
Abstract
In recent years, raising investigation activity in entropy-stabilized ceramic oxides has been driven by their innovative approach to designing a thermodynamically stable multi-element system, which provides a new focus on investigating complex functional materials. Especially, complex and singular physical properties of orthochromite have generated rising research in recent years. Of this family, the PrCrO3, DyCrO3, and HoCrO3 systems are particularly interesting since, below Néel temperature (TN), they present negative magnetization in the ZFC mode. Here, we have employed this approach and designed a medium-entropy (MECs) Pr1/3Dy1/3Ho1/3CrO3 (PDH) ceramic to study not only whether the magnetic characteristics prevail, but also the state of optical and dielectric properties of this new compound. In addition, the physical properties studied were compared with the single PrCrO3 (PC) compound. These compounds were synthesized through a facile combustion synthesis route. The X-ray diffraction analysis, energy-dispersive X-ray spectroscopy (EDS), and surface scanning elemental mapping reveal that a single phase is stabilized and equimolar cations are homogeneously distributed in the PDH-MECs. We found that the disorder of cations in the A-site of the structure has no significant effect on the optical transition in the visible electromagnetic region and dielectric properties with respect to the simple compound PC. In contrast, the magnetic properties are strongly affected in the MECs-PDH compound. We found that the TN is tuned through the O–Cr–O angles and below the antiferromagnetic transition. The results suggest that the negative magnetization in the ZFC mode is an intrinsic feature in these compounds since the negative ZFC susceptibility below TN predominates despite the cationic disorder. Furthermore, their negative magnetic mechanism in both the MECs-PDH and PC compounds are discussed in terms of the Γ2(FxCyGz) magnetic configuration.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the last years, many studies from the theoretical and experimental points of view have been performed in orthochromites materials since a rich variety of physical properties have been found [1,2,3,4,5,6,7]. The considerable attention to these physical properties envisions potential applications in data storage, magnetic refrigeration, and sensor devices [8,9,10,11]. The crystal structure of the RCrO3 (R = Y or rare earth) family is iso-structural with orthorhombic perovskite-derived structures (space group Pnma). The R substitution from La to Lu decreases the tolerance factor causing the cooperative octahedral rotation, and accordingly, the CrO6 octahedral tilting is progressively reduced, stabilizing the orthorhombic structure [12, 13]. This fact affects the superexchange coupling and, accordingly, the magnetic ordering, first through the Cr (3d) sublattice and, second through the rare earth sublattice (4f) at lower temperatures. In addition, the magnetic competition/interaction between ‘d’ and ‘f’ sublattices gives rise to a wide variety of interesting phenomena, such as magnetostriction-induced polarization, spin reorientation, magnetization reversal (in zero field cooling, ZFC and field cooling, FC mode), and exchange bias effect [14,15,16,17,18,19]. These physical phenomena have been found in simple compounds. They can also be induced and tuned by the substitution of isovalent and aliovalent cations in the A and B-sites of the perovskite lattice of orthochromites [4, 20,21,22]. At present, the presence of these phenomena has not yet been well understood, and it is evident to apply new strategies that shed light on mechanisms involved in these phenomena with the aim of the development of new paths in materials science technologies.
In the last years, the emergence of high-entropy ceramics (HECs) has attracted attention in advanced materials science [23, 24]. It has been reported that HECs can be a way to improve the physical properties through the substitution of 3, 5, or more cations in equimolar ratios in simple oxide sublattices or complex ceramic structures such as perovskite, garnet, spinel, and other layer-structural ceramics [24,25,26]. The number of chemical constituents in the equimolar ratio defines low, medium, and high configurational entropy ceramics [25]. Thus, in orthochromites ceramic compounds, as a result of the iso-structural behavior through the rare earth substitution, the physical phenomena can be shared in the same structure, and so, to reveal whether (the intrinsic physical properties) they depend on the rare earth molar ratio or the structural distortion caused by multiple cations substitution using the high-entropic ceramics (HECs) approach. Through this path, we have designed an entropic ceramic material from three equimolar chemical constituents at the A-site, with the chemical formula Pr1/3Dy1/3Ho1/3CrO3. The design of the entropic material is relevant since each of the individual systems, i.e., PrCrO3, DyCrO3, and HoCrO3 compounds synthesized as fine particles, have the particular characteristic of presenting below of TN negative magnetization in the ZFC mode [19, 27,28,29]. This behavior is unusual and little understood. Besides, several studies have addressed it as an artifact [30]. These particular magnetic characteristics in each system have motivated us to design a unique ceramic system where the three randomly distributed cations share the 4c Wyckoff atomic positions of the orthorhombic structure. Thus, in this work, a study of the chemical and structural characteristics is carried out, as well as the study of the optical, dielectric, and magnetic properties of the medium entropic compound Pr1/3Dy1/3Ho1/3CrO3, which is compared with the PC base system. The results shown below indicate that the dielectric properties are slightly modified with the equimolar substitutions of Pr, Dy, and Ho. However, not magnetic properties, which show that the O–Cr–O angles tune the TN, and the negative magnetization in the ZFC mode prevails below TN, and it is discussed in terms of the Γ2(FxCyGz) magnetic configuration.
Experimental procedure
Powder PrCrO3 and the medium-entropy ceramic Pr1/3Dy1/3Ho1/3CrO3 (hereafter called PC and MECs-PDH) samples were synthesized by the combustion method. Stoichiometric quantities of Pr6011 (Sigma Aldrich 99.9%), Ho(NO3)3.5H2O, Dy(NO3)3.xH2O, and Cr(NO3)3.9H2O were mixed to form the compounds under study according to the following reaction:
first, the stoichiometric Pr6O11 was previously dissolved in 1:1 HNO3 (Sigma-Aldrich 70%). Nitrate cations, and praseodymium previously dissolved were stirred, dissolved in deionized water, and gradually heated at 150 °C. The final solution containing the homogeneous cations was mixed with 10 mL (1 g: 10 mL) of 2-methoxiethanol (Sigma-Aldrich 99.8%). Then and then the cations in the solution were concentrated by evaporation and diluted (adding deionized water) several times until the excess of HNO3 was completely removed. Finally, the dried gel was burnt in self-propagation combustion to form a loose fine powder. The result combustion powder was calcinated at 1200 °C for 6 h in the air. The crystal structure of the samples was studied by X-ray diffraction (XRD) using a Bruker D2 phaser diffractometer, Bragg–Brentano (2θ-φ scan) para focusing geometry, applying Cu-Kα (10 mA, 30 kV) radiation. The XRD measurement was carried out with a 0.02° step in a range of 10–80° for 2θ at room temperature. The crystallographic phase was identified by comparison with the X-ray patterns of the ICSD database. Rietveld refinement was performed to determine the structural features for each composition using the TOPAS 3.1 software for powder diffraction data. Microstructure, EDX, and elemental mapping analysis in a JEOL 5400LV operated at 20 kV were carried out on MECs-PDH sample. A Perkin Elmer Lambda 365 UV–Vis spectrophotometer coupled with an integrating sphere accessory was used to find the optical band gap (Eg) for PC and PDH samples. The emission spectra were collected in the 200–1100 nm spectral range. The samples were palletized in stainless steel die (∅ = 6 mm) and sintered at 1350 °C for 8 h. Samples sintered were silver painted on the surface and cured in a furnace with a thermal program from room temperature to 500 °C at a heating rate of 10°/min. We use an LCR bridge (HP-4284A) to obtain the dielectric spectrum from room temperature to 550 °C in the 1–100 kHz frequency range. Magnetic measurements were obtained using SQUID based vibrating sample magnetometer (MPMS 3, Quantum Design). To minimize the remanent magnetization into the equipment’s superconductor magnet, and assure no magnetization perturbation remained in the sample, the external applied field was always switched back to zero in a 10 Oe/sec rate by using an oscillate approach at the end of each measurement.
Results and discussion
Crystal and microstructural features
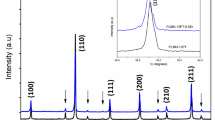
Figure 1 (Upper panel) shows the X-ray diffraction patterns of the medium-entropic MECs-PDH along with PC samples (Bottom panel). The diffraction peaks in both samples were Rietveld fit, assuming an orthorhombic perovskite distorted structure with the Pnma space group. The absence of extra peaks confirms the phase purity of the samples. Crystallographic data from the final refinement as well as the corresponding set of the reliability factors, such as weighted profile R-factor, expected R-factor, and goodness of fit (Rwp, Rexp, and χ2) are presented for both compounds in Table 1. It is seen that the Dy and Ho substitution in equimolar quantities decreases the ‘b’ and ‘c’ lattice parameters with an increase of the ‘a’ lattice parameter. The net result is a decrease in the volume cell from 230.64 for PC to 222.46 Å3 for the MECs-PDH sample. It is reasonable to expect a decrease in the volume cell for the MECs-PDH concerning the PC compound since 66% of the cations at the A-site have been occupied with smaller ionic radii (Dy = 1.083 Å, Ho = 1.072 Å) than that of the Pr (1.179 Å) cation. The tolerance factor (t) is determined from the ionic radii of the perovskite as \(t = \left( {r_{{\overline{R}^{3 + } }} + r_{{O^{2 - } }} } \right) / \sqrt 2 \left( {r_{{Cr^{3 + } }} r_{{O^{2 - } }} } \right)\). Here \(\overline{R}\) 3+ is the ionic radii average of rare earth cations, taking a coordination number of IX from Shannon’s tables [31]. Thus, the calculated tolerance factor is about 0.882, slightly smaller than 0.907 for the PC compound. In addition, the changes in the octahedral, CrO6 environment are evaluated through bond-length, bond-angle, and tilting in the [010] and [101] direction, and they are listed in Table 1 for PC and MECs-PDH samples, respectively. We deduce from those internal crystallography parameters that the cell volume decrease by substituting the smaller ionic radii is mainly led by the two bond-angles, Cr – O- Cr (see inset of Fig. 1), instead of bond-length. While a slight decrease in the average bond-length, < Cr–O > undergo from 1.973 for PC to 1.967 Å for MECs-PDH, a notable decrease occurs in the bond-angles, Cr–O–Cr from 159.6 (19) to 153.7(9) degrees in the [010] direction, and from 155.7 (16) to 150.60 (6) degrees in the [101] direction for PC and MECs-PDH samples, respectively. This fact increases the tilting from 10.2 to 13.5 [010] and 9.8 to 11.42 [101], respectively. Note that tilt values for MECs-PDH are close to those reported for DyCrO3 and HoCrO3 compounds [18, 32]. Figure 2 shows the EDS spectrum and the surface scanning elemental mapping on the MECs-PDH pellet sample to investigate the distribution and homogeneity of the elements. The analysis demonstrates that all elements are uniformly distributed without any segregation or aggregation in the form of clusters or precipitates. The theoretical and experimental weight percentage ratios listed in the inset of Fig. 2 show that they are within the stoichiometric ranges considering the accuracy of the EDS technique.

Fitting results of the Rietveld analysis at room temperature for the MECs-PDH (Upper panel) and PC samples (Bottom panel). Each Rietveld fitting shows the experimental ( +), theoretical (—), and the difference between the observed and calculated patterns. The inset shows the orthorhombic crystal structure showing the φ and θ tilting in the [010] and [101] direction.

SEM micrograph and EDS spectrum, elemental mapping images, and elemental chemical analysis for medium entropic MECs-PDH ceramic.
Optical characterization
The design of new materials requires knowledge of optical properties to provide good performance in optoelectronic device applications [33]. The interaction between light and matter can cause electrons to move from one energy level to another, creating an electrical signal or absorbing/emitting light. Thus, in this section, through the UV–Vis spectra, we analyze the optical transitions and energy band gap for the PC and the MECs-PDH samples. Figure 3a shows the UV–Vis reflectance spectra around the ultraviolet (200–800 nm) range for PC and the medium-entropy PDH samples. It can be noticed from the spectra that the reflectance in the visible region is higher for the MECs-PDH sample compared to the PC sample. In addition, the spectra show five main broad dips localized at ~365, ~ 454, ~ 612, ~ 692, and ~ 732 nm, being these signals stronger for the MECs-PDH sample (vertical dash line). The first signal at 365 nm is ascribed to the energy band gap (Eg). The more prominent two signals at ~ 454 and ~ 612 nm have been attributed to two optical transitions of Cr3+(d3) in an octahedral crystal field of CrO6 that correspond to: 4A2g → 4T1g and 4A2g → 4T2g in the visible spectrum. These two transitions have been observed in several orthochromites [5, 34,35,36], and all of them have been related to the effect of the crystal field in the Cr3+(d3) octahedral environment of the perovskite. At higher wavelengths shows two more weak transitions, which correspond to 4A2g → 2T1g (~ 612 nm) and 4A2g → 2Eg (~ 732 nm) related to spin-forbidden [5, 37, 38].

a The reflectance (R) variation as a function of the wavelength and b the Tauc’s relation graph (F(R) hν)2 vs energy plot) for the PC, and (Pr1/3Dy1/3Ho1/3)CrO3 samples.
The energy band gap (Eg) is evaluated by Kubelka–Munk (K-M) function [39, 40] for the MECs-PDH, and it is compared with PC samples, given by;
R is the reflectance, α is the absorption coefficient, and S is the dispersion coefficient. The Eg can be evaluated using the K-M equation, where the electronic properties can be found in a large number of semiconductor materials generally applied to highly light-scattering and absorbing particles. The dispersion coefficient is weakly energy-dependent and within the energy range contained by the absorption edge. It can be assumed that F(R) is proportional to α by;
where A is a proportional constant, hν is the photon energy, and the power factor “n” takes values of 2 for the direct band gap and ½ for the indirect band gap. R. Mguedla et al. [41], suggested a direct optical band gap in the PC ceramic compound by using and rearranging the Tauc´s relation (Eq. 5). Thus, the direct energy band gap is evaluated for both compounds through [F(R)*hν]2 vs. photon energy plot [42], as is shown in Fig. 3b. Extrapolating the drop curves to α = 0 yielded Eg values of 3.15 and 2.90 eV for MECs-PDH and PC, respectively. Note a slightly larger value, of around 0.25 eV, for the entropic system concerning the PC compound. This slight difference in the Eg should be related to the structural deformations caused by the equimolar disorder of the cations within the matrix. However, it is worth noting that the Eg values are in good agreement with that deduced from the experimental reflectance spectrum even with that calculated from density-functional theory for homolog compounds [33, 43, 44]. Once revealed the electronic properties through the energy band gap, it is imperative to reveal the dielectric properties in both compounds.
Dielectric properties
Frequency dependence of dielectric constant (ε’) and dielectric loss (tan δ) at 50, 100, 200, and 300 °C for MECs-PDH and PC samples are shown in Fig. 4a and b. In both compounds, the permittivity and dielectric loss curves show a very similar behavior where both magnitudes decrease rapidly with frequency, being more pronounced at low frequencies and high temperatures. In bulk ceramics, particularly orthochromites, this is due to space charge polarization at the grain boundaries generating a potential barrier. This fact led to higher values of the ε’ and tan δ. The dielectric dispersion at low frequency can be explained by the dominance of grain boundaries effect rather than by the grains. This behavior is attributed to the Maxwell–Wagner (MW) type interfacial polarization [36, 45, 46]. The giant ε’ values and higher Tan δ at low frequencies asserts the MW contribution in both systems. L. Boudad et al. [45], by impedance analysis, showed that the decrease and merging curves at higher frequencies could be due to the release of space charge polarization. Table 2 compares the magnitudes of the permittivity (ε), the dielectric loss (δ), and electric conductivity (σ) in both compounds. These results show that the cations configurational disorder does not have a significant effect on both physical parameters.

Dielectric constant (ε´) and loss tangent (Tan δ) as a function of frequency at 50, 100, 200, and 300 °C a P C and b MECs-PDH samples.
Magnetic properties
Temperature-dependence of the ZFC and FC magnetic susceptibility under applied fields of 0.1 and 1 kOe for PCO and MECs-PDH samples is shown in Fig. 5. Note that in both systems, unusual behavior in the ZFC magnetic ground states is displayed. For the PC sample, the susceptibility shows a large splitting in the ZFC and FC curves below ~ 239 K. It has been reported that the antiferromagnetic G-type structure with spin canting of the Cr+3 (S = 3/2) ion occurs below ~ 240 K for PC [19, 47, 48]. However, the equimolar Pr, Dy, and Ho substitution produces a decrease of the TN down to 171 K. On the other hand, the ZFC magnetic susceptibility curves show negative magnetization, which are kept up to 1 kOe, taking a value of ~ −8 emu/mol for the PC sample. On the contrary, the negative magnetization in the ZFC-mode is weaker, taking values of ~ –4.3 emu/mol, and positive magnetization values at 1 kOe are maintained up to 100 Oe. However, it is reversed to positive magnetization when the applied magnetic field reaches 1 kOe in the ZFC-mode for the MECs-PDH sample. The inverse susceptibility, χ−1–T for PC and -MECs-PDH, is shown in the inset of the bottom graph of Fig. 5.

Molar susceptibility (χ) as a function of temperature (T) at an applied magnetic field of 100 and 1000 Oe for PC and MECs-PDH ceramic compound. Insets show the inverse molar susceptibility (χ−1) with temperature (T) for both compounds.
Analyzing the paramagnetic state (T > TN), the values of the magnetic parameters are obtained from the linear fitting of χ−1- T using the Curie–Weiss law; χmol(T) = C/(T-Θcw) where C is the Curie Constant and Θcw is the Curie–Weiss temperature. The experimental values extracted from the fitting allow us to estimate the effective moments on the A-site through the following equations [49];
Equations 6 and 7 are used for PCO and MECs-PDH samples, respectively. The μeff obtained from the fitting and those calculated through both equations are tabulated in Table 3. For PCO, the effective moment on the A-site (Pr3+) is calculated (Eq. 6) from the experimental effective moment and assuming that the µCr3+ takes the theoretical value of 3.87 μB [50], which results in a value of µeff = 3.80 μB. The value is close to the magnetic ground state value for Pr3+ion (3.58 μB; S = 1) [50]. In the same way, the value of µCr3+ is calculated from the total effective moment. It assumes the theoretical value of µPr3+ (3.58 μB), which results in an effective moment value of 4.08 μB, also close to the theoretical value for Cr3+ ion (3.87 μB S = 3/2) [50]. For the case of MECs-PDH, where rare-earth cations share 1/3 on the A-site and using Eq. 7 assuming the magnetic ground state for the Cr3+ and the involved rare-earth, we obtain the theoretical magnetic moment on the A-site and the total effective moment of 8.27 and 9.13 μB, respectively. The experimental data for the MECs-PDH sample shows a total effective moment of 9.32 μB, which is quite close to the calculated theoretical value [50]. In a similar way to what was done for PCO, using the experimental magnetic moment and assuming the theoretical value of µCr3+, the magnetic moment on the A-site can be estimated, which results in 8.47 μB, which is also close to the calculated theoretical value [50]. The contribution of each ion to the total magnetic moment can also be obtained, assuming that the rest of the ions take the theoretical magnetic value. For example, for µPr3+, solving Eq. 7\({\upmu }_{{Pr^{3 + } }} = 3\left[ {({\upmu }_{eff} )^{2} - \left( {{\upmu }_{{Cr^{3 + } }} } \right)^{2} } \right]^{1/2} - {\upmu }_{{Dy^{3 + } }} - {\upmu }_{{Ho^{3 + } }}\) results in a value of 4.2 μB. Following the same procedure, we obtain 11.25, 11.22, and 4.29 μB. for Dy3+, Ho3+ and Cr3+ ions, respectively. These values are close to the theoretical effective moments, and they are listed in Table 3. This fact infers that the magnitude of the magnetic moments in the paramagnetic state of each one of the subcells corresponds to the magnetic moments of the rare earth cations as well as the Cr3+ (d3) ions distributed homogeneously and randomly in bulk. The paramagnetic state changes to antiferromagnetic ordering at ~ 239 and ~ 171 K and is observed as a drop in the χ−1-T curves for the PCO and MECs-PDH samples, respectively (Inset Fig. 5). At these temperatures, the G-type antiferromagnetic order of the Cr3+-magnetic moment occurs. It is well known in rare-earth orthochromites that TN values decrease almost linearly from 288 K for LaCrO3 to 110 K for LuCrO3, which is closely related to the ionic radius of the rare-earth at the A-site. It is accepted that the Cr -O -Cr bond angle is reduced from 180° due to cooperative octahedral tilting, which affecting the superexchange coupling and reducing the TN. As mentioned above, the equimolar Pr, Dy, and Ho substitutions produce a TN of ~ 171 K, which is close to ~ 175.6 K, taking into consideration the TN average of PrCrO3 (~ 239 K), DyCrO3 (~ 147 K), and HoCrO3 (~ 141 K) [35, 48, 51]. It is evident that Dy and Ho ions drive the drastic drop of the TN through Cr−O–Cr bond angles from 239 for PC to 171 K for the MECs-PDH through average structural deformation affecting the antiferromagnetic transition. From the geometrical point of view, the Rietveld refinement data shows a decrease of the volume cell from 230.64 Å3 for PC to 222.46 Å3 for MECs-PDH- accompanied by a tilt angle increase along to [0 1 0] in Pnma S.G. from 10.2 for PC to 13.15 for MECs-PDH. These structural and antiferromagnetic transitions agree with the GdCrO3 reporting a volume cell of ~ 223 Å3 with a tilt angle of about ~ 10.8, which leads to an antiferromagnetic order, TN ~ 170 K [32, 48, 52].
Analyzing the magnetic regimen below TN, Fig. 5 shows a large splitting below 239 K and 171 K in the ZFC and FC curves at a low applied magnetic field (100 Oe) for PCO and MECs-PDH, respectively. Although the negative magnetization in the ZFC mode has created controversy in other systems, it has been proven to be an artifact [53]. However, the result shown here demonstrates that this is not the case and is consistent with other studies in PC compounds [19, 54, 55]. A recent study [19] discussed that the negative magnetization from 2 to TN ~ 239 K in the ZFC mode is intrinsic behavior in the PC compound. There, we infer that the primary mechanism that causes this phenomenon is related to the Bertaut Γ2(FxCyGz) magnetic configuration as well as the poor correlation at low temperature between the paramagnetic rare earth sublattice and the antiferromagnetic order of the Cr-sublattice. This assumption motivates us to design a new material using the high entropy configurational approach. Thus, we find that DyCrO3 and HoCrO3 possess identical antiferromagnetic Γ2(FxCyGz) configurations as PC from 2 K to TN [56]. P. Gupta et al. [27] and Y. L. Sue et al. [29] report a large negative magnetization in the ZFC mode below 147 K for DyCrO3 and 141 K for HoCrO3, respectively. Here, we hypothesize, If the Γ2(FxCyGz) magnetic configuration is responsible for this behavior on the PC compound, then the equimolar MECs-PDH ceramic compound should keep the Γ2(FxCyGz) magnetic configuration. If so, this magnetic setting should strengthen or at least keep up the negative susceptibility below TN. In Fig. 5, the negative susceptibility infers two crucial statements in the equimolar MECs-PDH ceramic compound. The first suggests that the Γ2(FxCyGz) magnetic configuration is maintained homogeneously in the orthorhombic structure, and the second confirms that the involved mechanism inducing the negative magnetization in the ZFC mode is strongly related to the Γ2(FxCyGz) magnetic configuration. Recently, T. Sau et al. [57], studying the unusual behavior of SmCrO3 by neutron diffraction measurements, revealed that the Γ2(FxCyGz) magnetic configuration produces a local field in the z-direction (Gz), which could be responsible for the drop in magnetization around 40 K when spin reorientation occurs from Γ4(FzAyGx) to Γ2(FxCyGz). We infer that this local field, fundamentally in the Γ2(FxCyGz) magnetic configuration, is responsible for the negative magnetization in the FC mode and under mechanisms not yet well understood in the ZFC. Furthermore, the equimolar random distribution of net magnetic moments of Pr, Dy, and Ho weaken the opposing uniaxial magnetic component (Gz) since the negative magnetization is not kept at an applied magnetic field of 1 kOe compared with the PC compound. The experimental results shown here indicate that when turning on the applied magnetic field (below 100 Oe) in ZFC mode, the Pr, Ho, Dy sublattice, and the Cr sublattice are uncorrelated, feeling the same applied magnetic field. However, an internal field at the Cr-sites as a consequence of Γ2(FxCyGz) spin configuration exceeds the applied magnetic field during ZFC mode, leads to negative magnetization. As we have mentioned in previous studies [18, 19, 58], this hypothesis should be corroborated with neutron diffraction studies.
To complete the magnetic study, Fig. 6 shows magnetization hysteresis curves as a function of applied magnetic field (± 40 kOe) at 20 K for PC (upper panel) and MECs-PDH (bottom panel) samples. As expected, a robust ferromagnetic contribution as well as an exchange bias effect (EB), as a result of robust magnetic exchange anisotropy, is observed in the PC sample. The robust exchange anisotropy (EB effect) results from an exchange coupling between interfaces of weak ferromagnetic and antiferromagnetic domains in the bulk PC sample. The interfacial exchange anisotropy is observed as a shift of the coercive field (Hc) towards the right side of the upper M-H curve of Fig. 6. Notice that the positive EB is unusual since in homologous compounds, the EB is generally negative [18, 58]. This effect can be qualitatively explained by a strongly ferromagnetic coupling of uncompensated magnetic moments at the AFM-FM interface during the applied magnetic field [59]. However, the magnitude of these magnetic features is not maintained when equimolar ratios of Dy and Ho are shared with the Pr site even though all three sublattices have a ferromagnetic (Fx) contribution in the antiferromagnetic ordering of the Cr3+ sublattice. The decrease in both the weak hysteresis buckle in the magnetization curve and the exchange bias effect indicates that the weak ferromagnetic and the exchange anisotropy also decrease in the collinear antiferromagnetic ordering (Gz,Cy) in the MECs-PDH compound. This fact infers that the local deformation as a consequence of the difference in the ionic radii of cations in the A-site tends to cancel the robust ferromagnetic contribution (Fx) as well as the exchange anisotropy (EB) that occurs when only Pr exists in the orthochromite compound.

Magnetization as a function of an applied magnetic field at 20 K in the ZFC mode for PC and PDH-MECs. The inset (bottom panel) shows an expanded scale at a low applied magnetic field for the MECs-PDH sample.
Conclusions
Medium-entropy MECs-PDH and PC ceramics were successfully synthesized by combustion method. The volume cell decreased for the PDH-MECs sample concerning the PC compound. Surface scanning elemental mapping showed homogenous random cations distribution on A-sites of the MECs-PDH crystal structure. The cationic disorder does not have a significant effect on optical transitions in the electromagnetic visible range. However, it does have an energy band gap from 2.90 for PC to 3.15 for MECs-PDH. The permittivity, dielectric loss, and electric conductivity from RT to 300 °C showed that the configurational disorder of the cations does not have a significant effect on these physical properties. In contrast, the magnetic properties explored at low temperatures reveal that such cationic disorder affects the antiferromagnetic transition and the magnetic susceptibility below TN. The drop of the TN from 239 for PC to 171 K for the MECs-PDH is driven by the Dy and Ho substitution trough of the decreasing of the O – Cr – O bond angle. It is found that the negative ZFC prevails in the MECs-PDH sample, which strongly infers that this unusual magnetic feature is an intrinsic condition related to the Γ2(FxCyGz) spin configuration. In summary, the present study shows that the high entropy approach in designing new materials can provide new evidence of controversial magnetic phenomena that motivate researchers to develop a more comprehensive theory of reversal magnetization from practical and theoretical points of view.
Data and code availability
The data generated or used during the study are available from the corresponding author by request.
References
Ohkoshi SI, Abe Y, Fujishima A, Hashimoto K (1999) Design and Preparation of a Novel Magnet Exhibiting Two Compensation Temperatures Based on Molecular Field Theory. Phys Rev Lett 82:1285–1288. https://doi.org/10.1103/PhysRevLett.82.1285
Yamaguchi T, Tsushima K (1973) Magnetic symmetry of rare-earth orthochromites and orthoferrites. Phys Rev B 8:5187–5198. https://doi.org/10.1103/PhysRevB.8.5187
Pascual-Gonzalez C, Schileo G, Murakami S, Khesro A, Wang D, Reaney IM, Feteira A (2017) Continuously controllable optical band gap in orthorhombic ferroelectric KNbO 3 -BiFeO 3 ceramics. Appl Phys Lett 110:172902. https://doi.org/10.1063/1.4982600
Durán A, Escamilla R, Escudero R, Morales F, Verdín E (2018) Reversal magnetization, spin reorientation, and exchange bias in YCrO3 doped with praseodymium. Phys Rev Mater 2:014409. https://doi.org/10.1103/PhysRevMaterials.2.014409
Tavizon G, Barreto J, Mata-Ramírez J, Huerta L, Arenas J, de la Mora P, Duran A (2022) Magnetic and electrical properties by Ca2+ doping in SmCrO3 orthochromites. J Alloys Compd 890:161823. https://doi.org/10.1016/j.jallcom.2021.161823
Jacobs IS, Burne HF, Levinson LM (1971) Field-induced spin reorientation in YFeO3 and YCrO3. J Appl Phys 42:1631–1632. https://doi.org/10.1063/1.1660372
Kaneko M, Kurita S, Tsushima K (1977) Optical observation of spin reorientation in ErCrO 3. J Phys C Solid State Phys 10:1979–1985. https://doi.org/10.1088/0022-3719/10/11/032
Chu Y-H, Martin LW, Holcomb MB, Gajek M, Han S-J, He Q, Balke N, Yang C-H, Lee D, Hu W, Zhan Q, Yang P-L, Fraile-Rodríguez A, Scholl A, Wang SX, Ramesh R (2008) Electric-field control of local ferromagnetism using a magnetoelectric multiferroic. Nat Mater 7:478–482. https://doi.org/10.1038/nmat2184
Gajek M, Bibes M, Fusil S, Bouzehouane K, Fontcuberta J, Barthélémy A, Fert A (2007) Tunnel junctions with multiferroic barriers. Nat Mater 6:296–302. https://doi.org/10.1038/nmat1860
Scott JF (2007) Multiferroic memories. Nat Mater. https://doi.org/10.1038/nmat1868
Scott JF (2007) Applications of modern ferroelectrics. Science 315(80):954–959. https://doi.org/10.1126/science.1129564
Zhou J-S, Alonso J, Pomjakushin V, Goodenough J, Ren Y, Yan J-Q, Cheng J-G (2010) Intrinsic structural distortion and superexchange interaction in the orthorhombic rare-earth perovskites RCrO_{3}. Phys Rev B 81:1–5. https://doi.org/10.1103/PhysRevB.81.214115
Prado-Gonjal J, Schmidt R, Romero JJ, Ávila D, Amador U, Morán E (2013) Microwave-assisted synthesis, microstructure, and physical properties of rare-earth chromites. Inorg Chem 52:313–320. https://doi.org/10.1021/ic302000j
Dash BB, Ravi S (2016) Sign reversal of magnetization in Mn substituted SmCrO3. J Magn Magn Mater 405:209–213. https://doi.org/10.1016/j.jmmm.2015.12.047
Lee J-H, Jeong YK, Park JH, Oak M-A, Jang HM, Son JY, Scott JF (2011) Spin-canting-induced improper ferroelectricity and spontaneous magnetization reversal in SmFeO3. Phys Rev Lett 107:117201. https://doi.org/10.1103/PhysRevLett.107.117201
McDannald A, dela Cruz CR, Seehra MS, Jain M (2016) Negative exchange bias in single-phase. Phys Rev B 93:184430. https://doi.org/10.1103/PhysRevB.93.184430
Taheri M, Kremer RK, Trudel S, Razavi FS (2015) Exchange bias effect and glassy-like behavior of EuCrO 3 and CeCrO 3 nano-powders. J Appl Phys 118:124306. https://doi.org/10.1063/1.4931613
Durán A, Escamilla R, Escudero R, Morales F, Verdín E (2018) Reversal magnetization, spin reorientation, and exchange bias in doped with praseodymium. Phys Rev Mater 2:014409. https://doi.org/10.1103/PhysRevMaterials.2.014409
Mendivil LF, Alvarado-Rivera J, Tavizón G, Verdín E, Arenas-Alatorre J, Durán A (2021) Negative magnetization in the zero field-cooled and exchange-bias effect in Cu-doped PrCrO3. J Mater Sci Mater Electron 32:24484–24495. https://doi.org/10.1007/s10854-021-06926-y
Yoshii K, Nakamura A (2000) Reversal of Magnetization in La0.5Pr0.5CrO3. J Solid State Chem 155:447–450. https://doi.org/10.1006/jssc.2000.8943
Bora T, Ravi S (2015) Sign reversal of magnetization and tunable exchange bias field in NdCr1−xFexO3 (x=0.05–0.2). J Magn Magn Mater 386:85–91. https://doi.org/10.1016/j.jmmm.2015.03.060
Rajeswaran B, Khomskii DI, Zvezdin AK, Rao CNR, Sundaresan A (2012) Field-induced polar order at the Néel temperature of chromium in rare-earth orthochromites: Interplay of rare-earth and Cr magnetism. Phys Rev B 86:214409. https://doi.org/10.1103/PhysRevB.86.214409
Rost CM, Sachet E, Borman T, Moballegh A, Dickey EC, Hou D, Jones JL, Curtarolo S, Maria J-P (2015) Entropy-stabilized oxides. Nat Commun 6:8485. https://doi.org/10.1038/ncomms9485
Xiang H, Xing Y, Dai F, Wang H, Su L, Miao L, Zhang G, Wang Y, Qi X, Yao L, Wang H, Zhao B, Li J, Zhou Y (2021) High-entropy ceramics: Present status, challenges, and a look forward. J Adv Ceram 10:385–441. https://doi.org/10.1007/s40145-021-0477-y
Wright AJ, Luo J (2020) A step forward from high-entropy ceramics to compositionally complex ceramics: a new perspective. J Mater Sci 55:9812–9827. https://doi.org/10.1007/s10853-020-04583-w
Durán A, Sharma S, Brown F, Álvarez-Montaño VE (2022) Effect of sintering condition on crystal structure and dielectric properties of novel LuGa1/3Ti2/3O3+x/2 and RGa1/3Ti2/3O3+x/2 (R= Lu0.2Yb0.2Tm0.2Er0.2Ho0.2) high-entropy ceramic compounds: a comparative study. Mater Chem Phys 287:126307. https://doi.org/10.1016/j.matchemphys.2022.126307
Gupta P, Bhargava R, Das R, Poddar P (2013) Static and dynamic magnetic properties and effect of surface chemistry on the morphology and crystallinity of DyCrO3 nanoplatelets. RSC Adv 3:26427. https://doi.org/10.1039/c3ra43088j
Lei S, Liu L, Wang C, Wang C, Guo D, Zeng S, Cheng B, Xiao Y, Zhou L (2013) General synthesis of rare-earth orthochromites with quasi-hollow nanostructures and their magnetic properties. J Mater Chem A 1:11982. https://doi.org/10.1039/c3ta12281f
Su YL, Zhang JC, Li L, Feng ZJ, Li BZ, Zhou Y, Cao SX (2010) Novel magnetization induced by phase coexistence in multiferroic HoCrO 3 chromites. Ferroelectrics 410:102–108. https://doi.org/10.1080/00150193.2010.492729
Kumar N, Sundaresan A (2010) On the observation of negative magnetization under zero-field-cooled process. Solid State Commun 150:1162–1164. https://doi.org/10.1016/j.ssc.2010.03.016
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr Sect A 32:751–767. https://doi.org/10.1107/S0567739476001551
Late R, Rai HM, Saxena SK, Kumar R, Sagdeo A, Sagdeo PR (2016) Probing structural distortions in rare earth chromites using Indian synchrotron radiation source. Indian J Phys 90:1347–1354. https://doi.org/10.1007/s12648-016-0887-9
Mguedla R, A. Ben Jazia Kharrat, S. Kammoun, K. Khirouni, W. Boujelben, (2021) Optical studies of multiferroic HoCrO3 perovskite compound for optoelectronic device applications. Opt Mater 119:111311. https://doi.org/10.1016/j.optmat.2021.111311
Cruciani G, Ardit M, Dondi M, Matteucci F, Blosi M, Dalconi MC, Albonetti S (2009) Structural relaxation around Cr 3+ in YAlO 3 −YCrO 3 perovskites from electron absorption spectra. J Phys Chem A 113:13772–13778. https://doi.org/10.1021/jp9043072
Tiwari B, Surendra MK, Ramachandra Rao MS (2013) HoCrO 3 and YCrO 3: a comparative study. J Phys Condens Matter 25:216004. https://doi.org/10.1088/0953-8984/25/21/216004
Mguedla R, Ben Jazia Kharrat A, Saadi M, Khirouni K, Chniba-Boudjada N, Boujelben W (2020) Structural, electrical, dielectric and optical properties of PrCrO3 ortho-chromite. J Alloys Compd 812:152130. https://doi.org/10.1016/j.jallcom.2019.152130
Late R, Wagaskar KV, Shelke PB, Sagdeo A, Rajput P, Sagdeo PR (2020) Structural, optical, and dielectric investigations in bulk PrCrO3. J Mater Sci Mater Electron 31:16379–16388. https://doi.org/10.1007/s10854-020-04189-7
Alqahtani A, Husain S, Somvanshi A, Khan W (2019) Structural, morphological, thermal and optical investigations on Mn doped GdCrO3. J Alloys Compd 804:401–414. https://doi.org/10.1016/j.jallcom.2019.07.028
Kubelka P (1954) New contributions to the optics of intensely light-scattering materials part II: nonhomogeneous layers*. J Opt Soc Am 44:330. https://doi.org/10.1364/JOSA.44.000330
Yang L, Kruse B (2004) Revised Kubelka-munk theory I theory and application. J Opt Soc Am A 21:1933. https://doi.org/10.1364/JOSAA.21.001933
Mguedla R, Ben Jazia Kharrat A, Taktak O, Souissi H, Kammoun S, Khirouni K, Boujelben W (2020) Experimental and theoretical investigations on optical properties of multiferroic PrCrO3 ortho-chromite compound. Opt Mater 101:109742. https://doi.org/10.1016/j.optmat.2020.109742
Tauc J (1968) Optical properties and electronic structure of amorphous Ge and Si. Mater Res Bull 3:37–46. https://doi.org/10.1016/0025-5408(68)90023-8
Yaseen M, Ambreen H, Iqbal J, Shahzad A, Zahid R, Kattan NA, Ramay SM, Mahmood A (2020) Electronic, optical and magnetic properties of PrXO 3 (X = V, Cr): first-principle calculations. Philos Mag 100:3125–3140. https://doi.org/10.1080/14786435.2020.1812748
Singh KD, Pandit R, Kumar R (2018) Effect of rare earth ions on structural and optical properties of specific perovskite orthochromates; RCrO3 (R = La, Nd, Eu, Gd, Dy, and Y). Solid State Sci 85:70–75. https://doi.org/10.1016/j.solidstatesciences.2018.10.001
Boudad L, Taibi M, Belayachi W, Abd-Lefdil M (2022) Investigation of magnetic, dielectric, optical, and electrical properties of Fe half-doped PrCrO3 perovskite. J Solid State Chem 309:122933. https://doi.org/10.1016/j.jssc.2022.122933
Mguedla R, Ben Jazia Kharrat A, Moutia N, Khirouni K, Chniba-Boudjada N, Boujelben W (2020) Gd doping effect on structural, electrical and dielectric properties in HoCrO3 orthochromites for electric applications. J Alloys Compd 836:155186. https://doi.org/10.1016/j.jallcom.2020.155186
Gordon BM, Hornreich JD, Shtrikman RM, Wanklyn S (1976) Magnetization studies in the rare-earth ortochromites. V. TbCrO3 and PrCrO3. Phys Rev B 13:3012–3017
Sardar K, Lees MR, Kashtiban RJ, Sloan J, Walton RI (2011) Direct hydrothermal synthesis and physical properties of rare-earth and yttrium orthochromite perovskites. Chem Mater 23:48–56. https://doi.org/10.1021/cm102925z
Daniels LM, Weber MC, Lees MR, Guennou M, Kashtiban RJ, Sloan J, Kreisel J, Walton RI (2013) Structures and magnetism of the rare-earth orthochromite perovskite solid solution La x Sm 1–x CrO 3. Inorg Chem 52:12161–12169. https://doi.org/10.1021/ic402029u
Blundell S (2001) Magnetism in Condensed Matter. Oxford University Press, UK
Sibanda ET, Prinsloo ARE, Sheppard CJ, Mohanty P (2022) Structural and magnetic properties of DyCrO 3. AIP Adv 12:035342. https://doi.org/10.1063/9.0000337
Yoshii K (2001) Magnetic properties of perovskite GdCrO3. J Solid State Chem 159:204–208. https://doi.org/10.1006/jssc.2000.9152
Kumar A, Yusuf SM (2015) The phenomenon of negative magnetization and its implications. Phys Rep 556:1–34. https://doi.org/10.1016/j.physrep.2014.10.003
Venugopal RB, Vittal PB, Narsinga RG, Chou FC, Suresh BD (2015) Magnetization reversal in PrCrO3. Adv Mater Res 1086:96–100. https://doi.org/10.4028/www.scientific.net/AMR.1086.96
Zhang Y, Yao C, Fan Y, Zhou M (2014) One-step hydrothermal synthesis, characterization and magnetic properties of orthorhombic PrCrO 3 cubic particles. Mater Res Bull 59:387–393. https://doi.org/10.1016/j.materresbull.2014.07.049
Bousquet E, Cano A (2016) Non-collinear magnetism in multiferroic perovskites. J Phys Condens Matter 28:123001. https://doi.org/10.1088/0953-8984/28/12/123001
Sau T, Yadav P, Sharma S, Raghunathan R, Manuel P, Petricek V, Deshpande UP, Lalla NP (2021) High-resolution time of flight neutron diffraction and magnetization studies of spin reorientation and polar transitions in SmCrO3. Phys Rev B 103:144418. https://doi.org/10.1103/PhysRevB.103.144418
Mendivil LF, Alvarado-Rivera J, Verdín E, Díaz JA, Mata J, Conde A, Durán A (2020) Reversal magnetization and exchange bias effect of the nanocrystalline Yb1-xPrxCrO3 solid solution. Appl Phys A 126:574. https://doi.org/10.1007/s00339-020-03738-5
Nogués J, Lederman D, Moran TJ, Schuller IK (1996) Positive exchange bias in Fe-Fe bilayers. Phys Rev Lett 76:4624–4627. https://doi.org/10.1103/PhysRevLett.76.4624
Zhao Y, Weidner DJ, Parise JB, Cox DE (1993) Critical phenomena and phase transition of perovskite — data for NaMgF3 perovskite Part II. Phys Earth Plane Inter 76(1–2):17–34. https://doi.org/10.1016/0031−9201(93)90052−B
Acknowledgements
AD acknowledges National Autonomous University of México (UNAM) through the Support Program for Technological Research and Innovation Projects (PAPIIT), grants No. IN103623. The authors acknowledge V. Álvarez and F. Brown (UNISON) for assistance in the microscopic analysis; and E. Aparicio, P. Casillas, and I. Gradilla (CNyN) for technical support.
Author information
Authors and Affiliations
Contributions
AD: Experimental synthesis and global analysis, Writing – original draft, Writing, review & editing. ER: Formal magnetic analysis, review & editing. LFM: Rietveld analysis, review & editing. MG: Carry out Magnetic measurements, conception, analysis & review. EV Supervision, optical analysis, and review.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Handling Editor: David Cann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Durán, A., Reguera, E., Mendivil, L.F. et al. Negative magnetization in the medium-entropy Pr1/3Dy1/3Ho1/3CrO3 and PrCrO3 ceramics: comparative crystal structure, optic, dielectric and magnetic properties. J Mater Sci 58, 15021–15034 (2023). https://doi.org/10.1007/s10853-023-08955-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-023-08955-w