
Abstract
Introduction
Ductal carcinoma in situ (DCIS) of the breast includes a heterogeneous group of preinvasive tumors with uncertain evolution. Definition of the molecular factors necessary for progression to invasive disease is crucial to determining which lesions are likely to become invasive. To obtain insight into the molecular basis of DCIS, we compared the gene expression pattern of cells from the following samples: non-neoplastic, pure DCIS, in situ component of lesions with co-existing invasive ductal carcinoma, and invasive ductal carcinoma.
Methods
Forty-one samples were evaluated: four non-neoplastic, five pure DCIS, 22 in situ component of lesions with co-existing invasive ductal carcinoma, and 10 invasive ductal carcinoma. Pure cell populations were isolated using laser microdissection. Total RNA was purified, DNase treated, and amplified using the T7-based method. Microarray analysis was conducted using a customized cDNA platform. The concept of molecular divergence was applied to classify the sample groups using analysis of variance followed by Tukey's test.
Results
Among the tumor sample groups, cells from pure DCIS exhibited the most divergent molecular profile, consequently identifying cells from in situ component of lesions with co-existing invasive ductal carcinoma as very similar to cells from invasive lesions. Additionally, we identified 147 genes that were differentially expressed between pure DCIS and in situ component of lesions with co-existing invasive ductal carcinoma, which can discriminate samples representative of in situ component of lesions with co-existing invasive ductal carcinoma from 60% of pure DCIS samples. A gene subset was evaluated using quantitative RT-PCR, which confirmed differential expression for 62.5% and 60.0% of them using initial and partial independent sample groups, respectively. Among these genes, LOX and SULF-1 exhibited features that identify them as potential participants in the malignant process of DCIS.
Conclusions
We identified new genes that are potentially involved in the malignant transformation of DCIS, and our findings strongly suggest that cells from the in situ component of lesions with co-existing invasive ductal carcinoma exhibit molecular alterations that enable them to invade the surrounding tissue before morphological changes in the lesion become apparent.
Similar content being viewed by others


Introduction
Ductal carcinoma in situ (DCIS) of the breast is characterized by a proliferation of malignant-appearing epithelial cells of the ducts but without detachment of the basement membrane or evidence of invasion [1]. This disease lies within a spectrum of preinvasive lesions with a vast range of malignant potential. DCIS can progress rapidly to invasive cancer or it may change very slowly [2]. Therefore, an ability to identify which DCIS lesions are likely to progress to invasive carcinoma and over what time interval would greatly enhance treatment selection and outcome in breast cancer patients.
The current view of the malignant process is that cancer cells acquire malignant potential by accumulating alterations that permit them to overcome the strict rules of normal cell growth regulation imposed by their environment [3]. Breast cancer is a multistep process that manifests through a series of pathological stages, namely atypical ductal hyperplasia, DCIS and invasive ductal carcinoma (IDC), the latter being potentially lethal if subsequent development of distant metastasis occurs [4]. Molecular and pathological evidence suggests that DCIS can be precursor to invasive disease (although this is not without exception) [5–11]. However, it is not clear which cell populations progress to invasive disease and what molecular properties give them the capacity to spread to surrounding tissue.
Despite much research effort, the molecular basis of breast cancer tumorigenesis and progression [9, 12–17] has not completely been elucidated. Two major approaches have been used to address these issues: oligo/cDNA microarrays and laser microdissection. Microarrays allow researchers to examine the expression of several genes simultaneously, identifying gene sets that discriminate groups of cancer samples with common clinical or pathological characteristics and risk for progression to IDC. Laser microdissection is crucial in permitting the molecular analysis of defined, homogenous cell types from a specific solid tissue. Both methodologies have been used to discover novel prognostic markers and to predict disease outcomes [9, 16, 18, 19].
The pathological classification of DCIS does not accurately predict invasive disease. In the present study we compared the gene expression profiles of cells captured from in situ component lesions, pure DCIS, and in situ component of DCIS with co-existing IDC (DCIS-IDC), with the goal being to find molecular makers that can predict risk for invasive disease. We also examined epithelial cells of initial (non-neoplastic epithelial cells) and later stages (IDC cells) of ductal carcinoma progression.
The molecular characteristics of cells from the in situ component of DCIS-IDC are more similar to cells from IDC than to those from pure DCIS (the latter being morphologically identical), which strongly suggests that their molecular reprogramming precedes morphological alteration in the lesion. Moreover, we identified several candidate genes, including LOX [GenBank: NM_002317] and SULF-1 [GenBank: NM_001128206], which are putatively involved in the acquisition of the capacity to invade adjacent tissues of DCIS. These genes may serve as molecular markers that can identify those DCIS lesions that may become invasive.
Materials and methods
Samples
Fresh-frozen human breast tumor samples were retrieved from the Tumor Tissue Biobank of the Medical and Research Center – Hospital A C Camargo, São Paulo. Sections 5 μm thick from the fresh-frozen tumor blocks were cut onto glass slides, stained with hematoxylin and eosin, and reviewed by a pathologist. The hematoxylin and eosin stained sections were used to evaluate and select appropriate tumor areas corresponding to each histological component. The histological grade of the DCIS was assigned in accordance with the World Health Organization scale [20], and IDC was classified in accordance with the Scarff-Bloom-Richardson grading scheme [21, 22]. Forty-one samples were evaluated, consisting of four non-neoplastic breast samples, five pure DCIS (stage 0) samples, 11 in situ component of DCIS-IDC samples, and 10 IDC samples; these served as initial sample sets. An additional 11 in situ component of DCIS-IDC samples were evaluated as an independent sample set. The non-neoplastic samples were obtained from perilesional mammary specimens from patients obtained during resection of benign lesions. A pathologist subjected all slides representative of pure DCIS to a careful histopathological analysis in order to ensure the absence of any previously undetected microinvasions. At least 5 years of follow-up data were available for all patients.
The patients had a mean age of 49 years, and none of them had received preoperative systemic treatment. The patients' clinical characteristics are summarized in Table 1. The expression patterns of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor (HER)2/neu were positive in the majority of the samples. The research was approved by the Ethics Committee of the Medical and Research Center – Hospital A C Camargo, under protocol number 587/04. All participants gave written, informed consent.
Laser capture microdissection
Cells were laser captured using the PixCell II LCM system (Arcturus Engineering, Mountain View, CA, USA). About 4,000 cells were captured from 4 to 7 μm frozen sections, mounted onto glass slides, and stained with 100 μl of nuclear fast red (C.I.60760; Certistain®; Merck, Darmstadt, Germany) for microscopy. Only one type of cells was isolated from each sample group. Non-neoplastic breast epithelium samples were epithelial cells without contamination with stromal cells; pure DCIS samples were tumor cells captured from ducts of pure DCIS lesion; in situ component of DCIS-IDC samples were tumor cells captured from in situ component of the DCIS-IDC lesion; and IDC samples were infiltrative tumor cells captured from the invasive lesion.
A representative sample of cells from the pure DCIS depicting the different phases during the microdissection procedure is shown in Figure 1.

Breast epithelium captured from DCIS by LCM. Images of (a) pre-capture and (b) postcapture tissues, and (c) captured epithelial tumor cells. DCIS, ductal carcinoma in situ; LCM, laser capture microdissection.
RNA isolation and amplification
Laser capture microdissection (LCM) captured cells on CapSure™ HS LCM Caps (Arcturus Engineering) were resuspended in 10 μl of PicoPure RNA extraction buffer (Arcturus Engineering). Total RNA was extracted by using the PicoPure™ RNA Isolation kit (Arcturus Engineering #KT0204) and DNase treated using the RNase-Free DNase Set (Qiagen #79254; Qiagen-Germantown MD USA), in accordance with the manufacturer's instructions. A two-round linear amplification procedure, based on T7-driven amplification, was performed following a previously described protocol [23] with some modifications described below. The total RNA was first denatured at 70°C for 10 minutes in presence of 200 ng oligo (dT) [24]-T7 primer (5'-AAA CGA CGG CCA GTG AAT TGT AAT ACG ACT CAC TAT AGG CGC T (24)-3'; 57 base pairs) and snap cooled on ice.
Reverse transcription was performed by adding 1× first strand buffer and 0.01 mol/l dithiothrectol (Invitrogen Life Technology, Carlsbad, CA, USA), 2 μl diethylpyrocarbonate (DEPC; Sigma, St Louis, MO, USA) treated water, 40 U rRNasin (Promega, Madison, WI, USA), 1 mmol/l dNTP (Amersham Biosciences, Piscataway, NJ, USA), and 400 U SuperScript™ II Reverse Transcriptase (Invitrogen Life Technology) to a final volume of 20 μl. The reaction was incubated for 120 minutes at 42°C. Second-strand synthesis was performed by adding 53 μl of DEPC-treated water, 20 μl of 5× second strand buffer (Invitrogen Life Technology), 1 mmol/l dNTP, 1 U RNase H (Invitrogen Life Technology), 10 U Escherichia coli DNA ligase, and 40 U E. coli DNA polymerase I (Invitrogen Life Technology) to a final volume of 100 μl. The reaction was incubated for 2 hours at 16°C. Ten units of T4 DNA polymerase I (Invitrogen Life Technology) were added and incubated again at 16°C for 5 minutes.
The double strand cDNA (dscDNA) was stopped by adding 0.05 mol/l EDTA. UltraPure™ Phenol (Invitrogen, Carlsbad, CA, USA):chloroform:isoamyl alcohol (Merck), at a ratio of 25:24:1 and a pH of 8.0, was used for cDNA purification. The dscDNA was precipitated with absolute ETOH (Merck) and resuspended in 10 μl DEPC-treated water. The dscDNAs were subjected to in vitro transcription using reagents from Ribomax™ Large scale RNA production system T7 kit (Promega), in accordance with the manufacturer's recommendation. The amplified RNA (aRNA) was reverse transcribed into cDNA using 9 μg random hexamer (dN6; Amersham Biosciences, Little Chalfont, UK). cDNA synthesis was continued with the same conditions used in the first strand of the first round. The second strand was synthesized using Advantage® cDNA Polymerase (Clontech, Mountain View, CA, USA), and purification was performed in accordance with the methodology cited above.
The aRNA quality, in terms of purity and integrity, was assessed by absorbance at 260/280 nm using a GeneQuant pro spectrophotometer (Amersham Pharmacia Biotech, Little Chalfont, UK) and by electrophoresis in 1% UltraPure™ Agarose (Invitrogen Life Technology) gel with ethidium bromide staining (Sigma, St Louis, MO, USA), respectively. Only aRNA samples yielding a minimum of 15 μg and presenting a smear concentration between 300 and 700 base pairs (which guarantees high quality hybridization) were further processed. Total RNA from HB4a normal luminal epithelial mammary cells [24] was extracted and amplified following the same protocol and used as a reference for microarray hybridizations.
cDNA microarrays and probes
We used a customized cDNA platform (4.8K002 platform) comprising 4,608 cDNAs that represent human genes [25]. The labeled cDNA was generated in a reverse transcriptase reaction in the presence of 4 μg aRNA, 9 μg random hexamer primer (Invitrogen Life Technology), Cy3-labeled or Cy5-labeled dCTP (Amersham Biosciences, Little Chalfont, UK), and 400 U SuperScript II (Invitrogen Life Technology). The residual dye was removed using illustra AutoSeq™ G-50 (GE Healthcare, Little Chalfont, UK). Equal amounts of test and reference cDNA reverse colored Cy-labeled product were competitively hybridized against the cDNA probes in microarray slides. Dye swap was performed for each sample analyzed and used as replicate samples. Pre-hybridization was carried out in a humidified chamber at 42°C for 16 to 20 hours, and hybridizations were performed on GeneTac Hybridization Station (Genome Solutions, Ann Arbor, MI, USA) at 42°C.
Intensity signal capture and analysis
After hybridization, slides were washed as follows: 2× Saline-Sodium Citrate (SSC) for 10 minutes, 0.1 × SSC/0.1% SDS for 10 minutes (two times), and 0.1 × SSC for 10 minutes (two times) at 37°C. All solutions were pre-heated to 42°C. Hybridized arrays were scanned on the ScanArray™ Express (Packard BioScience Biochip Technologies, Billerica, MA, USA), and Cy5/Cy3 signals were quantified using the histogram method with ScanArray Express software (Perkin-Elmer Life Sciences, Boston, MA, USA). Fluorescent intensities of Cy5 and Cy3 channels on each slide were subjected to spot filtering and normalization. We first eliminated all saturated points (≥ 63,000; approximately 16 bits) and performed a local background subtraction, considering for analysis only those spots with positives values. Normalization was performed using locally weighted linear regression within and across arrays for inter-slide normalization. Local normalization has the advantage that it can help to correct for systematic spatial variations in the array [26]. After normalization, data for each gene were reported as the logarithm of the expression ratio used to represent the relative gene expression levels in the experimental samples. The raw data from hybridizations and experimental conditions can be obtained at the Gene Expression Omnibus [27] under accession number GSE11042. A detailed description of the platform array is available in accession number GPL1930.
Statistical analysis
For applying the concept for molecular divergence and to identify the most distinct group of samples, the general expression patterns were compared between the sample groups (non-neoplastic, pure DCIS, in situ component of DCIS-IDC, and IDC) using the analysis of variance (ANOVA) statistical test (positive false discovery rate – pFDR < 0.01) [28] followed by Tukey's test [29]. To select the putative genes involved in DCIS progression, two-by-two comparisons among non-neoplastic, pure DCIS, and in situ component of DCIS-IDC sample groups were performed; differentially expressed genes, whose expression levels exhibited at least twofold change, were selected. Downregulated and upregulated genes were analyzed separately. A Venn diagram was constructed to select the genes of interest.
To determine decay rate statistics for collections of genes belonging to the different gene sets, automated analysis of gene function was required. The functional assignments of the genes were obtained using the χ2 distribution of Onto-Tools (developed by Intelligent Systems and Bioinformatics Laboratory from Department of Computational Sciences of Wayne State University) [30]. Functional processes were considered as significant if the P value was under 0.01. Functional assessments, which were represented by only one gene in the platform, were not taken into consideration in order to avoid artifactual results.
For clustering samples based on gene expression profiles, we applied a nonsupervised hierarchical clustering based on Euclidean distance and average linkage. The reliability of the clustering was assessed by the Bootstrap technique using MEV (MultiExperiment Viewer – Boston, MA, USA) [31].
Quantitative RT-PCR
Quantitative RT-PCR reactions were performed using the ABI Prism™ 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Aliquots of cDNA from aRNA were used as templates. RT-PCR reactions were carried out using SYBR® Green PCR MasterMix (Applied Biosystems) in a total volume of 20 μl using the following program: 2 minutes at 50°C and 10 minutes at 95°C for the initial denaturing, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. The list of oligonucleotide sequences is shown in Table 2. Finally, dissociation curves were generated at 95°C for 15 seconds, 60°C for 30 seconds, and 95°C for 15 seconds. To evaluate the amplification of nonspecific products and primer-dimer formation, dissociation curves were analyzed and aliquots of each reaction were subjected to silver staining acrylamide gel electrophoresis. The efficiency of each pair of primers was calculated using standard curve dilutions (as described in the Applied Biosystems protocols). The analysis was conducted using the cDNA converted from the aRNA used in the microarray study. The reactions were performed in duplicate. Three internal control genes, namely HPRT1 [GenBank:NM_000194] [32], GAPDH [GenBank:AJ005371], and BCR [GenBank:NM_004327], were considered in gene expression normalization. Relative gene expression between sample groups was calculated using the Pfaffl model [33], employing the efficiency-corrected equation.
Results
Laser microdissection, RNA extraction, and microarray experiments
To generate a precise correlation between specific epithelial cells and their gene expression patterns, we integrated the use of LCM and T7-based RNA amplification with cDNA microarrays. This procedure permitted gene expression profile analysis with a cell based, rather than tissue based, resolution. To characterize the molecular alterations of cells from the in situ component of the two breast cancer lesions, namely DCIS and DCIS-IDC, we analyzed the gene expression profiles of both. We also used non-neoplastic epithelial cells and cells from IDC lesions as examples of zero and complete progression, respectively.
The Pearson correlation average between all hybridization replicates used in this study was 0.91. All replicates clustered together in dendrograms reporting suitable correction of the individual dye incorporation efficiency by normalization procedure and high experimental reproducibility.
Molecular divergence concepts based on general cell-based gene expression profile
To classify the groups of samples according to their molecular divergence, we used the number of differentially expressed genes as our distance measure; the larger the number of differentially expressed genes between one sample type and all the others, the more distant the sample was allocated and consequently more molecularly divergent. To accomplish this, an ANOVA test corrected by pFDR (<0.01) was performed on the four sample groups, identifying 764 differentially expressed genes (see Additional file 1). Next, Tukey's test was performed through two by two comparisons between the distinct groups. As expected, the non-neoplastic epithelial breast cells exhibited the most divergent expression profile (29%). A total of 221 genes out of 764 were only differentially expressed between non-neoplastic and the three tumor cell groups. In contrast, for cells from pure DCIS, the in situ component of DCIS-IDC and IDC, when compared with all of the other three, there were 12 (1.6%), 37 (4.8%) and 6 (0.3%) differentially expressed genes out of 764, respectively.
Using the same metrics, a new round of analysis was performed excluding the non-neoplastic sample group in order to identify which tumor cell population exhibits the highest molecular divergence level among the three analyzed groups. The ANOVA corrected by pFDR (<0.01) identified 90 variable genes among the three groups of neoplastic cells (see Additional file 2). Tukey's test revealed that cells from pure DCIS exhibited the most distinct gene expression profile, with 75 genes (83%) out of 90 being differentially expressed in comparison with the other two groups of tumor cells. For cells from the in situ component of DCIS-IDC and cells from IDC lesions, 16 genes (18%) and 6 genes (7%) out of 90 were found to be differentially expressed, respectively (Figure 2). A parallel analysis between molecular and morphological aspects yielded contradictory results, because – in terms of morphological features – IDC had the most distinct features and pure DCIS and the in situ component of DCIS-IDC lesions exhibited identical patterns. On the other hand, in terms of molecular features, cells from pure DCIS exhibited the most distinct molecular expression profile, and consequently cells from in situ component of DCIS-IDC retains high similarity to cells from IDC, which suggests alterations in gene expression programs of cells from in situ component of DCIS-IDC before their progression to IDC.

Comparison among the gene expression profile and morphological characteristics of progression of breast ductal carcinoma. (a) Non-neoplastic tissue (20×). (b) Pure DCIS (10×). (c) DCIS-IDC (arrow; hand lens). (d) IDC (hand lens). DCIS, ductal carcinoma in situ; DCIS-IDC, ductal carcinoma in situ with co-existing invasive ductal carcinoma; IDC, invasive ductal carcinoma.
Selection of putative genes involved in ductal carcinoma progression
DCIS progresses to malignant disease when some of the tumor cells acquire invasive capacity. Our previous data strongly suggested that the in situ component of DCIS-IDC already harbors molecular alterations that signal establishment of the invasive process. Based on these findings, we reasoned that the genes potentially involved in the earliest molecular step in acquiring the capacity to invade the surrounding tissue might be among the genes that are differentially expressed between cells from in situ component of DCIS-IDC and both pure DCIS and non-neoplastic groups.
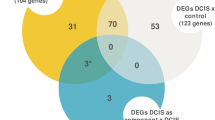
To identify these genes we conducted an ANOVA test corrected by pFDR (<0.01) among non-neoplastic, pure DCIS, and the in situ component of DCIS-IDC, identifying 785 altered genes (see Additional file 3). The two-by-two comparisons between samples from the three groups revealed 8, 215, and 161 genes between non-neoplastic versus pure DCIS, non-neoplastic versus in situ component of DCIS-IDC, and pure DCIS versus the in situ component of DCIS-IDC, respectively (Figure 3a; see Additional file 4). To eliminate genes involved in tumor formation from those potentially involved in tumor progression, we subtracted eight genes that were differentially expressed between cells from non-neoplastic and pure DCIS cells (Figure 3a). The common differentially expressed genes between in situ component of DCIS-IDC versus non-neoplastic or pure DCIS were selected in order to choose the more robust genes, yielding 147 genes classified as potentially implicated in DCIS progression. From those, 126 were upregulated and 21 were downregulated in pure DCIS (see Additional file 5), suggesting that the malignant process of tumor cells from pure DCIS to in situ component of DCIS/IDC occurs rather by downregulation than by upregulation of gene expression.

Putative genes involved in ductal carcinoma progression. (a) Venn diagram depicting the common and distinct genes in each comparison (downregulated and upregulated genes were analyzed separately). (b) Dendogram based on the expression profile of the 147 gene set. Green circles indicate non-neoplastic samples; yellow indicates pure DCIS, and red indicates in situ component of DCIS-IDC samples. (c) Legend of cluster support. (d) Scaled down representation of the entire cluster shown in panel b. Each row represents a single gene and each column a sample. Red indicates upregulation, green indicates downregulation, and black indicates no change in expression level compared with the reference sample. Gray indicates that no intensity was detected. DCIS, ductal carcinoma in situ; DCIS-IDC, ductal carcinoma in situ with co-existing invasive ductal carcinoma.
Functional annotation of this gene set, according to the Onto-Tools database, revealed a statistically significant enrichment of genes involved in cell adhesion (represented by C20orf42 [GenBank:AL118505], LPXN [GenBank:NM_004811], PCLKC [GenBank:NM_017675], DGCR2 [GenBank:NM_005137], AZGP1 [GenBank:NM_001185], CHST10 [GenBank:NM_004854], ITGB2 [GenBank:NM_000211], PLEKHC1 [GenBank:AK291738], PCDH10 [GenBank:NM_032961], and NEDD9 [GenBank:NM_006403]) and cellular defense (represented by CXCL9 [GenBank:NM_002416], MAPRE2 [GenBank:NM_014268], and C3AR1 [GenBank:NM_004054]). LPXN [GenBank:NM_004811] and NEDD9 [GenBank:NM_006403] have been reported as being involved in cancer progression. Over-expression of LPXN [GenBank:NM_004811] and NEDD9 [GenBank:NM_006403] resulted in increased migration in the bone-derived metastatic prostate cancer cell line [34] and in promotion of metastatic melanoma [35], respectively. These reports support our data, which reveal high expression of these genes in the in situ component of DCIS-IDC compared with pure DCIS cells. The 147 genes were organized into seven different biological classes in a hierarchical manner (Table 3).
The hierarchical clustering based on the expression pattern of this gene set resulted in two main branches with high level of reliability (Figure 3b,c). Non-neoplastic samples and 60% of the pure DCIS samples were discriminated from 100% of the in situ component of DCIS-IDC samples (Figure 3b). The ability of this gene set to generate an expression pattern (Figure 3d) that segregated the two groups of cells (pure DICS and in situ component of DCIS-IDC) relatively well, grouping together the samples representing cells from non-neoplastic and cells from pure DCIS lesion, strengthened the hypothesis that those genes might be involved in the early molecular alterations that are necessary for launching of the invasive process.
Confirmation of differential expression between pure DCIS and DCIS-IDC by quantitative RT-PCR
In order to evaluate the robustness of our microarray findings and to avoid choosing false differentially expressed genes because of technical limitations, we randomly selected eight genes from the 147 gene set (Table 4) and performed quantitative RT-PCR in duplicate experiments. The average of the internal control genes (HPRT1 [GenBank:NM_000194], GAPDH [GenBank: AJ005371] and BCR [GenBank:NM_004327]) was used in a normalization procedure.
Using the initial sample sets and the criteria of ≥ 2-fold change, five out of eight genes (62.5%) exhibited agreement in both methodologies. The genes C16orf5 [GenBank:NM_013339], GOSR2 [GenBank:NM_004287], and TXNL2 [GenBank:AL138831] were upregulated, and LOX [GenBank:NM_002317] and SULF-1 [GenBank:NM_001128206] were downregulated in pure DCIS in comparison with in situ component of DCIS-IDC. These five genes were also evaluated in an independent group of 11 in situ component of DCIS-IDC samples (Table 5). Three genes (60%; GOSR2 [GenBank:NM_004287], LOX [GenBank:NM_002317], and SULF-1 [GenBank:NM_001128206]) exhibited agreement with the previous data, strengthening the hypothesis that these genes are involved in the malignant process of DCIS.
Because we were interested in identifying genes that are involved with the acquisition of invasive capacity in DCIS, we reasoned that these genes should not exhibit differential expression between cells from non-neoplastic cells and pure DCIS cells, as neither of these cell types has established invasion capacity. Likewise, we would expect there to be no difference in expression level between cells from the in situ component of DCIS-IDC and IDC lesions, in which – despite of the morphological differences in the tissue – the capacity and program of invasion are already established. Therefore, the five genes confirmed in the initial sample set were evaluated in non-neoplastic and IDC samples. Based on our selection criteria, three genes (C16orf5 [GenBank:NM_013339], LOX [GenBank:NM_002317], and SULF-1 [GenBank:NM_001128206]) showed the expected gene expression behavior in the four sample groups, exhibiting slight or no difference between the group with no invasive program (non-neoplastic and pure DCIS) and that in which the invasive program is already established (DCIS-IDC and IDC; Figure 4a). The expression levels of these three genes exhibited statistically significant differences between the cells lacking the invasive program (non-neoplastic and pure DCIS) and cells possessing the invasive program (in situ component of DCIS-IDC and IDC; Figure 4b). This strongly indicates that LOX [GenBank:NM_002317] and SULF1 [GenBank:NM_001128206], confirmed in the initial and independent group of in situ component of DCIS-IDC samples, are potentially involved in the acquisition of invasive capacity in DCIS.

Gene expression behavior among samples that mimic breast cancer progression. (a) Gene expression difference between identical morphologic samples (pure DCIS and in situ component of DCIS-IDC) by analysis of variance. (b) Comparison between two groups (non-neoplastic [N] + pure DCIS and in situ component of DCIS-IDC + IDC] by quantitative RT-PCR. DCIS, ductal carcinoma in situ; DCIS-IDC, ductal carcinoma in situ with co-existing invasive ductal carcinoma.
Discussion
Characterization of the molecular events that are associated with DCIS progression has been among the major aims of the scientific community. Even though great efforts to decipher the molecular basis of DCIS have been made [2, 9, 13, 14, 16, 36–38], these molecular events remain poorly understood. Studies have been limited by the low availability of pure DCIS frozen samples. In this study, we combined the use of LCM, RNA amplification, and microarray technology to characterize the gene expression pattern of cells captured from pure DCIS and in situ component of DCIS-IDC, which retain identical morphological characteristics in the tissue. Non-neoplastic epithelial cells and IDC lesion cells were also analyzed.
The gene expression profile analysis of cells from three types of breast cancer lesions (pure DCIS, in situ component of DCIS-IDC, and IDC) yielded surprising results. We found that, rather than cells from IDC, cells from pure DCIS had the most divergent molecular aspects, which is in contrast to the morphological features. This finding is directly and indirectly supported by recent reports. Recent studies relating the expression of Her-2/neu, steroid receptors (estrogen receptor and progesterone receptor), Ki67, p53, and epidermal growth factor receptor in pure DCIS to in situ component of DCIS-IDC have suggested that both components have distinct molecular characteristics [39–42]. Gene expression analyses of the two distinct morphological components, the in situ component of DCIS-IDC and IDC, using matched and non-matched samples have identified very similar molecular profiles [2, 9, 16, 38]. Based on these findings, we speculate that either the acquisition of invasive capacity of the DCIS cells is driven by a very small number of genes that play a key role in the invasion process, or the molecular alteration occurs before the morphological modification of the lesion. The findings presented here support the latter hypothesis, strongly suggesting that the molecular alteration of cells from in situ component of DCIS-IDC is already established before the lesion exhibits morphological changes.
In practical terms, one of the major contributions of our study lies in using the molecular divergences between in situ components of the two types of lesions, which have identical morphological characteristics but distinct malignant potential, to identify gene markers that may predict the risk for progression from pure DCIS to invasive disease.
We validated five genes by quantitative RT-PCR in the initial sample sets, which showed 62.5% of agreement in both methodologies and ensured that our microarray data were robust. The GOSR2 [GenBank:NM_004287], C16orf5 [GenBank:NM_013339], and TXNL2 [GenBank:AL138831] genes were over-expressed in pure DCIS when compared with the in situ component of DCIS-IDC. The GOSR2 [GenBank:NM_004287] finding was also confirmed in an independent group of 11 in situ component DCIS-IDC samples. The GOSR2 [GenBank:NM_004287] gene encodes a membrane trafficking protein, which transports proteins among the medial- and trans-Golgi compartments and is involved in signal transduction and transporter activity. C16orf5 [GenBank:NM_013339] was confirmed in the initial set of samples, and in an independent set it exhibited the same tendency in relative expression level (1.4-fold change) but was eliminated by the cut-off adoption criterion (fold change > 2). C16orf5 [GenBank:NM_013339] has an uncommonly high content of proline residues (40% over 104 residues) at the amino-terminus of the protein and is highly expressed in brain [43]. This gene is also known as CDIP (cell death-inducing protein), a potential apoptosis inducer that is associated with caspase-8 cleavage, implicating the extrinsic cell death pathway in apoptosis mediated by CDIP [44]. The over-expression of TXNL2 [GenBank: AL138831] in pure DCIS appears to be dependent on the initial set of samples, because in the independent group of samples we found the change in its expression to be in the opposite direction (under-expression).
LOX [GenBank:NM_002317] and SULF-1 [GenBank:NM_001128206] were over-expressed in the in situ component of DCIS-IDC when compared with pure DCIS in both initial and independent sample sets. The LOX [GenBank:NM_002317] gene mediates metastasis of human breast cancer cells in a mouse model [45] and regulates in vitro breast cancer cell migration and cell-matrix adhesion through the regulation of Scr kinases and FAK [46], making it a candidate for predicting progression of DCIS. Other recent studies showed that LOX [GenBank:NM_002317] expression correlates positively with tumor progression and co-localization with hypoxic regions (defined by hypoxia inducible factor-1α expression) in DCIS and IDC primary tumors [47]. The gene SULF-1 [GenBank:NM_001128206] modulates heparin-binding growth factor signaling, and diminishes proliferation and mitogenecity in vitro in head and neck squamous carcinoma [48]. Evaluation of SULF-1 [GenBank:NM_001128206] expression levels in primary invasive breast tumors by RNA in situ hybridization indicated that this gene is down-regulated in the majority (60%) of samples, with a predominant association with lobular histology [49], which is a disagreement with our data. However, in human pancreatic adenocarcinoma tumors this gene is upregulated, and it is widely expressed in the human pancreatic adenocarcinoma cell line [50]. Despite incomplete concordance among our data and data from other groups for the SULF-1 [GenBank:NM_001128206] gene, over-expression in the in situ component of DCIS-IDC and IDC samples was unequivocal. Moreover, SULF-1 [GenBank:NM_001128206] and LOX [GenBank:NM_002317] exhibited statistically significant differences when the samples were grouped based on invasive behavior (non-neoplastic plus pure DCIS and in situ component of DCIS-IDC plus IDC), suggesting their putative involvement with the malignant transformation of DCIS.
The findings of the present study might have been influenced by the small number of pure DCIS, which retain very specific characteristic, such as high grade and HER2 positivity. Unfortunately, this sample group, because of difficulty in obtaining fresh tissue from this type of lesion, could not be evaluated as an independent group. Therefore, the actual role of these candidate genes in the malignant process of DCIS requires further investigation. Nevertheless, the evidence presented in this study identifies these genes as potential candidates for predicting risk for progression of pure ductal carcinoma.
Conclusions
Our findings strongly suggest that the cells from DCIS with the potential to become invasive exhibit modifications in the general gene expression pattern before the morphological alteration of lesion becomes visible. The SULF-1 [GenBank:NM_001128206] and LOX [GenBank:NM_002317] genes are candidate molecular markers that may be used to predict the risk for DCIS progression.
Abbreviations
- ANOVA:
-
analysis of variance
- aRNA:
-
amplified RNA
- DCIS:
-
ductal carcinoma in situ
- DCIS-IDC:
-
ductal carcinoma in situ with co-existing invasive ductal carcinoma
- DEPC:
-
diethylpyrocarbonate
- dscDNA:
-
double strand cDNA
- HER:
-
human epidermal growth factor receptor
- IDC:
-
invasive ductal carcinoma
- LCM:
-
laser capture microdissection
- RT-PCR:
-
reverse transcription polymerase chain reaction.
References
Olivotto I, Levine M: Clinical practice guideline for the care and treatment of breast cancer: the management of ductal in situ. CMAJ. 2001, 165: 912-913.
Porter D, Lahti-Domenici J, Keshaviah A, Bae YK, Argani P, Marks J, Richardson A, Cooper A, Strausberg R, Riggins GJ, Schnitt S, Gabrielson E, Gelman R, Polyak K: Molecular markers in ductal carcinoma in situ of the breast. Mol Cancer Res. 2003, 1: 362-375.
Gupta GP, Massagué J: Cancer metastasis: building a framework. Cell. 2006, 127: 679-695. 10.1016/j.cell.2006.11.001.
Allred DC, Mohsin SK, Fuqua SA: Histological and biological evolution of human premalignant breast disease. Endocr Relat Cancer. 2001, 8: 47-61. 10.1677/erc.0.0080047.
Zhuang Z, Merino MJ, Chuaqui R, Liotta LA, Emmert-Buck MR: Identical allelic loss on chromosome 11q13 in microdissected in situ and invasive human breast cancer. Cancer Res. 1995, 55: 467-471.
Claus EB, Stowe M, Carter D: Breast carcinoma in situ: risk factors and screening patterns. J Natl Cancer Inst. 2001, 93: 1811-1817. 10.1093/jnci/93.23.1811.
Amari M, Moriya T, Ishida T, Harada Y, Ohnuki K, Takeda M, Sasano H, Horii A, Ohuchi N: Loss of heterozygosity analyses of asynchronous lesions of ductal carcinoma in situ and invasive ductal carcinoma of the human breast. Jpn J Clin Oncol. 2003, 33: 556-562. 10.1093/jjco/hyg109.
Hwang ES, DeVries S, Chew KL, Moore DH, Kerlikowske K, Thor A, Ljung BM, Waldman FM: Patterns of chromosomal alterations in breast ductal carcinoma in situ. Clin Cancer Res. 2004, 10: 5160-5167. 10.1158/1078-0432.CCR-04-0165.
Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, Zhou YX, Varnholt H, Smith B, Gadd M, Chatfield E, Kessler J, Baer TM, Erlander MG, Sgroi DC: Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA. 2003, 100: 5974-5979. 10.1073/pnas.0931261100.
Chin K, de Solorzano CO, Knowles D, Jones A, Chou W, Rodriguez EG, Kuo WL, Ljung BM, Chew K, Myambo K, Miranda M, Krig S, Garbe J, Stampfer M, Yaswen P, Gray JW, Lockett SJ: In situ analyses of genome instability in breast cancer. Nat Genet. 2004, 36: 984-988. 10.1038/ng1409.
Burstein HJ, Polyak K, Wong JS, Lester SC, Kaelin CM: Ductal carcinoma in situ of the breast. N Engl J Med. 2004, 350: 1430-1441. 10.1056/NEJMra031301.
Perou CM, Sørlie T, Eisen MB, Rijn van de M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale AL, Brown PO, Botstein D: Molecular portraits of human breast tumours. Nature. 2000, 406: 747-752. 10.1038/35021093.
van 't Veer LJ, Dai H, Vijver van de MJ, He YD, Hart AA, Mao M, Peterse HL, Kooy van der K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH: Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002, 415: 530-536. 10.1038/415530a.
Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET: Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA. 2003, 100: 10393-10398. 10.1073/pnas.1732912100.
Nagaraja GM, Othman M, Fox BP, Alsaber R, Pellegrino CM, Zeng Y, Khanna R, Tamburini P, Swaroop A, Kandpal RP: Gene expression signatures and biomarkers of noninvasive and invasive breast cancer cells: comprehensive profiles by representational difference analysis, microarrays and proteomics. Oncogene. 2006, 25: 2328-2338. 10.1038/sj.onc.1209265.
Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, Walter M, Fehm T, Solomayer E, Riess O, Wallwiener D, Kurek R, Neubauer HJ: Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer Res. 2006, 66: 5278-5286. 10.1158/0008-5472.CAN-05-4610.
Livasy CA, Perou CM, Karaca G, Cowan DW, Maia D, Jackson S, Tse CK, Nyante S, Millikan RC: Identification of a basal-like subtype of breast ductal carcinoma in situ. Hum Pathol. 2007, 38: 197-204. 10.1016/j.humpath.2006.08.017.
Zhu G, Reynolds L, Crnogorac-Jurcevic T, Gillett CE, Dublin EA, Marshall JF, Barnes D, D'Arrigo C, Van Trappen PO, Lemoine NR, Hart IR: Combination of microdissection and microarray analysis to identify gene expression changes between differentially located tumour cells in breast cancer. Oncogene. 2003, 22: 3742-3748. 10.1038/sj.onc.1206428.
Nishidate T, Katagiri T, Lin ML, Mano Y, Miki Y, Kasumi F, Yoshimoto M, Tsunoda T, Hirata K, Nakamura Y: Genome-wide gene-expression profiles of breast-cancer cells purified with laser microbeam microdissection: identification of genes associated with progression and metastasis. Int J Oncol. 2004, 25: 797-819.
Tavassoli FA, Devilee P: Intraductal proliferative lesions In Pathology and Genetics. Tumours of the breast and female genital organs. World Health Organization Classification of Tumours. 2003, Lyon, France: IARC Press, 63-73.
Doussal VL, Tubiana-Hulin M, Friedman S, Hacene K, Spyratos F, Brunet M: Prognostic value of histologic grade nuclear components of Scarff-Bloom-Richardson (SBR). Cancer. 1989, 64: 1914-1921. 10.1002/1097-0142(19891101)64:9<1914::AID-CNCR2820640926>3.0.CO;2-G.
Howel L, Gandour-Edwards R, O'Sullivan D: Application of the Scarff-Bloom-Richardson tumor grading system to fine needle aspirates of the breast. Am J Clin Pathol. 1994, 101: 262-265.
Gomes LI, Silva RL, Stolf BS, Cristo EB, Hirata R, Soares FA, Reis LF, Neves EJ, Carvalho AF: Comparative analysis of amplified and nonamplified RNA for hybridization in cDNA microarray. Anal Biochem. 2003, 321: 244-251. 10.1016/S0003-2697(03)00466-4.
O'Hare MJ, Ormerod MG, Monaghan P, Lane EB, Gusterson BA: Characterization in vitro of luminal and myoepithelial cells isolated from the human mammary gland by cell sorting. Differentiation. 1991, 46: 209-221. 10.1111/j.1432-0436.1991.tb00883.x.
Brentani RR, Carraro DM, Verjovski-Almeida S, Reis EM, Neves EJ, de Souza SJ, Carvalho AF, Brentani H, Reis LF: Gene expression arrays in cancer research: methods and applications. Crit Rev Oncol Hematol. 2005, 54: 95-105. 10.1016/j.critrevonc.2004.12.006.
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP: Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002, 30: e15-10.1093/nar/30.4.e15.
NCBI Gene Expression Omnibus. [http://www.ncbi.nlm.nih.gov/geo]
Storey JD: A direct approach to false discovery rates. J Roy Stat Soc B. 2002, 64: 479-498. 10.1111/1467-9868.00346.
Neter J, Kutner MH, Nachtsheim CJ, Wasserman W: Tukey, multiple comparison procedure. Applied Linear Statistical Models. Edited by: Richard D. 1996, Burr Ridge, IL: Irwin Inc, 725-732. 4
The Intelligent Systems and Bioinformatics Laboratory. [http://vortex.cs.wayne.edu]
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J: TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003, 34: 374-378.
de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN: Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab Invest. 2005, 85: 154-159.
Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29: e45-10.1093/nar/29.9.e45.
Sahu SN, Nunez S, Bai G, Gupta A: Interaction of Pyk2 and PTP-PEST with leupaxin in prostate cancer cells. Am J Physiol Cell Physiol. 2007, 292: C2288-C2296. 10.1152/ajpcell.00503.2006.
Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, Brennan C, Hahn WC, Cordon-Cardo C, Wagner SN, Flotte TJ, Duncan LM, Granter SR, Chin L: Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006, 125: 1269-1281. 10.1016/j.cell.2006.06.008.
Seth A, Kitching R, Landberg G, Xu J, Zubovits J, Burger AM: Gene expression profiling of ductal carcinomas in situ and invasive breast tumors. Anticancer Res. 2003, 23: 2043-2051.
Hannemann J, Velds A, Halfwerk JB, Kreike B, Peterse JL, Vijver van de MJ: Classification of ductal carcinoma in situ by gene expression profiling. Breast Cancer Res. 2006, 8: R61-10.1186/bcr1613.
Dalgin GS, Alexe G, Scanfeld D, Tamayo P, Mesirov JP, Ganesan S, DeLisi C, Bhanot G: Portraits of breast cancer progression. BMC Bioinformatics. 2007, 8: 291-10.1186/1471-2105-8-291.
Farabegoli F, Champeme MH, Bieche I, Santini D, Ceccarelli C, Derenzini M, Lidereau R: Genetic pathways in the evolution of breast ductal carcinoma in situ. J Pathol. 2002, 196: 280-286. 10.1002/path.1048.
Mylonas I, Makovitzky J, Jeschke U, Briese V, Friese K, Gerber B: Expression of Her2/neu, steroid receptors (ER and PR), Ki67 and p53 in invasive mammary ductal carcinoma associated with ductal carcinoma in situ (DCIS) versus invasive breast cancer alone. Anticancer Res. 2005, 25: 1719-1723.
Park K, Han S, Kim HJ, Kim J, Shin E: HER2 status in pure ductal carcinoma in situ and in the intraductal and invasive components of invasive ductal carcinoma determined by fluorescence in situ hybridization and immunohistochemistry. Histopathology. 2006, 48: 702-707. 10.1111/j.1365-2559.2006.02403.x.
Steinman S, Wang J, Bourne P, Yang Q, Tang P: Expression of cytokeratin markers, ER-alpha, PR, HER-2/neu, and EGFR in pure ductal carcinoma in situ (DCIS) and DCIS with co-existing invasive ductal carcinoma (IDC) of the breast. Ann Clin Lab Sci. 2007, 37: 127-134.
Bhalla K, Eyre HJ, Whitmore SA, Sutherland GR, Callen DF: C16orf5, a novel proline-rich gene at 16p13.3, is highly expressed in the brain. J Hum Genet. 1999, 44: 383-387. 10.1007/s100380050183.
Brown L, Ongusaha PP, Kim HG, Nuti S, Mandinova A, Lee JW, Khosravi-Far R, Aaronson SA, Lee SW: CDIP, a novel pro-apoptotic gene, regulates TNFalpha-mediated apoptosis in a p53-dependent manner. EMBO J. 2007, 26: 3410-3422. 10.1038/sj.emboj.7601779.
Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ: Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006, 440: 1222-1226. 10.1038/nature04695.
Payne SL, Fogelgren B, Hess AR, Seftor EA, Wiley EL, Fong SF, Csiszar K, Hendrix MJ, Kirschmann DA: Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide-mediated mechanism. Cancer Res. 2005, 65: 11429-11436. 10.1158/0008-5472.CAN-05-1274.
Postovit LM, Abbott DE, Payne SL, Wheaton WW, Margaryan NV, Sullivan R, Jansen MK, Csiszar K, Hendrix MJ, Kirschmann DA: Hypoxia/reoxygenation: a dynamic regulator of lysyl oxidase-facilitated breast cancer migration. J Cell Biochem. 2008, 103: 1369-1378. 10.1002/jcb.21517.
Lai JP, Chien J, Strome SE, Staub J, Montoya DP, Greene EL, Smith DI, Roberts LR, Shridhar V: HSulf-1 modulates HGF-mediated tumor cell invasion and signaling in head and neck squamous carcinoma. Oncogene. 2004, 23: 1439-1447. 10.1038/sj.onc.1207258.
Narita K, Chien J, Mullany SA, Staub J, Qian X, Lingle WL, Shridhar V: Loss of HSulf-1 expression enhances autocrine signaling mediated by amphiregulin in breast cancer. J Biol Chem. 2007, 282: 14413-14420. 10.1074/jbc.M611395200.
Nawroth R, van Zante A, Cervantes S, McManus M, Hebrok M, Rosen SD: Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS ONE. 2007, 2: e392-10.1371/journal.pone.0000392.
Acknowledgements
We acknowledge Dr Ricardo Renzo Brentani for critical reading and comments.
We thank the researchers and technicians of Microarray Infrastructure's laboratories of the Ludwig Institute for Cancer Research and the Hospital AC Camargo. We also thank Luiz Paulo Camargo for Gene Expression Omnibus data submission. Grant support: CAPES – Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq #400818/2005-1, and Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP #98/14335-2. NC was supported by FAPESP #06/02670-0.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
NC performed all experiments and wrote the manuscript. CO reviewed the histological specimens and performed the LCM. CT carried out the bioinformatics analysis of the microarray data. EB participated in acquisition of microarray data. MM contributed with the breast cancer specimens and discussions. FS supervised the histological analysis and discussed the study design. HB supervised the bioinformatics analysis, helped in designing the study, and revised the manuscript. DC designed the study, interpreted the data, and wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13058_2008_2121_MOESM1_ESM.pdf
Additional file 1: Differentially expressed genes: non-neoplastic, pure DCIS, in situ component of DCIS-IDC, and IDC lesions. Presented is a table listing the differentially expressed genes among cells from non-neoplastic, pure DCIS, in situ component of DCIS-IDC, and IDC lesions. (PDF 61 KB)
13058_2008_2121_MOESM2_ESM.pdf
Additional file 2: Differentially expressed genes: pure DCIS, in situ component of DCIS-IDC, and IDC lesions. Presented is a table listing the differentially expressed genes among tumor cells from pure DCIS, in situ component of DCIS-IDC, and IDC lesions. (PDF 13 KB)
13058_2008_2121_MOESM3_ESM.pdf
Additional file 3: Differentially expressed genes: non-neoplastic, pure DCIS, and in situ component of DCIS-IDC lesions. Presented is a table listing the differentially expressed genes among cells from non-neoplastic, pure DCIS, and in situ component of DCIS-IDC lesions. (PDF 61 KB)
13058_2008_2121_MOESM4_ESM.pdf
Additional file 4: Differentially expressed genes resulting from of 2 × 2 comparisons. Presented is a table listing the differentially expressed genes resulting from of 2 × 2 comparisons: non-neoplastic × pure DICS; non-neoplastic × in situ component of DCIS-IDC; and pure DCIS × in situ component of DCIS-IDC. (PDF 26 KB)
13058_2008_2121_MOESM5_ESM.pdf
Additional file 5: 147 Genes differentially expressed between pure DCIS and in situ component of DCIS-IDC. Presented a table listing the 147 genes differentially expressed between pure DCIS and in situ component of DCIS-IDC with the respective fold changes. (PDF 21 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Castro, N.P., Osório, C.A., Torres, C. et al. Evidence that molecular changes in cells occur before morphological alterations during the progression of breast ductal carcinoma. Breast Cancer Res 10, R87 (2008). https://doi.org/10.1186/bcr2157
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr2157