
Abstract
Invasive breast cancer represents the endpoint of a developmental process that originates in the terminal duct lobular units and is believed to progress through stages of increasing proliferation, atypical hyperplasia, and carcinoma in situ before the cancer acquires invasive and metastatic capabilities. By comparison with invasive breast cancer, which has been studied extensively, the preceding stages of benign breast disease are more poorly understood. Much less is known about the molecular changes underlying benign breast disease development and progression, as well as the transition from in situ into invasive disease. Even less focus has been given to the specific role of stroma in this progression. The reasons for lack of knowledge about these lesions often come from their small size and limited sample availability. More challenges are posed by limitations of the models used to investigate the lesions preceding invasive breast cancer. However, recent studies have identified alterations in stromal cell function that may be critical for disease progression from benign disease to invasive cancer: key functions of myoepithelial cells that maintain tissue structure are lost, while tissue fibroblasts become activated to produce proteases that degrade the extracellular matrix and trigger the invasive cellular phenotype. Gene expression profiling of stromal alterations associated with disease progression has also identified key transcriptional changes that occur early in disease development. In this review, we will summarize recent studies showing how stromal factors can facilitate progression of ductal carcinoma in situ to invasive disease. We also suggest approaches to identify processes that control earlier stages of disease progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
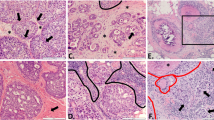
The human breast consists of a branched parenchymal network that produces milk during lactation and drains it to the nipple [1]. The milk-producing structures of the mammary gland are collections of multiple small acini at the distal ends of the ducts, and are known as terminal duct lobular units (TDLU) (Fig. 1a). Although many of the exact mechanisms and mediators underlying the development of human breast cancer remain unknown, the most commonly hypothesized model posits that invasive breast cancer initiates from the TDLU and progresses through stages of benign breast disease (BBD) in incremental steps of increasing cellular abnormalities marked by excessive proliferation and atypia [1–4] (Fig. 1b). According to this model, abnormal proliferation in TDLU initially leads to unfolded lobules and/or cystic structures (Fig. 1c a), which progress through stages where the epithelium becomes increasingly proliferative without acquiring atypical characteristics (proliferative disease without atypia, PDWA, Fig. 1c b–d). Atypical hyperplasia can manifest as either ductal or lobular forms (atypical ductal hyperplasia [ADH] or atypical lobular hyperplasia [ALH], respectively, Fig. 1c e, f), as can the early stages of carcinoma in situ (characterized as ductal carcinoma in situ [DCIS, Fig. 1c g] and lobular carcinoma in situ [LCIS]).

Pathological characteristics associated with breast cancer progression. a Normal terminal duct lobular unit (TDLU), the anatomic substructure from which breast cancer originates, containing numerous individual acini and surrounded by interlobular stroma. b Model of progression from normal epithelium through benign breast disease to invasive breast cancer, indicating relative lifetime risk for cancer development as compared to women with no proliferative disease. Women with proliferative disease without atypia (PDWA) have an increased relative risk (RR) of 1.3–1.9; women with atypical hyperplasia (atypia) have RR of 3.5–5.3; women with ductal carcinoma in situ (DCIS) have RR of 10–11. c Stages of progression from benign breast disease to invasive, metastatic breast cancer. a Nonproliferative disease depicting mammary cysts, fluid-filled structures derived from TDLU. b Proliferative disease without atypia (PDWA): adenosis, a proliferative lesion characterized by increased number or size of glandular structures. c PDWA: moderate hyperplasia, characterized by an increased number of epithelial cells in breast ducts and lobules with 5 or more cells above the basement membrane, often with bridging of the luminal space. d PDWA: florid hyperplasia, in which hyperplastic cells with unevenly distributed nuclei proliferate in solid clusters distending the lumens and often leading to their complete closing and loss e Atypical ductal hyperplasia, epithelial hyperplasia with bridging architecture and monotonous cytologic features suggesting expansion of a cell population. f Atypical lobular hyperplasia characterized by enlarged lobules partially involved with acini expanded with monotonous dyshesive epithelial cells. g Ductal carcinoma in situ, a malignant proliferation of epithelial cells remaining within the basement membrane; some surrounding inflammatory infiltrate is seen. h Invasive breast cancer- malignant cells have invaded the surrounding stromal tissues. i Lymph node metastasis
Multiple epidemiologic studies provide evidence supporting this model in which invasive breast cancer develops from benign disease [5–8]. Common genetic and epigenetic alterations have been identified in PDWA, atypia, DCIS and invasive breast cancer, often occurring progressively along this stepwise pathway [3, 4, 9, 10], with extensive similarities observed in gene expression profiles across atypia, DCIS and invasive cancer in the same breast [9]. Furthermore, the lifetime risk of subsequently developing invasive breast cancer (relative risk, RR) increases in a progressive fashion according to the histological subtype of benign disease: women with PDWA have RR of 1.3–1.9 [6–8], women with atypical hyperplasia have RR of 3.5–5.3 [6–8, 11], and women with DCIS have RR of 10–11 [1]. It should be emphasized however, that although benign breast disease is associated with significantly increased risk for subsequent disease progression, only a small proportion of benign disease lesions will actually develop into invasive cancer. Given the critical role of stroma in breast development and in transition from localized breast cancer to invasive disease [12], it is likely that the stromal microenvironment is also involved in progression of benign disease to carcinoma. In this review, we will describe the characteristics of benign disease progression, evaluate the potential contribution of microenvironmental signals to the progression of benign disease to invasive cancer, and examine recent studies that identify microenvironmental alterations in premalignant disease. We propose that a better understanding of the specific features of the stromal microenvironment that contribute to progression of benign disease to invasive cancer will yield new prognostic markers better identifying those women at greatest risk for subsequent development of invasive cancer, will improve early detection of disease, and will identify novel points for therapeutic intervention to reduce disease incidence.
Modeling Progression from Benign Disease to Invasive Cancer
The best investigated benign breast lesions are ALH, ADH, and DCIS [1]. It should be noted that the terminology “ductal” and “lobular” used to define distinct breast lesions does not imply the site of origin within the mammary gland or the types of cells from which it is likely to have formed; rather the classification is based on the discrete architecture, cytology and immunohistochemistry (IHC) of the lesion. Moreover, the vast majority of mammary tumors and their precursors, independent of their histological type, are believed to originate from TDLUs and not, as previously believed, from other microanatomical sites of the normal breast [13–15]. However, as the ductal type lesions encompass almost 80% of all diagnosed breast cancers worldwide [16], the majority of investigations of genetic and molecular changes in breast cancer development and progression are based on this type of preinvasive and invasive tumor; accordingly, we will focus here on discussion of ductal-type lesions.
ADH lesions are derived from outgrowths of luminal epithelial cells and are morphologically related to low-grade DCIS [1, 4, 17], with the distinction based on the degree of occupied space. The proliferating cells become monomorphic with similar-sized nuclei and few mitotic figures [5]. The average proliferation rate in normal TDLUs is about 2%, whereas it increases to 5% in ADH and 15% in DCIS [4]. DCIS is a group of preinvasive cancerous lesions that arise as a result of neoplastic proliferation of luminal epithelial cells that do not cross the basement membrane (BM) [1]. Traditionally, the transition from DCIS to invasive carcinoma is considered to involve disruption of the BM and the surrounding layer of myoepithelial cells (Fig. 2). However, the Sontag-Axerold model proposes a different pathway in which both DCIS and invasive cancer originate from a common progenitor cell [18, 19]. Both DCIS and invasive cancer are histologically and biologically diverse, composed of many different subtypes [2]; in high grade DCIS the myoepithelial cell layer and the BM become discontinuous, with proliferation of fibroblasts, increased angiogenesis, and infiltration of lymphocytes [20–22]. It may be that well-differentiated DCIS gives rise to low grade invasive cancer, whereas poorly-differentiated DCIS evolves through a distinct pathway, progressing into high grade invasive cancer [23]. Alternatively, it may be that atypia (or perhaps even an earlier proliferative lesion) may represent the key step from which cancer develops. In support of this possibility, biopsies of breast tissue removed prophylactically from BRCA1/2 carriers were found to contain various proliferative benign lesions including atypia in over 50% of cases [24, 25]. Moreover, PDWA and atypia are frequently found in random periareolar fine needle aspirations from high risk women compared to normal risk women [26]. If DCIS represents a symptom of a propensity to develop invasive cancer, rather than an obligate precursor, then it becomes paramount to define which benign lesions are true precursors of invasive cancer, and to identify the signals that drive disease progression.

Stromal alterations in breast cancer progression. a In phenotypically normal tissue, epithelial structures consist of central luminal epithelial cells encircled by myoepithelial cells and enclosed by a continuous basement membrane, while the primarily collagenous stroma contains fibroblasts, immune cells, and vasculature. b Progression to carcinoma in situ is characterized by proliferative epithelial cells enclosed in a still-continuous basement membrane, increased numbers of fibroblasts and immune cell infiltrate, and enhanced angiogenesis. c Invasive breast carcinoma is defined by breakdown of the basement membrane, loss of myoepithelial cells, and invasion of the tumor cells into the surrounding stroma and the vasculature
Microenvironmental Components that Drive Progression from Benign Disease to Invasive Cancer
Myoepithelial Cells
In the nonmalignant mammary gland, myoepithelial cells surround luminal and alveolar epithelial cells of the mammary duct lobular network, separating the luminal cells from the basement membrane, and playing key roles in mammary gland development and reorganization [27, 28]. They help regulate luminal cell polarity, ductal morphogenesis, produce basement membrane components and aid milk ejection during lactation. One of the key characteristics of progression to invasive cancer is alteration in and loss of the myoepithelial cells. Even though the myopeithelial cell layer remains intact in DCIS, the myoepithelial cells themselves differ substantially from those found in normal tissue: DCIS-associated myoepithelial cells have decreased expression of genes involved in normal cell function, including thrombospondin, laminin, and oxytocin receptor, and increased expression of genes that drive increased proliferation, migration, invasion and angiogenesis, including CXCL12 and CXCL14 [29]. Phenotypic alterations of myoepithelial cells, as evidenced by decreased staining for myopeithelial specific markers, were also found in an IHC study that compared DCIS cases to normal tissue [30]. Therefore, even if the myoepeithelial cell layer remains intact in DCIS, the cells themselves may have already acquired changes that facilitate tumorigenic progression of the luminal epithelial cells. It may be possible to design therapeutic approaches to counter the tumor potentiating activities of altered myoepithelial cells. Experimental mouse models of progression from DCIS to invasive cancer using the MCF10DCIS.com human cell line revealed that co-injection of human fibroblasts, either from normal tissue or invasive cancer, promoted progression into invasive carcinoma, whereas additional injection of normal human myoepithelial cells overcame this tumor-promoting effect [31]. This may be due in part to a loss of the tumor suppressing characteristics of normal myoepithelial cells. Normal myoepithelial cells secrete maspin and other proteinase inhibitors that suppress cancer cell proliferation and invasion [27, 32, 33], while tumor myoepithelial cells produce matrix metalloproteinases (MMPs) and cathepsins that degrade the basement membrane and facilitate tumor cell invasion [34]. Tumor myoepithelial cells are deficient for production of laminin-1, a critical component of the basement membrane, which as a result renders them unable to aid polarization and organized growth of mammary epithelial cells [35].
Fibroblasts
Fibroblasts are key players in the maintenance of normal tissue structure and in the progression to malignancy [36]. Early experiments revealed that carcinoma-associated fibroblasts (CAFs) can promote tumorigenic conversion of initiated epithelial cells, while fibroblasts derived from normal tissue suppress this transition [37]. Hu et al. have previously shown that fibroblasts promote, and myoepithelial cells suppress, progression form DCIS to invasive cancer in a mouse xenograft model [31]. Subsequent investigations provided information about the key signals from CAFs that drive tumor progression. A study using a co-implantation xenograft model revealed that secretion of CXCL12 by CAFs promoted angiogenesis and increased cancer cell proliferation through interaction with CXCR4 expressed by tumor cells [38]. A separate study with a similar design revealed that coimplantation of CAFs with MCFDCIS cells leads to activation of COX-2, a mediator of inflammation that is a negative prognostic indicator in invasive cancer [39, 40]. Inhibition of COX-2 completely blocked the increased growth of tumors with co-injected fibroblasts and inhibited transition from DCIS to invasive cancer. These studies indicate a critical role for fibroblast activation and accumulation in breast cancer progression.
ECM-degrading Proteinases
Loss of the basement membrane is one of the key steps in the transition from DCIS to invasive cancer. Matrix metalloproteinases (MMPs) are proteolytic enzymes able to degrade nearly all the components of the basement membrane, as well as to activate growth factors, degrade cell-cell and cell-ECM adhesion molecules, and activate zymogen forms of other MMPs [41, 42]. DCIS has been found to have higher expression of MMP-2 and MMP-9 when compared to normal and hyperplastic tissue [43]. Other studies have observed MMP-1, -2, -3, -9, and -11 in the stroma around preinvasive lesions [23, 44–46]. The expression of heparanase-1, another matrix degrading enzyme able to degrade heparin sulfate proteoglycans, has been found to correlate with higher grade in situ tumors, suggesting its role in progression from DCIS into invasive cancer [47]. Analysis of stromal protease expression by transcriptional profiling of DCIS and invasive cancer using laser capture microdissection (LCM, Fig. 3) identified matrix related genes, MMP11, MMP13 and urokinase-type plasminogen activator (PLAU), among stromal genes upregulated in invasive cancer [46]; increased expression of MMP1 and MMP12 expression in the stroma has also been associated with poor prognosis for patients with invasive cancer [48]. Such studies provide insight into which MMPs and other matrix-degrading proteinases may be potentially useful therapeutic targets. However, increased selectivity of any new candidate proteinase inhibitors will be important in order to avoid the problems of previous clinical trials in which broad-spectrum MMP inhibitors performed very poorly in patients with advanced disease due to adverse side effects [49].

Transcriptional profiling of stromal alterations. Slides from breast tissue biopsies are microdissected to separate the proliferative epithelial cells from the surrounding stroma. RNA is extracted from the tissue slides and analyzed by microarray; comparison of transcriptional profiles from patients who subsequently progressed to invasive breast cancer with profiles from patients who did not progress can reveal transcripts prognostic for breast cancer progression
Transcriptional Dissection of Stromal Contribution to Breast Cancer Progression
Development of new methodologies for separation of breast stroma from the epithelial lesions by LCM (Fig. 3) or by sorting of cells on the basis of surface markers, combined with transcriptional profiling to identify patterns of gene expression differences, has provided substantial new information about stromal signals that control breast cancer progression. A recent study by Finak et al. [48] used IBC tissue biopsies and matched uninvolved tissue from patients for which clinical outcome data were available. LCM-isolated IBC tumor stroma and matched normal stroma were subjected to microarray profiling, and the results were used to derive a prognostic predictor gene set that could be validated using separate profiling data and that was shown to predict outcome prior to detectable metastasis [48]. Another study in which both tumor and stroma from IBC patients were analyzed for transcriptional profiles of genes encoding ECM and ECM-modifying proteins identified a good prognosis gene set which showed increased expression of serine protease inhibitors, and a poor prognosis set, which showed upregulation of integrins and MMPs and downregulation of laminins [50].
Another LCM/microarray-based study comparing expression profiles of epithelium and stroma from IBC and matched adjacent normal tissue found that gene signatures from uninvolved normal epithelium and stroma were not predictive of tumor outcome, and also that the uninvolved epithelial and stromal tissue was not substantially different from expression profiles of epithelial and stromal tissue derived from reduction mammoplasty [51]. The results of this study suggest that stromal effects on tumor progression are likely to be specific to the tumor microenvironment rather than systemic alterations.
Profiling of tumors derived from the stroma has also provided insight into the role of stroma in the progression to IBC. Two types of fibroblastic tumors, solitary fibrous tumor (SFT) and desmoid-type fibromatosis (DTF), have been investigated by transcriptional profiling, and gene sets which differentiated normal from SFT or DTF were also found to be predictive of outcome for IBC [52]. When profiles of IBC biopsies (obtained from samples containing both stroma and epithelial tissue) were grouped by similar gene expression, one subgroup of IBC patients showed significantly elevated expression of genes that were associated with DTF, the majority of which were involved in a pro-fibrotic ECM interaction or stimulation (e.g., collagens, MMPs, transforming growth factor-β, and myofibroblast-associated genes). This set was termed a fibrotic stromal response group and corresponded with lower tumor grade, increased estrogen receptor expression, and better survival prognosis [52, 53]. A distinct IBC subgroup was found to be associated with SFT-associated transcriptional alterations, mostly ECM/basement membrane specific genes, and was correlated with poor prognosis. A third type of stromal response signature, designated a macrophage/colony stimulating factor-1 (CSF-1) signature, also showed relevance to breast cancer subsets [54]. These were higher grade tumors, progesterone and estrogen receptor negative, and positive for TP53 mutations, with survival prognosis varying between the cancer subsets. The fibroblast (DTF) and macrophage (CSF1) stromal response signatures were also found in specific subsets of DCIS, as identified by both gene expression and IHC [55]. Interestingly the macrophage signature corresponded with clinicopathologic characteristics of DCIS similar to those found in IBC, namely higher grade and negative hormone receptor status. Such studies emphasize the role of stroma in breast cancer progression and outcome prognosis and point to the need for similar studies evaluating stromal gene expression changes at stages preceding invasive cancer to identify signatures that may influence transition from benign to invasive disease.
Analysis of transcriptional profiles of epithelial tissue derived from ADH, DCIS, and invasive cancer revealed broad similarities between premalignant and malignant disease, suggesting a common clonal origin of the different stages of benign disease [9]. Moreover, most of the alterations were observed in ADH and persisted through DCIS and IBC, supporting the concept that characteristics necessary for development of IBC are already present in premalignant lesions. A follow-up study performed to analyze gene expression in the tumor microenvironment during breast cancer progression identified a large number of transcriptional alterations in both the epithelium and in the stroma in DCIS and IBC as compared to normal tissue, with the differences suggesting that the majority of the stromal alterations seem to occur before the DCIS stage rather than in progression from DCIS to invasive cancer [56]. These findings suggest that paracrine and endocrine signaling, rather than cell-cell interactions, may be the main factors influencing stromal changes, as the basement membrane in DCIS is mostly uninterrupted [56, 57].
Further supporting the importance of stroma in progression of DCIS to IBC, another transcriptional profiling study of cells from either pure DCIS, the in situ component of DCIS-IBC or IBC suggested that the molecular changes in the epithelial cells occurred before the morphological alterations associated with progression [58, 59]. One of the proposed explanations of this observation is that the transition from in situ to invasive carcinoma strongly depends on the signals from myoepithelial cells, fibroblasts and myofibroblasts. That the microenvironment is a key mediator of disease progression is also in line with previous findings suggesting that the most dramatic transcriptional alterations occur along with development of DCIS, and that fewer alterations are found in the DCIS to IBC transition [56]. Also supporting the concept that the epithelial cells of DCIS and IBC are very similar in their gene expression characteristics, a recent study has demonstrated that the MCF10DCIS cells are able to spontaneously progress into IBC-like cells, although normal myoepithelial cells are able to block this progression [31].
Allinen et al. performed the first gene expression profiling study of subtypes of stromal cells from normal, DCIS and IBC samples [29]. They used a cell type-specific purification procedure based on distinct cell surface markers and magnetic bead separation methods and performed serial analysis of gene expression (SAGE) to identify dramatic changes in the microenvironment of DCIS and IBC as compared to normal tissue. Myoepithelial cells and myofibroblasts exhibited the most substantial transcriptional alterations, including many genes encoding secreted and cell surface proteins. Although gene expression changes were found in all cell types, genetic alterations, analyzed using single nucleotide polymorphism (SNP) arrays, were only detected in epithelial cancer cells, which suggested that the underlying processes were likely due to epigenetic regulation rather than genetic mutations. In agreement with these conclusions, DNA methylation profiling studies provided evidence of consistent epigenetic alterations in stromal cells when assessing luminal epithelial, myoepithelial, and fibroblast cells from normal mammary tissue, DCIS, and IBC [60], or of epithelial and stromal cells from HER2 positive cancers [61]. The challenge now is to identify the specific signals that lead to activation of the stroma, how the epigenetic effects are induced, and how they are maintained.
Future Directions
Transcriptional profiling has proved a powerful tool for identifying disease categories or processes associated with disease progression, and critical information has been obtained when applied to stroma derived from patients with IBC or to stromal components of patients with DCIS. A major gap in our knowledge is the identification of the stromal factors that control transition from benign disease to carcinoma in situ and on to invasive disease, which will require use of clinical cohorts of patients with benign disease for which clinical outcome is known. Of course, validation of candidate processes involved in disease progression will require experimental systems that model the relevant transitions. Studies of progression from DCIS to invasive disease have been facilitated by the use of well characterized cell lines, in particular the MCF10A series [62, 63] which provides a tool to study molecular changes that occur at the different stages of breast cancer development from benign to atypical hyperplasia, through carcinoma in situ, and on to malignant cells able to form tumors with metastatic capabilities. The cell series originated in breast tissue obtained from a woman with extensive fibrocystic disease [64, 65], and was used to create a model series of derived cell lines including premalignant benign proliferating cells with potential for neoplastic progression (MCF10AT lines) [66, 67], ductal carcinoma in situ (MCF10DCIS.com) [68] and invasive carcinoma (MCF10CA1 lines) [63]. MCF10DCIS.com cells in particular have been employed to model the DCIS to invasive cancer transition; similar modeling of the atypia to DCIS (and invasive cancer) transition may be possible using MCF10AT lines, or new cell lines may be developed from patients with benign disease. The specific role of stromal signals in these processes may be investigated using recently developed humanization models in which cleared fat pads of immunocompromised mice are populated with human stromal cells prior to epithelial cell implantation [69–72].
Abbreviations
- ADH:
-
atypical ductal hyperplasia
- ALH:
-
atypical lobular hyperplasia
- BBD:
-
benign breast disease
- BM:
-
basement membrane
- CAF:
-
carcinoma-associated fibroblast
- CSF-1:
-
colony stimulating factor-1
- CTGF:
-
connective tissue growth factor
- DCIS:
-
ductal carcinoma in situ
- DTF:
-
desmoid-type fibromatosis
- ECM:
-
extracellular matrix
- IBC:
-
invasive breast carcinoma
- IHC:
-
immunohistochemistry
- LCIS:
-
lobular carcinoma in situ
- LCM:
-
laser capture microdissection
- PDWA:
-
proliferative disease without atypia
- RR:
-
relative risk
- SAGE:
-
serial analysis of gene expression
- SFT:
-
solitary fibrous tumor
- SNP:
-
single nucleotide polymorphism
- TDLU:
-
terminal duct lobular unit
References
Arpino G, Laucirica R, Elledge RM. Premalignant and in situ breast disease: biology and clinical implications. Ann Intern Med. 2005;143(6):446–57.
Allred DC, Wu Y, Mao S, et al. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res. 2008;14(2):370–8.
Santen RJ, Mansel R. Benign breast disorders. N Engl J Med. 2005;353(3):275–85.
Allred DC, Mohsin SK, Fuqua SA. Histological and biological evolution of human premalignant breast disease. Endocr Relat Cancer. 2001;8(1):47–61.
Lopez-Garcia MA, Geyer FC, Lacroix-Triki M, et al. Breast cancer precursors revisited: molecular features and progression pathways. Histopathology. 2010;57(2):171–92.
Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med. 1985;312(3):146–51.
London SJ, Connolly JL, Schnitt SJ, et al. A prospective study of benign breast disease and the risk of breast cancer. JAMA. 1992;267(7):941–4.
Tamimi RM, Rosner B, Colditz GA. Evaluation of a breast cancer risk prediction model expanded to include category of prior benign breast disease lesion. Cancer. 2010;116(21):4944–53.
Ma XJ, Salunga R, Tuggle JT, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA. 2003;100(10):5974–9.
O’Connell P, Pekkel V, Fuqua SA, et al. Analysis of loss of heterozygosity in 399 premalignant breast lesions at 15 genetic loci. J Natl Cancer Inst. 1998;90(9):697–703.
Degnim AC, Visscher DW, Berman HK, et al. Stratification of breast cancer risk in women with atypia: a Mayo cohort study. J Clin Oncol. 2007;25(19):2671–7.
Radisky ES, Radisky DC. Stromal induction of breast cancer: inflammation and invasion. Rev Endocr Metab Disord. 2007;8(3):279–87.
Wellings SR, Jensen HM, Marcum RG. An atlas of subgross pathology of the human breast with special reference to possible precancerous lesions. J Natl Cancer Inst. 1975;55(2):231–73.
Wellings SR, Jensen HM. On the origin and progression of ductal carcinoma in the human breast. J Natl Cancer Inst. 1973;50(5):1111–8.
Weigelt B, Geyer FC, Reis-Filho JS. Histological types of breast cancer: how special are they? Mol Oncol. 2010;4(3):192–208.
Bombonati A, Sgroi DC. The molecular pathology of breast cancer progression. J Pathol 2010; Oct 14. [Epub ahead of print].
Guray M, Sahin AA. Benign breast diseases: classification, diagnosis, and management. Oncologist. 2006;11(5):435–49.
Sontag L, Axelrod DE. Evaluation of pathways for progression of heterogeneous breast tumors. J Theor Biol. 2005;232(2):179–89.
Kuerer HM, Albarracin CT, Yang WT, et al. Ductal carcinoma in situ: state of the science and roadmap to advance the field. J Clin Oncol. 2009;27(2):279–88.
Burstein HJ, Polyak K, Wong JS, et al. Ductal carcinoma in situ of the breast. N Engl J Med. 2004;350(14):1430–41.
Guidi AJ, Fischer L, Harris JR, et al. Microvessel density and distribution in ductal carcinoma in situ of the breast. J Natl Cancer Inst. 1994;86(8):614–9.
Damiani S, Ludvikova M, Tomasic G, et al. Myoepithelial cells and basal lamina in poorly differentiated in situ duct carcinoma of the breast. An immunocytochemical study. Virchows Arch. 1999;434(3):227–34.
Hannemann J, Velds A, Halfwerk JB, et al. Classification of ductal carcinoma in situ by gene expression profiling. Breast Cancer Res. 2006;8(5):R61.
Kauff ND, Brogi E, Scheuer L, et al. Epithelial lesions in prophylactic mastectomy specimens from women with BRCA mutations. Cancer. 2003;97(7):1601–8.
Hoogerbrugge N, Bult P, de Widt-Levert LM, et al. High prevalence of premalignant lesions in prophylactically removed breasts from women at hereditary risk for breast cancer. J Clin Oncol. 2003;21(1):41–5.
Fabian CJ, Kamel S, Zalles C, et al. Identification of a chemoprevention cohort from a population of women at high risk for breast cancer. J Cell Biochem Suppl. 1996;25:112–22.
Adriance MC, Inman JL, Petersen OW, et al. Myoepithelial cells: good fences make good neighbors. Breast Cancer Res. 2005;7(5):190–7.
Polyak K, Hu M. Do myoepithelial cells hold the key for breast tumor progression? J Mammary Gland Biol Neoplasia. 2005;10(3):231–47.
Allinen M, Beroukhim R, Cai L, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6(1):17–32.
Hilson JB, Schnitt SJ, Collins LC. Phenotypic alterations in ductal carcinoma in situ-associated myoepithelial cells: biologic and diagnostic implications. Am J Surg Pathol. 2009;33(2):227–32.
Hu M, Yao J, Carroll DK, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13(5):394–406.
Sternlicht MD, Kedeshian P, Shao ZM, et al. The human myoepithelial cell is a natural tumor suppressor. Clin Cancer Res. 1997;3(11):1949–58.
Sternlicht MD, Barsky SH. The myoepithelial defense: a host defense against cancer. Med Hypotheses. 1997;48(1):37–46.
Kleer CG, Bloushtain-Qimron N, Chen YH, et al. Epithelial and stromal cathepsin K and CXCL14 expression in breast tumor progression. Clin Cancer Res. 2008;14(17):5357–67.
Gudjonsson T, Ronnov-Jessen L, Villadsen R, et al. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci. 2002;115(Pt 1):39–50.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401.
Olumi AF, Grossfeld GD, Hayward SW, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59(19):5002–11.
Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48.
Hu M, Peluffo G, Chen H, et al. Role of COX-2 in epithelial-stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc Natl Acad Sci USA. 2009;106(9):3372–7.
Singh-Ranger G, Salhab M, Mokbel K. The role of cyclooxygenase-2 in breast cancer: review. Breast Cancer Res Treat. 2008;109(2):189–98.
Hadler-Olsen E, Fadnes B, Sylte I, et al. Regulation of matrix metalloproteinase activity in health and disease. FEBS J 2010; Oct 21. doi:10.1111/j.1742-4658.2010.07920.x. [Epub ahead of print].
Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 2010; Oct 21. doi:10.1111/j.1742-4658.2010.07919.x. [Epub ahead of print].
Zhao YG, Xiao AZ, Park HI, et al. Endometase/matrilysin-2 in human breast ductal carcinoma in situ and its inhibition by tissue inhibitors of metalloproteinases-2 and -4: a putative role in the initiation of breast cancer invasion. Cancer Res. 2004;64(2):590–8.
Lebeau A, Nerlich AG, Sauer U, et al. Tissue distribution of major matrix metalloproteinases and their transcripts in human breast carcinomas. Anticancer Res. 1999;19(5B):4257–64.
Wiechmann L, Kuerer HM. The molecular journey from ductal carcinoma in situ to invasive breast cancer. Cancer. 2008;112(10):2130–42.
Schuetz CS, Bonin M, Clare SE, et al. Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer Res. 2006;66(10):5278–86.
Maxhimer JB, Pesce CE, Stewart RA, et al. Ductal carcinoma in situ of the breast and heparanase-1 expression: a molecular explanation for more aggressive subtypes. J Am Coll Surg. 2005;200(3):328–35.
Finak G, Bertos N, Pepin F, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14(5):518–27.
Radisky ES, Radisky DC. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15(2):201–12.
Bergamaschi A, Tagliabue E, Sorlie T, et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol. 2008;214(3):357–67.
Finak G, Sadekova S, Pepin F, et al. Gene expression signatures of morphologically normal breast tissue identify basal-like tumors. Breast Cancer Res. 2006;8(5):R58.
West RB, Nuyten DS, Subramanian S, et al. Determination of stromal signatures in breast carcinoma. PLoS Biol. 2005;3(6):e187.
Beck AH, Espinosa I, Gilks CB, et al. The fibromatosis signature defines a robust stromal response in breast carcinoma. Lab Invest. 2008;88(6):591–601.
Beck AH, Espinosa I, Edris B, et al. The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin Cancer Res. 2009;15(3):778–87.
Sharma M, Beck AH, Webster JA, et al. Analysis of stromal signatures in the tumor microenvironment of ductal carcinoma in situ. Breast Cancer Res Treat. 2010;123(2):397–404.
Ma XJ, Dahiya S, Richardson E, et al. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009;11(1):R7.
Howell A, Landberg G, Bergh J. Breast tumour stroma is a prognostic indicator and target for therapy. Breast Cancer Res. 2009;11 Suppl 3:S16.
Castro NP, Osorio CA, Torres C, et al. Evidence that molecular changes in cells occur before morphological alterations during the progression of breast ductal carcinoma. Breast Cancer Res. 2008;10(5):R87.
Schnitt SJ. The transition from ductal carcinoma in situ to invasive breast cancer: the other side of the coin. Breast Cancer Res. 2009;11(1):101.
Hu M, Yao J, Cai L, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37(8):899–905.
Fiegl H, Millinger S, Goebel G, et al. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 2006;66(1):29–33.
Miller FR. Xenograft models of premalignant breast disease. J Mammary Gland Biol Neoplasia. 2000;5(4):379–91.
Santner SJ, Dawson PJ, Tait L, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65(2):101–10.
Soule HD, Maloney TM, Wolman SR, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6075–86.
Pauley RJ, Soule HD, Tait L, et al. The MCF10 family of spontaneously immortalized human breast epithelial cell lines: models of neoplastic progression. Eur J Cancer Prev. 1993;2 Suppl 3:67–76.
Miller FR, Soule HD, Tait L, et al. Xenograft model of progressive human proliferative breast disease. J Natl Cancer Inst. 1993;85(21):1725–32.
Strickland LB, Dawson PJ, Santner SJ, et al. Progression of premalignant MCF10AT generates heterogeneous malignant variants with characteristic histologic types and immunohistochemical markers. Breast Cancer Res Treat. 2000;64(3):235–40.
Miller FR, Santner SJ, Tait L, et al. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst. 2000;92(14):1185–6.
Wu M, Jung L, Cooper AB, et al. Dissecting genetic requirements of human breast tumorigenesis in a tissue transgenic model of human breast cancer in mice. Proc Natl Acad Sci USA. 2009;106(17):7022–7.
Proia DA, Kuperwasser C. Stroma: tumor agonist or antagonist. Cell Cycle. 2005;4(8):1022–5.
Kuperwasser C, Dessain S, Bierbaum BE, et al. A mouse model of human breast cancer metastasis to human bone. Cancer Res. 2005;65(14):6130–8.
Kuperwasser C, Chavarria T, Wu M, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci USA. 2004;101(14):4966–71.
Financial Support
This work is supported by grants from the National Cancer Institute (CA122086, CA128660, and CA132879), from the Susan B. Komen foundation (FAS0703855), and by the Mayo Clinic Breast Cancer Specialized Program of Research Excellence (SPORE) grant CA116201 (PI James Ingle) from the National Institutes of Health. Amy C. Degnim is supported by the CA90628-08 Paul Calabresi Award for Clinical-Translational Research (K12) via the Mayo Clinic Cancer Center.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cichon, M.A., Degnim, A.C., Visscher, D.W. et al. Microenvironmental Influences that Drive Progression from Benign Breast Disease to Invasive Breast Cancer. J Mammary Gland Biol Neoplasia 15, 389–397 (2010). https://doi.org/10.1007/s10911-010-9195-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-010-9195-8