncertainties are reported at the 1-sigma level
The net effect of the Monte Carlo corrections may be observed in the average ratios reported in Fig. 1, which change from (0.96 ± 0.02) to (1.02 ± 0.02) depending on whether corrections are applied or not. Moreover, minimum and maximum deviation from unity decrease considerably in the corrected dataset, thus making in practice all empirical efficiencies fall within the uncertainty levels claimed by ISOCS. For this experimental setup, it may be concluded that both efficiency modelling approaches yield statistically similar results at the 1-sigma level.
DTM results and the statistical analyses
As majority of the partners carried out yield measurements using stable Fe, Ni and Sr, the original concentration of the aforementioned elements in the studied resin is an important parameter when original amount is significant compared to the amount of carrier added. Samples #1, #5, and #8 yield corrections were carried using 63Ni standard addition for 63Ni yield, 85Sr for 90Sr and 99mTc for 99Tc, respectively. In these cases, the original amount of stable element does not affect the yield corrections. However, only four out of nine partners carried out analysis of original Fe, Ni, and Sr compositions of the resin material. The results in Table 3 show that three out of four Fe results are in good agreement whereas Sample #3 Fe content is approximately half of the values reported by the other partners. No calculation error was detected and the reason for lower Fe amount remained unknown. The original Fe amount in 0.039 g of resin (Sample #3 mass) calculated with the reported Fe concentration (13 mg g−1) results in 0.5 mg of Fe whereas the Fe amount in the same amount of resin calculated with averaged Fe concentration of Samples #4, 5 and 7 (34 mg g−1) results in 1.5 mg of Fe. The amount of Fe in the radiochemical analysis of Sample #3 was planned to be 4 mg and therefore 3.5 mg of Fe was added after acid digestion. Therefore, the correct Fe amount in Sample #3 after Fe addition was 5 mg and not the intended 4 mg causing increased yield percentage. The yield results are shown in Table 4. The Fe yield percentages were generally between 60 and 100%. Sample #1 and #2 Fe yields were estimated and for Sample #6 yield determination difficulties were reported for both Fe and Ni.
The reported original Ni amounts in the resin (Table 3) were all below limit of detection except for Sample #7, for which largest amount of material had been digested. In case of Sample #5 (0.075 g of resin), the original Ni amount calculated from Sample #7 Ni results (0.25 mg g−1) is 0.02 mg. Since 3.2 mg of Ni was added, the calculated original amount of 0.02 mg of Ni did not affect the yield calculations. However, for Sample #9 100 μg of carrier amounts were reported (no Ni results submitted) and in this case yield calculations would have been affected by the original Ni amount. A general trend of 60–100% Ni yields is seen similarly to Fe results (Table 4).
All reported original Sr amounts in the resin were below limits of detection with maximum of 0.04 mg g−1. Minimum of 100 μg, but mainly few mg, of Sr carrier amounts were reported and therefore the original Sr amount did not affect the yield calculations. Similarly to Fe and Ni yield results (Table 4), Sr yields varied between 60 and 100%.
The radionuclides in the purified fractions were measured mainly using LSC. However, contrarily to the previous years, one partner utilised Inductively Coupled Plasma Mass Spectrometry (ICP-MS) in determination of 90Sr. Mass spectrometric applications together with LSC in determination of DTM radionuclides are further described in Discussion section.
The 3H and 14C yields were determined experimentally with analysis of either spiked or a similar in-house-made reference material. For 3H, 90–100% yields were reported whereas 14C yields varied 70–100%. The 3H and 14C analysis results with replicates and 2 k uncertainties are shown in Table 5. Statistical analysis of these results were not carried out as low number of data entries were reported (i.e., ≤ 3). The 3H results show that two out of three results (Sample #3 and #7) were below limit of detection. These two samples were analysed using thermal oxidations whereas the only result above limit of detection was Sample #4, which was analysed using alkaline distillation. The results indicate that alkaline distillation does not suffer from 3H contamination in the analytical equipment as in pyrolyser, in which memory effect is possible and it may increase with age. The 14C results show that the reported results vary from 1400 to 6400 Bq g−1. It can be concluded that the results produced using the pyrolysers are 3–4 times higher compared to the wet oxidation acid stripping (Sample #4). One possible reason for the deviation is 14C memory effect in the pyrolysers. However, the activities are significantly higher than an average memory effect. One possible reason for lower 14C result in Sample #4 is incomplete oxidation of 14C to CO2.
The 99Tc results in Tables 4 and 5 show that one result is below limit of detection and the other is 0.2 Bq g−1. The reported Tc yields were 60% (measured using Re surrogate) and 94% (measured using 99mTc). The volatility of Tc is especially challenging causing special attention in acid digestion acids and temperatures.
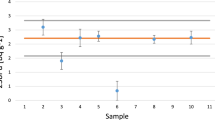
The final 55Fe, 63Ni, and 90Sr results presented by Leskinen et al. [4] were re-assessed to include all the submitted results in the calculations of assigned values. The reason for the re-assessment was that some of the results were submitted after the project deadline and therefore not included in previous calculations. The number of data entries (one per partner) with standard and robust statistics result are shown in Table 6. The results show that the median, average and assigned value of 90Sr results are the same (i.e., 170 Bq g−1) whereas a small decrease from 35,200 to 34,300 Bq g−1 in the corresponding 55Fe values are seen. Largest difference between the median, average and assigned value is seen for 63Ni results (i.e., 48,400 Bq g−1median and 43,700 Bq g−1 average). The largest robust standard deviation percentage was calculated for 63Ni (33% compared with 19% and 20%) indicating that the 63Ni results contained higher number of results which deviated from the general trend. This phenomenon can be visually seen in Figs. 2, 3, 4, in which the reported 55Fe, 63Ni, and 90Sr results and assigned values with 2 k uncertainties are presented. The reported 55Fe results are all within the 2 k uncertainty of the assigned value whereas three 63Ni results are outside. Figure 4 shows that one result for 90Sr is just above the upper uncertainty of the assigned value.

Reported 55Fe activity concentrations and assigned value with 2 k uncertainties

Reported 63Ni activity concentrations and assigned value with 2 k uncertainties

Reported 90Sr activity concentrations and assigned value with 2 k uncertainties. An outlier Sample #2 is not included
The 55Fe, 63Ni and 90Sr performance assessment results, namely z score results, are shown in Table 7. The results show that all 55Fe z score results are below 2 and therefore in acceptable range. The 63Ni results include two results, namely Sample # 1 and 2, which are in warning range (i.e., 2.0 <|z|< 3.0) and one result, namely Sample #6, is in unacceptable range (i.e., z ≥ 3). The 90Sr results show that all except the one outlier (Sample #2) are in acceptable range. Sample #2 might be an outlier because the separation yield was only estimated and not measured for the present separation.
ETM results and statistical analysis
The final ETM values presented by Leskinen et al. [4] were re-assessed to include all the submitted results in the calculations of assigned values similarly to the DTM results. Only those results which were above detection limits are statistically analysed in this paper. The number of data entries (one per partner) with standard and robust statistics are shown in Table 8. Individual results per participant (i.e., by sample number) are also presented visually along with their assigned values and associated 2 k uncertainties in Figs. 5, 6, 7, 8, 9, 10, 11. The averages and assigned values of 54Mn, 57Co, 125Sb, and 137Cs are identical. For 57Co, only four data entries above limits of detection were reported. The median, average and assigned values are close to each other with only 7% relative robust standard deviation. Activity concentrations of 54Mn, 60Co, 134Cs, and 137Cs systematically exhibit higher values in the case of Samples #1 and #2. These observations are also reflected in the performance assessment data presented in Table 9. With the exception of 54Mn, the computation of robust statistics for the aforementioned radionuclides entails higher number of iterations than for the rest of the nuclides (i.e., 13–24 compared to the others below 10). The latter observation is also true to a lesser extent in the case of 58Co, where only Sample #2 is associated with an activity value considerably above the rest. Inspection of the gamma-emitter performance assessment results (namely, z score results) from Table 9 readily indicates that all z scores of 54Mn, 57Co, 60Co, 125Sb, 134Cs, and 137Cs fall within the acceptable range, whereas one 58Co result does not (z ≥ 3 for Sample #2).

Reported 54Mn activity concentrations and assigned value with 2 k uncertainties

Reported 57Co activity concentrations and assigned value with 2 k uncertainties

Reported 58Co activity concentrations and assigned value with 2 k uncertainties

Reported 60Co activity concentrations and assigned value with 2 k uncertainties

Reported 125Sb activity concentrations and assigned value with 2 k uncertainties

Reported 134Cs activity concentrations and assigned value with 2 k uncertainties

Reported 137Cs activity concentrations and assigned value with 2 k uncertainties
Additionally, it is worth mentioning that some partners submitted preliminary identification of 7Be, 109Cd, 95Nb, 110mAg, 152Eu and 231Th in the resin sample [4]. Discussions in the preliminary meeting concluded that at least 7Be, 109Cd, 152Eu and 231Th were most likely false positives [4]. All the aforementioned radionuclide identifications were re-checked by the partners and determined to be false positives [4]. Reasons for the false identifications are exposed in the discussion section.
Discussion
The first critical step in the analysis of the non-volatile and volatile DTMs is quantitative release of the analytes of interest from the solid matrix into aqueous medium. The main non-volatile DTMs of interest in this study were 55Fe, 63Ni and 90Sr and therefore acid digestions did not require special attention of temperature nor for the acid mixtures. Another method for matrix destruction is alkali fusion, which was not used in this project whereas pre-treatment of drying and ashing were performed by some partners. However, volatilities of 3H, 14C and 99Tc need to be considered already in the first critical step, i.e., acid digestion and combustion. Actually, the volatilities of 3H and 14C are exploited in combustion. Analysis of gamma-emitters can be carried out non-destructively and therefore gamma spectrometric analyses does not necessitate pre-treatment. The following sub-sections present critical considerations in the DTM and ETM analyses including discussion on radionuclide specific considerations and analytical techniques.
Critical considerations in the 3H and 14C analyses
3H and 14C are pure beta emitters with energy maxima of 18.6 keV and 156 keV, respectively. The critical considerations of 3H and 14C analyses have been discussed in previous papers [5, 6]. Reliable 14C analysis requires quantitative release and conversion of carbon to CO2 and efficient trapping into a trapping solution. The release can be carried out either using oxidative acid digestion or thermal oxidation. The same phenomenon is also relevant in 3H analysis, in which the analysis is carried out with thermal oxidation or distillation. The yield determinations need to be carried out using spiked samples or representative materials, which are often prepared in-house as there are no commercially available reference materials. Anomalies may arise due to differences in 14C and 3H speciations between studied material and spiked samples/representative materials. For example, spiked 14C and 3H may be more easily oxidised compared to the same radionuclides present in more tightly bound in the studied material. In cases when oxidative power of the acid mixture is partly consumed by destruction of the organic matrix (i.e., graphite, organic resin etc.), it is especially important to utilise enough oxidative power in the acid digestion. The main measurement method for both 3H and 14C is LSC [33] whereas accelerator mass spectrometry (AMS) has been used widely in 14C dating applications.
In this study, a trend is seen between results produced between wet oxidation and combustion i.e., combustion resulted in 3–4 times higher 14C activity concentrations compared to wet oxidation whereas wet oxidation was the only method with which above limit of detection 3H result was obtained. However, a small number of 3H and 14C analyses were carried out and therefore confirmative conclusions are impossible.
Critical considerations in the 99Tc analysis
99Tc is a pure beta emitter, emitting soft beta rays (Emean = 84.6 keV, Emax = 290 keV) [34] and decaying into stable 99Re [35]. 99Tc is a fission product of thermal fission of 238U and 239Pu, with a relative high fission yield of 6% [35]. There are no stable isotopes of technetium and 97Tc, 98Tc and 99Tc are the most long-lived isotopes with half-life of 2.6 × 106 years, 4.2 × 106 years and 2.1 × 105 years, respectively [36]. Due to high mobility of 99Tc in water, high fission yield and long half-life [36], 99Tc is an important nuclide to assess in regard to nuclear waste.
There are several radioanalytical methods for measuring the activity of 99Tc i.e., solid phase extraction and imaging plate and beta-counting [37], co-precipitation, solid phase extraction and LSC [38], acid leaching and ashing before separation by anion resin and counting by low background beta counter and ICP-MS [39], purification and decontamination before utilising measurement by ICP-MS [40], to mention a few.
In the determination of 99Tc by LSC, the 99Tc is purified and decontaminated for interference prior to activity measurement [41]. Low level environmental samples will require larger volumes of sample, up to several hundred litres of water, and a high degree of concentration before purification [36]. With radioactive waste, somewhat depending on the activity in the material, the sample volume can be less than one gram [42].
There are several nuclides that will interfere with the beta counting of 99Tc, such as 14C, 36Cl, 45Ca, 60Co, 137Cs and uranium isotopes [41, 43], thus emphasising the need for purification of the 99Tc before measurement of its activity.
The challenges with 99Tc analyses are choice of yield monitor, the volatility of 99Tc [44] and use of eluting agent [45]. There are different options for yield tracers i.e., isotopic Tc (95mTc, 97mTc, 97Tc, 98Tc and 99mTc) and 185Re and 187Re, which are in the same group as Tc in the periodic table [36]. With the use of 99mTc as yield monitor, the 99mTc requires time to decay before counting 99Tc with LSC [33]. With 185Re and 187Re as yield monitor, care must be taken as Tc and Re have somewhat different chemical properties [36]. Re-isotopes are used as yield tracers when measuring the activity of technetium with non-radiometric methods.
It has been shown that for environmental samples, the pretreatment strategy affects the 99Tc yield [44]. When using ashing as the destruction method for the matrix, it is reported that the highest recovery of 99Tc is found when wetting the matrix with ammonia before ashing [44]. It was also found that the temperature did not influence the recovery as much as choice of wetting agent. This is in good correlation with reported measurements of 99Tc in radioactive waste i.e., the volatility of 99Tc is best combated by using an alkaline wetting agent during heating, as 99Tc has increased volatility in acidic medium [38, 42, 46, 47].
Wigley et al. [44] studied the effect of different leaching agents had on the recovery of 99Tc from environmental samples (seaweed, lobster and sediment). The most successful leaching agent was 8 M HNO3 in comparison with aqua regia, HCl and a mixture of HCl and H2O2. Wigley et al. [44] also studied the effect of leaching time on recovery. The results revealed that a leaching time of about 120 min gives the best results for recovery.
Beals [45] studied how various concentrations and volumes of HNO3 as eluting agent affected the recovery of 99Tc from aqueous samples. The results showed that by increasing 4 M HNO3 volume from 20 to 30 ml, the recovery increased approximately from 70 to 90%.
In this study, only two 99Tc analyses were carried out. Sample #3 was acid digested in 8 M HNO3 at below 100 °C and stable Re was utilised in the yield determination. After cooling to room temperature, separation of the resin residue and addition of an oxidizing agent H2O2, the acid solution was treated with ion exchange resin without evaporation to a lower volume. Oxidising agent was added in order to have Tc as TcO4−, which has high affinity on strong anion exchange resin in diluted acid, alkaline or neutral media [33]. The 8 M HNO3 was chosen in order to obtain the sample in correct medium for ion exchange resin directly after acid leaching without evaporation and consequently loss of volatile 99Tc. Sample #8 was analysed in sequence together with 55Fe, 63Ni and 90Sr. The sample #8 was acid digested in HNO3, HCl and H2O2 mixture. 99mTc was utilised in the yield determinations and measured gamma spectrometrically. Both sample #3 and #8 99Tc activities were measured using LSC. The partner analysing Sample #3 reported that they were currently building their capabilities in 99Tc analysis whereas Sample #8 was analysed by a partner with longstanding experience in 99Tc analysis in environmental samples. The yields (i.e., 60% for Sample #3 and 94% for Sample #8) may reflect the experience.
Critical considerations in the 55Fe and 63Ni analyses
As 55Fe and 63Ni have been analysed as one of the main DTMs of interest in each three years of the intercomparison exercises, their critical considerations have been discussed in previous publications [5, 6]. An overarching challenge in the radiochemical analysis of 55Fe, which decays via electron capture emitting Auger electrons and low energy X-rays, is quenching in the LSC measurements due to the easily quenched low energy signal. Additionally, the original stable Fe concentration in the studied sample plays a significant role in the radiochemical procedures especially in cases when large volumes of low activity level samples are studied, such as in the case of activated concrete in the second year of this project [2, 6]. A mixture of low solubility sample and lack of original chemical composition of the material can easily lead to inconsistent results [2, 6]. Modifications in the carrier amounts and volumes of ion exchange and chromatographic resins are also needed if the material originally contains significant amounts of the elements of interest (e.g. stable iron in activated steel) or interfering elements. However, in this study no difficulties were reported for 55Fe analysis and the statistical analysis showed well comparable results with all z scores in acceptable range.
63Ni is a relative low energy (Emax 67 keV) pure beta emitter, which purification was carried out in this study with Ni-resin treatment or dimethylglyoxime (DMG) precipitation. Both of these methods are based on precipitation of Ni with DMG. In Ni-resin, the DMG has been coated on an inert support and the separation is based on an on-column precipitation of Ni whereas in DMG precipitation, DMG reagent is placed in contact with the sample in a batch mode. In all analyses, 55Fe was removed from the 63Ni fraction prior to DMG using a precipitation, an ion exchange resin and/or a chromatographic resin. The main interference in 63Ni analysis is 60Co because Co is chemically similar to Ni and therefore it can also form complexes with DMG and consequently cause interferences in the LSC spectrum. Significant amounts of 60Co can be present especially in activated steel samples and careful purification of 63Ni from 60Co is needed [5]. Presence of 60Co in the 63Ni fraction can be easily detected using gamma spectrometry and if necessary, the DMG treatment can be repeated. Additionally, spectral corrections are possible [5]. In this study, difficulties in 60Co removal were not reported. However, the statistical analysis of the results showed that two results were in warning range and one result in unacceptable range. Interference by 60Co would mean increased signal for 63Ni. The only result above the general trend was Sample #1, where the processing methodology consisted an initial AgCl precipitation for potential Ag removal followed by two chromatographic resin purifications (i.e., TRU and Ni-resin) and yield correction using standard addition. However, it is not known if the deviation from the general trend is caused by the methodology, yield correction, or possible interference by 60Co. Sample #2 and #6 were below the general trend and more specifically #6 in unacceptable range and #2 in warning range. The partner, which analysed Sample #6, reported difficulties in yield measurements and calculations. Similar reporting was also given for 55Fe results even though deviation from the general trend is not seen in this case.
Critical considerations in the 90Sr analysis
90Sr is a strong pure beta emitter (Emax 546 keV) which disintegrates into 90Y which is also a pure beta emitter (Emax 2280 keV). 90Sr is a major fission product of 235U after 137Cs. Therefore, this radionuclide can be found in significant quantities in spent nuclear fuel or in radioactive waste generated from nuclear reactors such as in spent ion exchange resins. As a non-volatile radionuclide, 90Sr extraction requires a complete destruction of matrix which was accomplished in the present work in acid conditions by using open or closed digestion systems. The critical considerations in 90Sr analysis consist of its purification and measurement. As a pure beta emitter, 90Sr needs to be isolated from the matrix and interferences by radiochemical separation prior to its activity measurement. The challenges of the purification steps concern mainly the elimination of calcium, its chemical analog, and also of the all beta and gamma emitters and its daughter 90Y for radiometric measurements such as for LSC or gas proportional counting [48]. For mass spectrometry detection such as for ICP-MS, another challenge deal with the removal of isobaric interferences generated by 90Zr, 50Ti40Ar or 50Cr40Ar.
This year was the first time when the 90Sr was measured in the framework of the intercomparison exercises organised by the three-year NKS project for nuclear decommissioning. Most radiochemical procedures are based on the implementation of the extraction chromatographic resin dedicated to 90Sr purification denoted as Sr-resin®. This resin contains 4,4′(5′)-di-t-butylcyclohexano 18-crown-6 in 1-octanol impregnated on an inert polymeric support which makes it especially selective towards the majority of interferences [49]. In particular, yttrium, calcium and most of metals are not fixed in concentrated nitric acid whereas strontium is trapped in the cavity of the crown ether and thus strongly retained. The major interfering elements are barium, plutonium and neptunium which have a behavior close to strontium on Sr-resin®. The presence of large amounts of Ca in samples can also interfere towards the selectivity of Sr-resin® by reducing the uptake of strontium. This was not the case for the studied sample since Ca was measured at low concentration (around 0.3 mg g−1). As the present spent ion exchange resin did not contain high amounts of interfering radionuclides, samples #4 and #5 were only purified by using Sr-resin®. For samples #2, #3, #7, #8 and #9, additional purification steps based on precipitation, ion exchange chromatography or extraction chromatography were added prior to purification with Sr-resin®. Precipitations at basic pH were implemented by using sodium hydroxide or ammonia for samples #2, #8 and #9: this step enables to precipitate the majority of metals such as iron, yttrium, and plutonium, whereas strontium remains in solution. In the case of sample #8, an additional precipitation based on carbonate was added after hydroxide precipitation. For sample #3, a preliminary purification based on an anion exchange resin was implemented i.e., in acid conditions, strontium was not retained contrary to iron. Sample #7 was purified with TRU-resin® prior to separation on Sr-resin® so as to eliminate potential traces of barium, plutonium and neptunium. Partner analysing Sample #7 also applied another separation strategy to isolate 90Sr i.e., precipitation based on nitric acid followed by ammonia precipitation and oxalate precipitation was used to purify another aliquot of sample #7 [50].
After purification, the samples were all analysed to determine the separation yield and the 90Sr activity, except Sample #2 for which the yield was estimated. The separation yields were assessed from stable strontium measured by ICP-OES, ICP-MS, MP-OES, gravimetry or EDTA titration. One laboratory used 85Sr tracer which was measured by gamma spectrometry. As mentioned in the Part “DTM results and the statistical analysis”, the separation yields were satisfactory with values higher than 60%. The challenges towards the determination of 90Sr activity depend on the type of measurement techniques applied. In the present intercomparison exercise, two strategies were considered, namely the direct measurement of 90Sr and the indirect measurement of 90Sr through 90Y ingrowth. 90Sr was directly measured with LSC (samples #7 and #9) or gas flow proportional counters (sample #7) or ICP-MS (sample #9). After ingrowth (generally more than 2 weeks), 90Y was measured by using two different counting modes with LSC devices: liquid scintillation counting (sample #3) and Cherenkov counting (samples #2, #4 and #8). In the case of sample 5, after 2–3 weeks ingrowth, 90Y was isolated from 90Sr by performing ammonia and sulphate precipitations before LSC measurement. Even if the time for equilibrium adds a delay for the measurement (14 days for sample #4 and 20 days for sample #8), Cherenkov counting has a great advantage as no scintillation cocktail is needed. After separation, the samples #4 and #8 were respectively dissolved in HNO3 0.05 M and < 0.5 M and no scintillation cocktail is added. Scintillation cocktails necessary for liquid scintillation measurement of 90Sr are concerned by REACH regulation [51] and should, shortly, be banned from laboratories or at least subject to derogation. In terms of measurement, Gačnik et al. [52] have compared limits of detection on 4 different samples measured by direct 90Sr measurement with scintillation cocktail and by 90Y indirect measurement with Cherenkov and they were 2 to 3 times lower for 90Sr direct measurement. However, the sensitivity is mainly dependent on the type of counter (background count rate) and the time of acquisition [53] and can be optimised. The measurement of 90Y by Cherenkov effect is an interesting alternative to the implementation of scintillation cocktail and satisfactory results were obtained with this method in this intercomparison exercise. Partner analysing Sample #7 mentioned that the ratio between 90Sr and 90Y was measured at different time delays after 90Y ingrowth and compared to the theoretical ratio of 90Sr/90Y to check the selectivity of the separation, as presented in [54]. For example, a problem with unsuitable values of 90Sr/90Y ratios was observed preliminarly, caused by low activity level measured closed to detection limit. To solve the issue, the amount of purified samples taken was increased from 5 to 20 ml of digested solution.
It can be noticed that despite the variety of purification and measurement methods, the 90Sr results are relatively consistent (robust standard deviation percentage of 18%), which demonstrates the analytical capabilities of the laboratories involved in the project and corroborates the strategies optimised by the laboratories.
Critical considerations between the liquid scintillation counting and mass spectrometry in DTM analyses
Different mass spectrometric techniques are being increasingly used in determination of DTM radionuclides, in addition to or instead of traditional radiometric detection techniques. Mass spectrometry is applicable for the radioisotopes which have relatively long physical half-lives. In LSC and Geiger-Müller (GM) counting, it is of great importance to exclude the other beta emitting radionuclides, especially those with similar decay energies, which interfere the determination of the analyte. Furthermore, stable elements in the measurement sample, causing radiation quenching might be another great concern in LSC and GM measurements. Different challenges occur in mass spectrometry, where isobaric and isometric interferences from isotopes and molecules with similar mass, complicating the determination of DTM content in the sample, might be the most important considerations. In general, radiochemical separation method is needed for producing adequately pure measurement samples for both radiometric and mass spectrometric determinations.
The measurement technique dictates the chemical and physical form of the purified DTM fraction. In LSC measurements, the sample solution must be compatible with the liquid scintillation cocktail. If the solution is not fully soluble in the LSC cocktail, it will cause a decreased counting efficiency due to higher quench in the sample. In GM counting, the sample is prepared in a solid form. If mass spectrometry is used as a counting method, sample preparation depends on the used mass spectrometer type. For example, ICP-MS requires a different sample form than RIMS (Resonance Ionisation Mass Spectrometry).
Beyond sample preparation and purity requirements, each detection method has its advantages and disadvantages related to availability, detection limits and counting times. Low background GM counter has a slightly better detection limit than LSC and a comparable cost, but the counting time is considerably longer, thus making GM counting unsuitable for rapid determination. Measurement time in LSC is normally hours per sample, while in mass spectrometry it is minutes per sample. While LSC has one of the lowest detection limits, it has high availability and relatively low cost and counting time. Detection limit of LSC is typically on mBq scale, and it can be the same or lower, even on µBq scale, in ICP-MS [48]. This makes ICP-MS extremely suitable for analysing low level environmental and nuclear decommissioning samples, when the detection limit of LSC might be too high for these samples, and ICP-MS is quite commonly accessible technique. RIMS, TIMS (Thermal Ionisation Mass Spectrometry), and AMS have among the best detection limits, but these techniques are not commonly available, and their costs are often high.
The detection limits of all techniques vary at some extent, according to the analysed radionuclide, sample matrix and other factors (e.g. radioactive and stable impurities in the sample, and instrumental properties). Among DTM radionuclides discussed in this article, ICP-MS can be used for determining 63Ni, 90Sr, and 99Tc [55] and lately also for 36Cl and 41Ca [56]. Some reported detection limit values for these radionuclides obtained by LSC and ICP-MS, to give an idea about their level, are: 20 mBq and 25.6 Bq g−1 (the latter value without chemical purification and with interference from 63Cu) for 63Ni, 19 mBq and 3.9 mBq g−1 for 90Sr, 19 mBq and 8.5 mBq g−1 for 99Tc, 14 mBq and 99 mBq g−1 for 36Cl, 15 mBq and 0.3 Bq g−1 for 41Ca, respectively [56,57,58,59]. However, the range of detection limits for different mass spectrometer setups and sample types is much wider, see, e.g., an overview on the application of mass spectrometric methods in determination of nuclear decommissioning radionuclides by Croudace et al. [55].
AMS has further applications in detection of DTMs 3H, 14C, 36Cl, 41Ca, and 59Ni [55, 56, 60, 61]. AMS has been also tested successfully for determination of 55Fe in activated pressure vessel steel samples without preceding radiochemical separation [62]. This pilot study proved that AMS is not necessarily a slow and expensive determination method for DTM radionuclides, and it provides lower background count level compared to other mass spectrometric determination methods. However, AMS instruments are not easily available for all researchers. For more complex decommissioning sample materials, a chemical separation of analytes is needed in most cases for removing isobaric interferences and lowering detection limit, prior to determination of DTMs by mass spectrometric methods.
In addition to the used determination method, there is a wide selection of calculation methods for uncertainty and limit of detection. In our previous intercomparison campaigns we found out, that a variety of approaches for calculating combined uncertainty and detection limit were used among the participating laboratories. The uncertainty calculations have been executed by GUM (Guide to the Expression of Uncertainty in Measurement) method [63], Kragten method [64], and by summing a total uncertainty from known or estimated uncertainties for different analytical steps. Limit of detection has been calculated by Currie’s method [65], ISO 11929–1:2019 standard method [66], or by French standards NF M60-322 [67] and NF M60-317 [68] among our intercomparison participants. As we discussed previously [6], the use of different calculation methods and sometimes lack of detailed calculation information can complicate the comparison of the analytical results between the laboratories. If we take into account the use of different determination techniques with their specific features, as well as different practises for yield determination, the comparison of results and methods among the intercomparison participants gets even more difficult.
Critical considerations in the ETM analysis
At the time of the measurement campaigns of the partners, the gamma spectra of resin samples were largely dominated by photopeaks arising from 60Co, 134Cs, and 137Cs. The presence of other nuclides with substantially less activity stressed out the need for reliable gamma spectra analysis software as well as a careful characterisation of laboratory background radiation spectra. Although the use of a lead shield surrounding the detector can significantly reduce the intensity of background spectra, the presence of X-rays from lead in the energy region below 87 keV may in practice deteriorate the quality of the spectra. Such X-rays were identified as the root cause of false positives (such as 231Th and 109Cd) during initial screening of the participants' results [3]. In view of this, multi-layered shielding arrangements between the lead and the detector are highly recommended.
Even though the nuclides listed in Table 2 are not subject to interference effects, when dealing with complex spectra of unknown samples it is always advisable to scrutinise the consistency among activity estimates obtained via various gamma lines of the same nuclide. Not only should such estimates be statistically comparable, but furthermore the lack of photo-peak counts corresponding to gamma-lines with high yields may provide a hint about a false positive. During an early stage of this work, for example, one participant indicated the presence of 152Eu in the sample [3]. The root cause for this false positive was that the analysis software had associated the 122-keV peak of 57Co with 152Eu. A careful re-assessment of the results showed that other abundant gamma-energies from 152Eu were missing altogether from the spectrum, thus revealing the issue.
The quality of the photopeak efficiency calibration has a direct impact on activity estimates. Efficiency calibration can be attained by different methods. For those involving empirical measurements, involved efficiency transfer calculations must cater not only for the geometric change between calibration setup and actual sample, but also for the possible attenuation of photons inside the sample and its container. Whenever feasible, the assessment of absolute efficiencies via independent methods is highly recommended. Only in cases where the calibration standard matches the sample specification (pertaining geometry and materials) closely, the efficiency transfer step may be omitted. Even though close sample-detector arrangements bring about the advantage of lower counting times, the downside resides in that the efficiency values can be considerable sensitive with regards to small vertical displacements of the sample (due to the presence of the vial bottom), as well as relocation of the resin in powder form into a non-uniform heap. For large (i.e., more than 10 cm) sample-detector distances, this geometrical sensitivity decreases considerably. Near geometries also put more strain on the quality of models and computational tools used for true coincidence summing corrections for reliably quantifying the extent of efficiency loss in the actual measurement configuration.
Summary and conclusions
The DTM and ETM analyses in spent ion exchange resin were carried out in the final year of the NKS intercomparison exercises on real decommissioning waste. Contrary to the material studied during the second year, namely activated concrete, the spent ion exchange resin did not suffer from low solubility nor low activities. Additionally, the spent ion exchange resin contained a wide range of radionuclides originating from spent fuel, activated coolant and corrosion products.
This year was the first time 90Sr and 99Tc were analysed. In general, no major difficulties were observed for 90Sr determination. A variety of purification and measurement methods were applied but most of the 90Sr results were consistent resulting in 170 ± 30 Bq g−1 assigned value. The relatively low robust standard deviation (i.e., 19%) highlights the analytical capabilities of the laboratories involved in the project to characterise 90Sr accurately in decommissioning waste. Significant challenges are involved in determination of 99Tc as diligent care must be taken due to the volatility of 99Tc. In this study, only two partners carried out the 99Tc analysis giving consistent results even though one result was below limit of detection (i.e., < 2.5 Bq g−1) and the other ten times lower i.e., 0.2 Bq g−1.
3H, 14C, 55Fe and 63Ni analyses have been carried out in all three years of the intercomparison project. Low number (i.e. three) of data entries for 3H and 14C prevented statistical analysis of the results and also conclusive statements in the analysis results. All partners submitted 55Fe results whereas eight out of nine partners submitted 63Ni results. All 55Fe results were consistent with assigned value of 34,300 ± 6800 Bq g−1. The robust standard deviation was also relatively good in 55Fe results (i.e., 20%) whereas higher scatter was seen for 63Ni results (i.e., 33%). The z scores assessed against the 63Ni assigned value 44,900 ± 14,600 Bq g−1 showed that two results were in warning range and one in unacceptable range.
Pertaining specific activity estimates of gamma emitters, the initial screening of the participants' results was mainly targeted at assessing what nuclides were present in the samples provided. Given that gamma analysis software products are not completely fail proofed, when analysing samples with characteristics that deviate significantly from every-day specimens, it is of particular relevance to carefully assess the consistency among activity estimates of the same nuclide obtained from different gamma lines, provided that such approach is feasible. In the case of this work, this approach helped in the identification of false positive results others than those originating in the X-rays produced in the lead shielding. An adequate shielding design may help in minimising background radiation whilst keeping the low-energy region of the spectrum free from undesirable disturbances. The samples studied in this work did not require acid digestion, but in any case good agreement was found among all participants irrespective of whether the sample had been pre-treated or not. There was only one instance of a sample being rejected on grounds of an unacceptably high z score, namely Sample #2 in 58Co analysis. Unless statistics pose severe constraints on the maximum counting time available, it is advisable not to resort to very close sample-detector arrangements. This is not recommended because such setups may pose more strain on the quality of TCS corrections, but also because the absolute detection efficiency becomes more sensitive to geometrical parameters such as vial dimensions and the shape of the actual sample compared against the one used as a calibration reference.
The third year of intercomparison exercise on DTM analysis in spent ion exchange resin can be concluded to be the most versatile matrix studied within the three years because it included significant amounts of both DTMs and ETMs. Additionally, excluding a couple of clear blunders in the analyses, the general trend in statistical analysis results showed good performances by the partners. Even though the three-year project of DTM analyses in decommissioning waste has ended, the intercomparison exercises were continued in 2022 focusing on alpha emitters in the sample spent resin sample as studied in 2021 and reported in this paper.
References
Leskinen A, Tanhua-Tyrkkö M, Kekki T, Salminen-Paatero S, Zhang W, Hou X,Stenberg Bruzell F, Suutari T, Kangas S, Rautio S, Wendel C, Bourgeaux-Goget M, Stordal S, Isdahl I, Fichet P, Gautier C, Brennetot R, Lambrot G, Laporte E (2020) Intercomparison exercise in analysis of DTM in decommissioning waste. NKS-429, Roskilde, Denmark
Leskinen A, Tanhua-Tyrkkö M, Salminen-Paatero S, Laurila J, Kurhela K, Hou X, Stenberg Bruzell F, Suutari T, Kangas S, Rautio S, Wendel C, Bourgeaux-Goget M, Moussa J, Stordal S, Isdahl I, Gautier C, Laporte E, Guiliani M, Bubendorff J, Fichet P (2021) DTM-Decom II—Intercomparison exercise in analysis of DTM in decommissioning waste. NKS-441, Roskilde, Denmark
Leskinen A, Dorval E, Salminen-Paatero S, Hou X, Jerome S, Jensen KA, Skipperud L, Vasara L, Rautio S, Bourgeaux-Goget M, Moussa J, Stordal S, Isdahl I, Gautier C, Baudat E, Lambrot G, Giuliani M, Colin C, Laporte E, Bubenforff J, Brennetot R, Wu S-S, Ku H, Wei WC, Li YC, Luo QT (2022) DTM-Decom III—Intercomparison exercise in analysis of DTM beta and gamma emitters in spent ion exchange resin. NKS-457, Roskilde, Denmark
International Standard ISO 13528:2015(E), Statistical methods for use in proficiency testing by interlaboratory comparison. ISO, Geneva, Switzerland
Leskinen A, Salminen-Paatero S, Gautier C, Räty A, Tanhua-Tyrkkö M, Fichet P, Kekki T, Zhang W, Bubendorff J, Laporte E, Lambrot G, Brennetot R (2020) Intercomparison exercise on difficult to measure radionuclides in activated steel: statistical analysis of radioanalytical results and activation calculations. J Radioanal Nucl Chem 324:1303–1316
Leskinen A, Gautier C, Räty A, Kekki T, Laporte E, Giuliani M, Bubendorff J, Lautila J, Kurhela K, Fichet P, Salminen-Paatero S (2021) Intercomparison exercise on difficult to measure radionuclides in activated concrete—statistical analysis and comparison with activation calculations. J Radioanal Nucl Chem 329:945–958
Leskinen A, Hokkinen J, Kärkelä T, Kekki T (2022) Release of 3H and 14C during sampling and speciation in activated concrete. J Radioanal Nucl Chem 331:859–865
Committee on Radiation Protection and Public Health (2014) Radiation protection aspects of primary water chemistry and source-term management. NEA/CRPPH/R(2014)2
International Atomic Energy Agency, Nuclear Data Section. Live Chart of Nuclides. URL https://www-nds.iaea.org/relnsd/vcharthtml/VChartHTML.html. Data last accessed on 15.05.2022
Hou X, Østergaard LF, Nielsen SP (2005) Determination of 63Ni and 55Fe in nuclear waste samples using radiochemical separation and liquid scintillation counting. Anal Chim Acta 535(1–2):297–307
Baard J, Zijp W, Nolthenius H (1989) Nuclear data guide for reactor neutron metrology. Kluwer Academic Publishers, United States
Arribére M, Ribeiro Guevara S, Suárez P, Kestelman A (2001) Threshold reaction cross sections of nickel isotopes, averaged over a 235U fission neutron spectrum. Nucl Sci Eng 139(1):24–32
l’Annunziata M (2012) Handbook of radioactivity analysis. ISBN 987-0-12-384873-4
Baudat E, Gautier C, Fichet P, Destrel E, Colin C, Giuliani M, Lambrot G (2021) Optimization of 90Sr precipitation in nitric acid using design of experiments for radioactive waste characterization method. J Radioanal Nucl 328:637–650
Gautier C, Laporte E, Lambrot G, Giuliani M, Colin C, Bubendorff J, Crozet M, Mougel C (2020) Accurate measurement of 55Fe in radioactive waste. J Radioanal Nucl Chem 326:591–601
Gautier C, Colin C, Garcia C (2015) A comparative study using liquid scintillation counting to determine 63Ni in low and intermediate level radioactive waste. J Radioanal Nucl Chem 308:261–270
Hazan I, Korkisch J (1965) Anion-exchange separation of iron, cobalt and nickel. Anal Chim Acta 32:46–51
Hou XL, Østergaard LF, Nielsen SP (2005) Determination of 63Ni and 55Fe in nuclear waste and environmental samples. Anal Chim Acta 535:297–307
Hou X (2007) Radiochemical analysis of radionuclides difficult to measure for waste characterization in decommissioning of nuclear facilities. J Radioanal Nucl Chem 273:43–48
Hou X, Togneri L, Olsson M, Englund S, Gottfridsson O, Forsström M, Hironen H (2016) Standardization of radioanalytical methods for determination of 63Ni and 55Fe in waste and environmental samples. NKS-356 report ISBN 978-87-7893-440-6
Lee C, Suh M, Jee K, Kim W (2007) Sequential separation of 99Tc, 94Nb, 55Fe, 90Sr and 59,63Ni from radioactive wastes. J Radioanal Nucl Chem 272(1):187–194
Lee C, Lee M, Han S, Ha Y-K, Song K (2011) Systematic radiochemical separation for the determination of 99Tc, 90Sr, 94Nb, 55Fe and 59,63Ni in low and intermediate radioactive waste samples. J Radioanal Nucl Chem 288:319–325
Lee C, Ahn H, Lee J, Ha Y-K (2016) Rapid separation of 99Tc, 90Sr, 55Fe, 94Nb and 59,63Ni in radioactive waste samples. J Radioanal Nucl Chem 308:809–816
Leskinen A, Salminen-Paatero S, Räty A, Tanhua-Tyrkkö M, Iso-Markku T, Puukko E (2020) Determination of 14C, 55Fe, 63Ni and gamma emitters in activated RPV steel samples—a comparison between calculations and experimental analysis. J Radioanal Nucl Chem 323:399–413
Passo C, Cook G (1994) Handbook of environmental liquid scintillation spectrometry—a compilation of theory and methods. PMC0387 12/94. Packard, United States
Triskem International method, (2001) Iron-55 in water, Analytical procedure, method number FEW01, Revision 1.2
Triskem International method (2003) Nickel 63/59 in water, Analytical procedure, method number NIW01, Revision 1.0
Triskem International method (2003) Strontium 89/90 in water, Analytical procedure, method number SRW01, Revision 1.4
Triskem International method (2014) Nickel 63/59 in water, Analytical procedure, method number NIW01, Revision 1.3
Shi K, Hou XL, Roos P, Wu WS (2012) Stability of technetium and decontamination of Ru and Mo in determination of 99Tc in environmental solid samples by ICP-MS. Anal Chem 84:2009–2016
Leppänen J, Pusa M, Viitanen T, Valtavirta V, Kaltiaisenaho T (2015) The Serpent Monte Carlo code: status, development and applications in 2013. Ann Nucl Energy 82:142–150
Kaltiaisenaho T (2020) Photon transport physics in Serpent 2 Monte Carlo code. Comput Phys Commun 252:107143
Hou X (2018) Liquid scintillation counting for determination of radionuclides in environmental and nuclear applications. J Radioanal Nucl Chem 318:1597–1628
Bihari Á, Szücs Z, Mogyorósi M, Pintér T (2010) Combined determination of 99Tc and 108mAg in L/ILW liquid waste. J Radioanal Nucl Chem 286:759–763
Harvey BR, Ibbett RD, Williams KJ, Lovett MB (1991) The determination of technetium-99 in environmental materials. MAFF-AEMR-8/1991. Lowestoft, United Kingdom
Shi K, Hou X, Roos P, Wu W (2012) Determination of techentium-99 in environmental samples: a review. Anal Chim Acta 709:1–20
Kameo Y, Katayama A, Hoshi A, Haraga T, Nakashima M (2010) Simple determination of 99Tc in radioactive waste usin Tc extraction disk and imaging plats. Appl Radiat Isot 68(1):139–143
Banavali AD, Raimondi JM, Moreno EM, McCurdy DE (1995) The determination of technetium-99 in low-level radioactive waste. Radioact Radiochem 6(3):26–35
Bartosova A, Rajec P, Klimekova A (2006) Determination of technetium-99 in soils and radioactive wastes using ICP-MS. Chem Pap 6(2):125–131
Suh MY, Lee CH, Han SH, Kim JS, Park YJ, Kim WH (2003) Separation and determination of technetium-99 in spent PWR nuclear fuels. Bull Korean Chem Soc 24(11):1686–1688
Barrera J, Tarancón A, Bagán H, Garcia JF (2016) A new plastic scintillation resin for single-step separation, concentration and measurement of technetium-99. Anal Chim Acta 936:259–266
Hepiegne P, Dall`ava D, Clement R, Degros JP (1995) The separation of 99Tc from low and medium-lever radioactive wastes and its determination bu inductively coupled plasma spectrometry. Talanta 42(6):803–809
Beckman Coulter (2002) Isotope booklet for liquid scintillation counters logarithmic output
Wigley F, Warwick PE, Croudace IW, Canorn J, Sanchez AL (1998) Optimised method for the routine determination of Technetium-99 in environmental samples by liquid scintillation counting. Anal Chim Acta 380:73–82
Beals DM (1995) Determination of technetium-99 in aqueous samples by isotope dilution inductively coupled plasma-mass spectrometry. Westingjouse Savannah River Company, Aiken, United States of America
Chiu JH, Chu TC, Weng PS (1991) Radiochemical determination of technetium-99 in LLW by chelation with sodium diethyl dithiocarbamate (NaDDC) and extraction with chloroform. J Radioanal Nucl Chem 150(2):493–507
Wang L, Tang T, Yang T, Yang Y, Yang L (2013) Determination of technetium-99 from complex matrix. J Radioanal Nucl Chem 296(2):739–742
Hou X, Roos P (2008) Critical comparison of radiometric and mass spectrometric methods for the determination of radionuclides in environmental, biological and nuclear waste samples. Anal Chim Acta 608:105–139
Horwitz EP (1993) New chromatographic materials for determinations of actinides, strontium, and technetium in environmental, bioassay, and nuclear waste samples. ZZ931, Argonne National Laboratory, United States of America
Baudat E, Gautier C, Fichet P, Destrel E, Colin C, Giuliani M, Lambrot G (2021) Optimization of Sr-90 precipitation in nitric acid using design of experiments for radioactive waste characterization method. J Radioanal Nucl 328:637–650
ECHA -REACH. Substance Infocard - 4-Nonylphenol, branched and linear, ethoxylated https://echa.europa.eu/fr/substance-information/-/substanceinfo/100.239.148 Data last accessed on 6.7.2022
Gacnik J, Sarap NB, Mazej D, Prosen H, Strok M (2019) Liquid scintillation counter calibration approach for 90Sr detection and testing performance of TK100 resin. Appl Radiat Isot 151:111–115. https://doi.org/10.1016/j.apradiso.2019.04.039
O’Hara MJ, Burge SR, Grate JW (2009) Automated radioanalytical system for the determination of 90Sr in environmental water samples by 90Y Cherenkov radiation counting. Anal Chem 81(3):1228–1237
Marchesani G, Trotta G, De Felice P, Bortone N, Damiano R, Nicolini M, Accettulli R, Chiaravalle AE, Iammarino M (2022) Fast and sensitive radiochemical method for Sr-90 determination in food and feed by chromatographic extraction and liquid scintillation counting. Food Anal Methods 15:1521–1534
Croudace IW, Russell BC, Warwick PW (2017) Plasma source mass spectrometry for radioactive waste characterisation in support of nuclear decommissioning: a review. J Anal At Spectrom 32:494–526
Russell B, Goddard SL, Mohamud H, Pearson O, Zhang Y, Thompkins H, Brow RJC (2021) Applications of hydrogen as a collision and reaction cell gas for enhanced measurement capability applied to low level stable and radioactive isotope detection using ICP-MS/MS. J Anal At Spectrom 36:2704–2714
Takagai Y, Furukawa M, Kameo Y, Suzuki K (2014) Sequential inductively coupled plasma quadrupole mass-spectrometric quantification of radioactive strontium-90 incorporating cascade separation steps for radioactive contamination rapid survey. Anal Methods 6:355–362
Su TY, Tsai TL, Wu HC, Men LC (2015) Determination of ultratrace-levels of 99Tc in low-level radioactive waste samples using ICP-QMS. J Radioanal Nucl Chem 303:1245–1248
Russell B, Mohamud H, Miranda MG, Ivanov P, Thompkins H, Scott J, Keen P, Goddard S (2021) Investigating the potential of tandem inductively coupled plasma mass spectrometry (ICP-MS/MS) for 41Ca determination in concrete. J Anal At Spectrom 36:845–855
Hampe D, Gleisberg B, Akhmadaliev S, Rugel G, Merchel S (2013) Determination of 41Ca with LSC and AMS: method development, modifications and applications. J Radioanal Nucl Chem 296:617–624
Nottoli E, Bourles D, Bienvenu P, Labet A, Arnold M, Bertaux M (2013) Accurate determination of 41Ca concentrations in spent resins from the nuclear industry by accelerator mass spectrometry. Appl Rad Isot 82:340–346
Merchel S, Rugel G, Lachner J, Wallner A, Walther D, Ziegenrücker R (2021) Evaluation of a sensitive, reasonable, and fast detection method for 55Fe in steel. J Radioanal Nucl Chem 330:727–735
BIPM, IEC, IFCC, ISO, IUPAC, IUPAP and OIML (2008) Guide to the expression of Uncertainty in Measurement, 2nd ed. JCGM
Kragten J (1994) Calculating standard deviations and confidence intervals with a universally applicable spreadsheet technique. Analyst 119:2161–2165
Currie LA (1968) Limits for qualitative detection and quantitative determination. Appl Radiochem Anal Chem 40(3):586–593
International Standard ISO 11929-1 (2019) Determination of the characteristic limits (decision threshold, detection limit and limits of the coverage interval) for measurements of ionizing radiation—Fundamentals and application—Part 1: Elementary applications. ISO, Geneva, Switzerland
NF M60–322 (2005) Technologie du cycle du combustible nucléaire—Déchets—Détermination de l’activité du fer 55 dans les effluents et déchets par scintillation liquide, après séparation chimique préalable. Association Française de Normalisation, Paris, France.
NF M60–317 (2001) Technologie du cycle du combustible—Déchets—Détermination de l’activité du nickel 63 dans les effluents et déchets par scintillation liquide, après séparation chimique préalable. Association Française de Normalisation, Paris, France.
Acknowledgements
The authors would like to thank the Nordic Nuclear Research NKS-B programme (www.nks.org) for funding the DTM Decom III project in which the intercomparison exercise was carried out. The authors would also like to thank the other participating laboratories, namely Technical University of Denmark, Norwegian University of Life Sciences, Fortum Power and Heat Oy, Institute for Energy Technology Kjeller and Taiwan Power Company Radiation Laboratory for provision of data and collaboration. National fundings were given by Finnish Research Programme on Nuclear Waste Management KYT 2022. A-LABOS-EX-PR-SC-01 project is thanked for the CEA self-funding. CEA LASE laboratory operators Gabriel Lambrot, Margaux Giuliani, Christèle Colin, Elodie Laporte, Jacques Bubendorff and René Brennetot are thanked for their experimental contributions.
Funding
Open Access funding provided by Technical Research Centre of Finland (VTT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing financial interests or personal relationships with people or organisations that could have inappropriately influenced the work presented in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leskinen, A., Dorval, E., Baudat, E. et al. Intercomparison exercise on difficult to measure radionuclides in spent ion exchange resin. J Radioanal Nucl Chem 332, 77–94 (2023). https://doi.org/10.1007/s10967-022-08687-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-022-08687-2