
Abstract
Shape-changing materials open an entirely new solution space for a wide range of disciplines: from architecture that responds to the environment and medical devices that unpack inside the body, to passive sensors and novel robotic actuators. While synthetic shape-changing materials are still in their infancy, studies of biological morphing materials have revealed key paradigms and features which underlie efficient natural shape-change. Here, we review some of these insights and how they have been, or may be, translated to artificial solutions. We focus on soft matter due to its prevalence in nature, compatibility with users and potential for novel design. Initially, we review examples of natural shape-changing materials—skeletal muscle, tendons and plant tissues—and compare with synthetic examples with similar methods of operation. Stimuli to motion are outlined in general principle, with examples of their use and potential in manufactured systems. Anisotropy is identified as a crucial element in directing shape-change to fulfil designed tasks, and some manufacturing routes to its achievement are highlighted. We conclude with potential directions for future work, including the simultaneous development of materials and manufacturing techniques and the hierarchical combination of effects at multiple length scales.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Complex multifunctional materials are common within the natural world across all length scales and taxa: from the high tensile strength of spider silk [1] and the compressive properties of bone [2], to the iridescence of the butterfly wing [3] and the fast actuation of cuttlefish skin [4]. These materials display intricate architectures across the nano-, micro- and mesoscales, allowing for an impressive array of tailored functional materials properties. Natural materials have long been an inspiration to the materials chemist, physicist and engineer, and using the combined toolkits of synthetic chemistry and advanced fabrication techniques, many of these materials have been successfully replicated and, in some cases, improved upon in the synthetic world (e.g. a gecko-inspired adhesive which, unlike the original lizards, also adheres underwater) [5].
However, an area where nature still outperforms our current synthetic capabilities is that of active, self-shaping and stimuli-responsive materials. Material structures found in nature must be more than just a static object, framework or skin. Natural structures need to be triggered under certain conditions, or by external stimuli, for basic biological functions to occur, and where possible exhibit multiple capabilities. What is remarkable, however, is that the variety of ways that this can be achieved across the plant and animal kingdom takes place with relatively limited chemical diversity and with large structural changes triggered under mild, ambient conditions. This forms a severe contrast to the variety of synthetic molecules and extreme environments available to human designers and suggests the existence of a wide solution space once key biological principles are extracted and understood.
Naturally, the properties of actuating materials inform the potential for further development and refinement of response, and ultimately applications. Many studies of shape memory alloys and piezoelectric materials, some of the most prominent shape-changing materials, exist—–see Mohd Jani [6] and Irschik [7]—but these differ radically from the natural solutions seen around us. Therefore, in keeping with the biomimetic theme of this review, the materials focused on here will be ‘soft’ actuators— soft materials capable of changing their shape in response to stimuli. Soft materials are usefully characterised by Doi as having fundamental ‘building blocks’ on the order of 10–10,000 Å and thus having relatively slow response times between 1 and 104 s , a high response to stimuli that necessitates non-linear description and being easily driven out of equilibrium [8]. The subsection of soft materials that behave as actuators, capable of moving themselves or their surroundings, forms the topic of this review. As examples, this includes materials such as muscle and unfolding petals.
While soft materials depart from the traditional materials of engineering and may seem to lack robustness, they also offer novel advantages, as detailed by Quake and Scherer [9]. They are efficient to manufacture at small scale and they can be easily integrated with hard elements as required, for instance as a valve seal. In devices, soft materials give increased compatibility with human and animal users, and greater adaptability which confers the ability to operate in unpredictable, extreme environments [10, 11]. Such behaviour has many prospective applications, from biomedical devices [12] and passive building materials [13], to microfluidics [14], product design [15] and soft robotics [10, 16].
In this article, the paradigm of self-shaping and actuating materials found in nature will be introduced, to lay the groundwork for a review of the current literature on soft synthetic analogues. Candidate materials for biomimicry of response will be identified, along with their mechanisms, limitations and triggers. It will be seen that control of structure, and therefore available manufacturing possibilities, will be crucial to realise the goal of synthetic morphing materials.
Benchmarking natural materials against synthetics
There are many examples of actuation response of natural materials to changes in environmental humidity. This can range from the simple curling of a wet leaf as it dries, to more complex processes such as self-burial of seeds [17]. The passive opening and closing of the scales of a pinecone is an elegant example of hygroscopic actuation for seed dispersal, accompanied by a simple synthetic model. The reversible movement of the pine cone scales is driven by differences in structural orientation of the cellulose microfibrils within cell walls across the structure. The microfibrils have a greater resistance to extension along their axis of alignment, and so by varying this angle relative to the body of the cell, deformation of the volume can be channelled preferentially in one direction. Cells making up the outside of the scale are orientated to elongate on exposure to humidity, whereas the inner layer are more resistant to elongation [18].
This leads to a behaviour which has been compared to that of a thermally responsive bimetallic strip, which undergoes bending due to the differing thermal responses of the two constituent metals bound together [18, 19]. This is a familiar and tractable model in engineering and physics with a long history. First modelled by Timoshenko in 1925 [20], bimetallic strips were a common control switching element before being replaced with modern relays. The form has, from the very early work on shape-changing hydrogels by Hu [21], been a simple test-case for soft shape-changing materials. Since then research in the area has blossomed, using techniques including chemical variation [21], variation in cross-linking density [22], and alignment of restrictive elements [23] to achieve controlled movement, as discussed in more detail later in this manuscript.
Cellulose catapults: a natural example
In an effort to overcome limitations in chemical diversity and operating temperature, nature programmes the sequential arrangement and orientations of individual structural units, while simultaneously tailoring their function and attachment strategies to maximise the design for survival and longevity. A key element is the transmission and amplification of small degrees of strain across the length scales. As will be described below, this can translate a relatively simple and modest response to external stimuli into large-scale movements such as twisting, bending and opening.
Many plants exploit the principle of cellulose orientation directing moisture driven deployment, from the opening of eudicot seedpods [24] to the unfurling of the desert resurrection plant [19]. More complex behaviour is produced with more elaborate cellulose arrangements, and closer examination of this illuminates principles that may be helpful for reproducing their properties.
Erodium circodia, commonly known as redstem filaree (among many other names), is a small Mediterranean plant related to the geranium. Filarees disperse by firing their seeds around half a metre, powered only by the arrangement and drying of cellulose and lignin. The awn, or seed delivery mechanism, of E. circodia is a thin support which attaches the seeds to the main body of the plant, and as it desiccates curls into a spring, storing elastic energy. At a critical point, this material cracks and the elastic energy is released. This behaviour is illustrated in Fig. 1, as recorded and analysed by Evangelista et al. [17].

The capability of natural shape-change. a Representative launch trajectory of the filaree awn, as recorded by Evangelista et al., shown by multiple exposures at 4-ms intervals. Movement is from left to right. Scale bar 10 cm. b Trajectory predicted by modelling, matching the initial launch trajectory [inset box, seed (green) and tip (red) positions shown at 2-ms intervals] and final distance thrown (blue star). Reproduced from Evangelista et al. with permission from the Company of Biologists. [17] Copyright 2011
Within the awn, each constituent cell is encircled by cellulose in a helical arrangement. Importantly, the axis of the helix is not aligned with the axis of the cell, causing the cell itself to bend such that it packs in a helical form. Thus, the off-axis alignment of the cellulose nanostructure instigates a similar directionality in the cellular alignment, which results in the macroscopic coiling effect seen [25]. As the material dehydrates, the coil becomes tighter, storing more and more tension until the material eventually fractures. This has been successfully mathematically modelled by Aharoni et al. [26] and in a simplified version reproduced in a physical model by Abraham et al. [25]. This latter work demonstrates that one does not need to reproduce all the elegant details of the natural solution to draw on the concept.
This is all achieved with cellulose microfibrils embedded in a matrix of polysaccharides, aromatics and structural proteins—materials far removed from the inorganic chemical palette of most modern engineers. For comparison purposes, we compared the range of an equivalent volume to the filaree seed and awn (Fig. 1), composed of a variety of natural and synthetic shape-changing materials, if this were to be launched under the propulsion of its own storage energy. This provides a way to parametrise the energy stored in a fixed unit volume, while building in a penalty for greater weight. Thus, the energy density per unit weight is converted into an easy-to-visualise range of lengths, highlighting the capability of natural, low-density materials. Comparative values are shown in Fig. 2.

Comparing the capabilities of various natural and synthetic shape-changing materials, using the example of ballistic seed dispersal, which incorporates both the capacity for elastic energy storage and an allowance for density. They are compared to the natural example of the filaree, which has an energy density of approximately 750 kJ/kg and a mass of 5 × 10−3g, enabling projection 2.62 m (without drag). Literature values and calculations of energy density within the Hookean region are used to extrapolate range for plant (solid green), animal (dashed coral), soft and hard synthetic (dotted light and dark grey, respectively) materials. Derivation is based on the calculations of Evangelista et al., simplified to neglect drag and assume a constant volume. [17] Full details of sources and values are given in Supplementary Information
Our figures are derived from some simple assumptions, and the application of conservation of energy and Newtonian mechanics. We assume a cylindrical volume of which undergoes uniform contraction. Values for the stored energy contained within such a volume are drawn from a variety of sources (see supplementary information) and are drawn from both active contributions (for materials such as muscle and shape memory polymer) and passively stored elastic energy (for materials such as gels and spider silk, for which the highest strain value which remains in the Hookean region is used). These are, where necessary, converted into energy per unit mass using available measures of density (sources in supplementary information). We then assume an equivalent percentage of stored strain energy as in the filaree is converted into rectilinear motion—2 % as calculated by Evangelista et al. Finally, we take a constant launch angle and, neglecting drag, use standard calculations of projectile motion to find the range of a synthetic awn and seed.
This is not presented as a detailed analysis or a design strategy. But nevertheless it provides a simple comparison between actuating materials of different classes in terms of energy available for self-motion and highlights some features of interest. The potential of soft biological materials such as filaree and tendon is clear in this context. Their advantage derives from their low density—a crucial point for self-shaping materials that are required to move their own weight, as in many realistic applications. In synthetic competitors, both shape memory polymer and hydrogel are comparable to the best plant and animal material energy storages shown, with only dragline spider silk—a highly anomalous material which has resisted many attempts to analyse and reproduce—exceeding the capacity of our most successful synthetic by this (admittedly artificial) metric, Kevlar. Both Kevlar and spider silk are liquid crystalline in solution and achieve alignment during the process of spinning extrusion, from which their properties derive [27].
These back-of-the-envelope considerations suggest some initial areas of interest for a researcher looking into self-shaping materials through a biological lens. Gels suggest the potential of low-density, soft materials; memory polymers show a promising high-energy density; and Kevlar and spider silk suggest the importance of molecular alignment.
It can be seen that of the animal tissues that exist inside the organism (thus exempting spider silk), pure elastin has the best performance. However, in nature, this must be used as a composite, due to the need for biological tissues to meet multiple performance requirements. This reduces their energy storage capacity somewhat. Analogously, the high theoretical performance of carbon nanotubes is unlikely to translate wholly into a practical material. However, performance can reasonably be expected to match or exceed the performance of CNT yarn, which again compares well with elastin. The ability of these efficient molecules to improve performance, even at low proportions in the material, hints at the potential of nanoinclusions and other composites.
We therefore see that to emulate the complex actuation observed in the stork’s bill awn and other natural morphing systems, we require knowledge of the inherent properties of the material (the swelling matrix), the stimuli (the conditions or trigger required, for example the magnitude of dehydration the plant will experience), the reinforcement architecture (the direction of the fibrils and their hierarchical assembly), and the cost in energy for actuation. Without waiting for 4.5 billion years for evolution to produce protein machines to conduct this assembly, we should also consider methods of manufacturing these structural features.
These topics will inform the rest of the review, which will address some prototypical natural morphing materials and related synthetics, run through triggering stimuli including hygroscopic, chemical, heat, light and electrical and magnetic inputs, and finally discuss methods of modifying such materials in order to direct the response in a programmed direction. We conclude with some promising future directions.
Natural morphing materials and synthetic substitutes
A vast amount of actuating soft materials exist, including shape memory polymers (SMPs), hydrogels, elastomers and liquid crystals. This library only expands when the potential for composites or alternative processing is considered. The addition of secondary materials may enhance properties such as tensile strength, compressive strength, toughness and elasticity, or confer response to external stimuli. Therefore, this section aims instead to focus on a few key natural actuating materials, discuss the principles at both the micro- and macroscale by which each operates, sketch an option for artificial emulation and its mechanism, and provide an overview of their potential uses and limitations. We will highlight bio-inspired and otherwise notable uses of the material, alone or as a composite, and where relevant applications.
Numerous reviews are available for a deeper look at the topics united in this review. Broad sketches of actuating polymers and polymer gels are provided by Behl, Ahn and Geryak [28–30], and composites by Meng; [31] more specific reviews will be highlighted as topics are discussed. We are indebted to Ionov for his informative division of polymeric actuators according to their working principles [32], from which we draw inspiration for our comparison of natural actuators with similar artificial systems.
Skeletal muscle—active contraction
The most obvious example of a natural actuator is muscle, a tissue specialised to decrease in length when free to move or increase in tension when constrained. This is achieved using hierarchical principles, triggered by the presence of calcium ions, and produces macroscale contraction from staggered fibrous structures.
Within each muscle cell are filaments known as myofibrils, formed from small cylindrical units, sarcomeres, interspersed with Z-discs (Fig. 3). Hundreds of thin filaments of the protein actin are attached to the capping Z-discs at each end of the sarcomere, and thick filaments of myosin float freely, interdigitated with the actin filaments at both ends. The release of Ca2+ ions causes the myosin heads at both ends of the thick filament to pull on the anchored actin filaments, reducing the distance between Z-units and thus the length of the sarcomeres by around 70 % [33].

The hierarchical structure of muscle, an active contracting material. A skeletal muscle fibre is surrounded by a plasma membrane called the sarcolemma. Inside, the muscle fibre is composed of many fibrils, where each fibril is composed of sarcomeres, the individual units of contraction. A ‘Z-disc’ at either end is attached to thin actin filaments (shown here in green), with thick myosin filaments in between (shown as thick purple lines). Motion is achieved by the grasping of actin by myosin, which draws the filaments together. Reproduced from Anatomy and Physiology under Creative Commons Attribution License 4.0 from OpenStax. Copyright 2016
This effect is magnified to the microscopic level by multiple, aligned myofibrils, formed from sarcomeres packaged within stabilizing proteins, notably the stretchy, massive protein titin, which gives the muscle elastic properties and keeps the arrangement in place [34]. Thousands of these sub-units make up a single myofibril, which are aligned inside the muscle cells. Muscle cells are then bunched into fascicles, which define the direction of contraction of the muscle tissue, and inform the complex motions enabled by muscles. These range from the 486 ms−2 acceleration achieved by the chameleon’s tongue [35] to the exquisite shape control of cuttlefish papillae [4]. The degree of macroscale muscle contraction varies between approximately 20–40 % [36].
Artificial systems in which molecular-scale structures change position or orientation upon the application of an energy source, causing a change in dimensions while retaining approximately constant volume, include liquid crystals [37] and ionic-polymer metal composites, [38] among other electroactive polymers (EAPs) [39]. This latter is a very large category containing multiple methods of actuations, from the piezoelectric behaviour of some organic polymers to the high-performance behaviour of carbon nanotube supercapacitors. Due to limitations of space and their potential for anisotropic structuring, we focus on liquid crystal elastomers (LCEs) as a synthetic analogue for muscle. However, the reader is directed to further reviews for more information on EAPs and related systems [40–42].
Shape-change of these materials is an active process, necessitating constant stimulation, and thus power use, to retain deformation. This permits control, both remote and local, but limits efficiency and independence of operation. However, this is also a feature of well-established synthetic actuation methods such as hydraulic or piezoelectric control and so should not be necessarily be regarded as a disqualification.
Comparisons between liquid crystals and biological muscles have a long history, beginning with the original proposal and theoretical analysis by De Gennes in 1975 and continuing to the present day [43–46]. This well-understood state of matter exhibits a shift in stiffness [47], volume and optical properties as a result of changes in the internal order of rod-like sub-unit mesogens. LCs are found throughout nature, from spider silk to retinal proteins [27], although as yet the reason for the prominence of this state is not clear. However, studies of insect wing muscles have yielded crystalline forms under X-ray [48], demonstrating that at least some kinds of muscle structures benefit from high levels of order. It is also known that cell membranes are composed of liquid crystalline structures with various phases. Their function is the subject of ongoing research, but it is thought they may play a role in cell division and response to external influences [49]. As synthetic actuators, liquid crystals are bound to polymers, which are cross-linked to form a structural role analogous to titin in muscle; [34] the backbone provides a flexible anchor for the mesogenic groups as they reorientate, allowing the recovery of some strain. As the sub-units are aligned the polymer network is stretched: as the external stimulus is removed, it recovers a more entropically favourable coiled configuration. The combined system is known as a LCE and was initially developed by Küpfer and Finkelmann [50]. Figure 4a shows a schematic of a liquid crystal elastomer reminiscent of a muscle (i), and the polymer sub-unit which would make it up (ii).

Sketches of the structure of LCEs and their analogies to muscles. a A striated artificial muscle after de Gennes (1997) (i) based on a triblock copolymer RNR (ii). Shown here in a lamellar phase with the elastomer part R cross-linked. Similarities to the structure of muscle fibrils (shown in Fig. 3) are clear, with the domain R replacing the Z-disc and the aligned mesogenic units performing similarly to the interpenetrating fibrils, although changing shape via extension rather than contraction. b Options for attachment of mesogenic liquid crystalline (LC) groups (denoted by shaded ovals) to elastomeric polymer backbone: (i) main-chain LC polymer; (ii) side-on side-chain LC polymer; and (iii) end-on side-chain LC polymer. Any of the configurations illustrated in b may be used in block N of a, demonstrating the diversity of LCE chemistry possible. Adapted from Li et al., with permission from John Wiley and Sons [205]. Copyright 2004
Mesogenic units may be connected via an element on the short side (‘end-on’), in the middle of the longer side (‘side-on’), or within the backbone itself (shown in Fig. 4b) and may be joined to an existing backbone, polymerised, or cross-linked and polymerised in one step [51]. Commonly used mesogenic units include azo-containing groups, which under photostimulus shift from a rod-like transisomer to a kinked cis-version. This change in shape results in a photo-induced change in packing, and therefore volume. This was demonstrated by Tsutsumi and compared to theory by Hogan et al. [52, 53]. Mesogens with acrylate or methacrylate moieties, which are readily polymerised, may be varied to adapt the material properties. ABA triblock copolymers allow the construction of alternative structures, such as dilute gels [54]. Options for components and synthetic routes are discussed by Ohm [51], and other useful reviews may be found in Jiang et al. [55], Chambers et al. [56], and White et al. [37]. Existing issues and open questions are highlighted by Urayama [57].
The direction of contraction depends on the axis of alignment for the mesogenic units, known as the director, and analogous to the myofibril direction. This may be constant throughout a material, creating simple uniaxial deformation of up to 300 %, or vary in orientation creating twists [58] and out of plane effects [59]. Programmable orientation of the director in discrete volume elements across an LCE was demonstrated in 2015, unlocking arbitrary deformation through ‘voxelation’ [60]. This enables the exertion of greater forces, as the 55 % contraction attainable in a single element of the material is amplified by local forces to give a 3000 % stroke. Additional larger scale structure may be imparted by soft lithography, such as the work by Buguin et al. in using soft lithography to create forest of thermo-responsive LCE micropillars, suggested as tiny artificial pumps or switchable surfaces [61].
Tendons—release of stored energy
A second large-scale division of actuation materials is those that are capable of storing energy and, when triggered, causing deformation. This may result from the simple removal of the initial stimulus (in the case of elastomers), or require an additional trigger to escape a local energy minimum and release the internal stored energy. The first case is analogous to many elastic tissues in animals, for which tendons will be our example. The second is rarely seen in nature, but forms a large class of interesting engineering materials known as SMPs.
Tendons are viscoelastic tissues composed of collagen, which connect muscle to bone and in some cases, depending on their anatomical role, may also exhibit energy-conserving elastic properties [62]. Like muscles, they too show hierarchical structures with liquid crystalline order (Fig. 5). The smallest element of collagen, the tropocollagen molecule, consists of three intertwined helices which are then cross-linked into fibres, wound into fibrils, and grouped into fascicles to form a tendons [63]. It is notable that the strain of the fibre is exceeded by that of the fibril and subsequently of the fascicle: in other words, the hierarchical structure ensures the properties of the whole are greater than the sum of the parts [64].

The hierarchical structure of tendon, an energy storing material. a Simplified tendon structure. Tendon is made of a number of parallel fascicles containing collagen fibrils (marked F), which are themselves assemblies of parallel molecules (marked M). b The tendon fascicle can be viewed as a composite of collagen fibrils (having a thickness of several hundred nanometres and a length in the order of 10 μm) in a proteoglycan-rich matrix, subjected to a strain ε T. c Some of the strain will be taken up by a deformation of the proteoglycans (pg) matrix. The remaining strain, εF, is transmitted to the fibrils (F). d Triple-helical collagen molecules (M) are packed within fibrils in a staggered way. Reprinted from Fratzl, with permission from Elsevier. [64] Copyright 2003
Healthy tendons have been suggested to be auxetic, showing an absence of Poisson’s contraction when subjected to uniaxial stress. This unusual property is thought to derive from the varying angle of collagen fibrils throughout the fascicle, which was observed to vary depending on the biological role of the tendon, and affects its elastic response [65]. This is notably efficient: under stresses of 20 MPa and frequencies of 1–2 Hz, tendon tissues were shown to return up to 93 % of strain energy [66].
Shape memory polymers are somewhat analogous in that they derive their ability to reshape from an external applied deformation, which is translated into stored energy in the material and upon release enables a return to the original shape (see Fig. 6). However, in the case of SMPs, they may be ‘set’ into an intermediary shape and remain there indefinitely, until the input of sufficient additional energy.

The molecular mechanism of a dual shape memory polymer throughout a thermal cycle. Black dots netpoints; blue lines molecular chains of low mobility below transition temperature; red lines molecular chains of high mobility above transition temperature. Reproduced from Zhao et al. with permission from Elsevier. [70] Copyright 2015
At a microscale, the behaviour arises from a system of netpoints, which may be physical or chemical bonds, and connecting chains which switch their flexibility under different conditions, the most common of which is a change in heat [67]. In a typical application, the system is heated above the glass temperature of the chain sections, when they become flexible. It is then deformed by an external force while cooling occurs to below T g, ‘locking’ the chains in place in a high-energy state. When the temperature is raised above T g, the chains regain mobility and release strain to gain entropy, and return to their original maximum entropy state, corresponding to the relaxed macroscopic shape.
Recent experimental work on feathers and hair have identified shape memory behaviour in keratin-based natural materials [68, 69]. As seen in human hair styling, deformation can be set in by, for instance, wrapping around a curler while wet, but the original form will be recovered when wetted again. The authors hypothesise that in these materials, a combination of crystalline regions, chemical bonds, and hydrogen bonds act as netpoints to hold the molecular shape. Hydrogen bonds are removed from their original position when water penetrates the hair, leaving just chemical and crystalline netpoints to hold the polymers in place. These may be distorted away from their lowest energy configuration during drying and form temporary hydrogen bonds to lock in the temporary shape, but upon the addition of more water, the netpoints relax again into the original shape. Xiao and Hu suggest that different ratios between chemical and crystalline netpoints derive from the protein makeup of the hair and cause different fixity ratios of hairs from different individuals [68]. However, there is still much work to be done before the body of research on natural SMPs is as rich as that existing on tendon.
Synthetic SMPs have been demonstrated that show multiple shapes, cycling between three or more intermediary stages with varying stimulus intensity (multi-way SMP), and that can reversibly cross between two programmed states, rather than just returning to the lowest energy state (2 W-SME) [70]. In addition to heat, SMPs have been developed which respond to light [71], magnetism [72] and hydration with cooling [73]. A major advantage of SMPs over other shape-changing materials mentioned so far is their programmability, which enables them to take multiple forms and functions. Existing limitations for SMPs have been given as a low-energy storage modulus, limiting their force and stroke, a slower response time than hard materials, and a deterioration in response to repeated cycling [74].
The family of SMPs is large; as a start, any copolymer combination with different glass temperatures for each section fulfils this requirement, with the polymer of highest T g acting as the netpoint. Beyond the possibilities of chemical diversity, the thermal, structural and shape memory properties for a given combination may be modified by adjusting the molecular weight of each polymer component [75], the molecular architecture and temperature history [76]. Post-synthesis manufacturing techniques also affect response, as shown by Zhuo et al. when varying the voltage and solution concentration of electrospun polyurethane nanofibers [77].
Further possibilities are opened by the addition of composite reinforcement materials, such as nanocellulose [78] or CNTs [79, 80] although it should be noted that composite reinforcement is far from a magic bullet and may in fact degrade SMP performance [81]. The reader is referred to a number of excellent general reviews of SMPs and their composites [82–85] for more exhaustive assessments of previously studied polymers and their properties. A focus on electrically active SMPs may be found in Liu et al. [86].
Plant tissues—volume-change
As seen in the earlier example, plants achieve large, complex actuation as a result of relatively modest changes in volume. Examples abound, from the opening of leaves and tracking of the sun, to the projectile dispersion of seeds and spores and the clasping of climbing tendrils.
Swelling-based actuation occurs over a timescale of milliseconds to hours and crosses length scales from stomata on the leaf surface, composed of just two cells, to the entire plant structure [87]. The pulvinus, an organ found at the base of leaves and inside the stem, consists of internal cells that swell and contract [88]. This enables plants to track the sun with leaves or flowers, and modulate their temperatures and rates of photosynthesis. A wide number of angiosperms have been studied, including alpine flowers [89] and the Cornish mallow [90].
Volume-changes may be combined with other structural features to achieve faster responses through the sudden release of energy stored in other formats. Fracture-based launching was discussed in the context of the filaree awn and may also be seen in flowers [91]. The Sporangium fern exploits cavitation effects caused by a drop in pressure as volume is redistributed to catapult its spores onwards [92, 93]. The Venus flytrap exhibits a bistable structure which, when stored elastic energy from turgor pressure crosses a threshold, triggers rapid curvature in a perpendicular direction on the plane [94]. This enhances the response rate of a turgor-based system sufficiently to capture highly mobile prey.
Approaches to plant-based actuation and emulation are thoroughly covered by Fratzl and Burgert [95]. A common starting point has been selective reinforcement of materials that swell in response to hydration, often hydrogels—three-dimensional polymer networks, as little as 0.5 % w/w, swollen in solvent. Interestingly, responsive hydrogels made of pectin are found in the vasculature tissue of plants, which react to increasing ion concentration with corresponding changes in volume that modify the uptake of liquids [96]. Hydrogels as a class are typically brittle, slow to reach maximum deformation, and exert a limited force. However, their ease of processing and modification, as well as their demonstrated efficacy in natural systems, makes them a well-used model system with potential uses in biomedical fields [12, 97, 98].
Hydrogels may change volume by as much as 1100 times [99] in response to chemical, hygroscopic, heat, or light triggers, and may also exhibit shape memory characteristics [70, 100]. Properties depend on cross-link density, choice of polymer, copolymer or blend, nature of cross-link bonding [101], and solvent. In addition, structured or unstructured reinforcement may be added. Well-studied hydrogel chemistries include synthetics such as acrylamide, PEG and PVA, and biopolymers such as cellulose [102] and gelatin [103]. A general review of the theory used to understand stimuli-responsive hydrogels, and the multitude of work on the subject, can be found in Koetting [104].
While they are easy to work with and share similarities with some biological materials, hydrogels hold two major drawbacks; their slow response time and their lack of mechanical strength. Their need for hydration is also a challenge. However, the recent success of dye-sensitised solar cells, which are achieving commercial success despite currently relying on a corrosive liquid electrolyte, suggests this does not necessarily disqualify a technology [105]. As Calvert has commented, fruits such as oranges require hydration and yet may be transported across the world without issue [106].
Swelling response is diffusion-based and shows a power law relationship between time and length scale [107]. Response rate can be increased with the addition of microchannels [108], mimicking the operation of vasculature and allowing integration into microfluidic devices. Addition of graft chains, creating a comb-type hydrogel, has also been demonstrated to improve the response speed of some nanocomposites by around a factor of three [109].
Hope for strong hydrogel-based devices is inspired by the properties of gels seen in nature. Synthetic hydrogels typically have fracture energies of about 10 Jm−2; cartilage, meanwhile, withstands an additional two orders of magnitude, fracturing at around 1000 Jm−2 [106]. Many strategies are currently being employed to strengthen hydrogels, including double networks, composite addition and structural modification, but there is a long way to go before their potential can be fully realised [110].
Possible solutions are improvements in gel nanostructure or microstructure. For example, the 20 % of 20 nm collagen fibres found in the cornea are theorised to be the source of its 4 MPa tensile strength [106]. A similar concept was demonstrated with fibre reinforcement of an epoxy-based hydrogel, increasing its breaking stress by a factor of 20 [111]—despite to retain other properties simultaneously, the fibres would need to be significantly smaller, as seen in the cornea.
Modification of nanostructure has been able to produce hydrogels with a large swelling ratio, rapid response rate and elastic properties that permit extensive deformation without fracture [112]. In a two-step synthesis, functionalised nanogels of less than 100 nm in diameter are created, which are then subsequently joined together. This approach generates heterogeneous mesh-like structures relatively easy, suggesting biomimetic hierarchical nanostructures need not require extensive manufacturing.
Beyond the modification of structure, the addition of nanocomposites to hydrogels may confer still more properties [113]. Nanoparticles (NP) such as silicates and metal oxides can promote cross-linking, increase strength and modulate shear response. In particular, metal oxide NPs can add an orthogonal degree of responsiveness [114], improve conductivity [115] and give additional properties such as antimicrobial activity [115]. Cellulosic polymers have been used to sensitize hydrogels to temperature, pH and redox potential [116]. More information on hydrogels can be found in Ionov [110, 117].
Remote control: stimuli to shape-change
A major influence on the potential applications and development of responsive materials is, of course, how they can be triggered into movement. Again, natural systems operate with a limited palette of stimuli, with the vast majority of actuation being achieved by hygroscopic and chemical gradients. These are familiar to us in everyday life: the wilting and blooming of flowers and leaves arises from hydroscopic triggers, while muscle contraction is mediated by Ca++ concentration within the myocytes.
In the artificial world, the prospects for remote control and integration with existing systems make light and electrical and magnetic fields attractive candidates for controlling response, and raise the possibility of moving beyond natural capabilities.
Hygroscopic
Hydrogels are the prototypical material for hygroscopic response, changing in size over 1100-fold when solvent particles have fully infiltrated their polymer networks and caused expansion through hydrophilic effects [99]. However, other polymers and LCEs [118] can also show this property as the networks hydrate. Overall global swelling of polymer networks may be controlled by the use of different solvents [119], while local swelling may be modulated via modulation of vasculature [120], chemical composition [121] or altering of cross-link density [122], for example via ionoprinting [123]. In the case of LCEs, deformation is anisotropic and determined by the direction of mesogenic alignment [118]. Variation in swelling across a hydrogel sample may be used to move the hydrogel itself [124], surroundings [125] or inclusions [126].
In shape memory networks with polyurethane, the shape memory effect may be triggered by percolation of water or other low molecular weight solvents through the network. The presence of the water lowers the glass temperature of the cross-linking polymer to below ambient, whereupon the material returns to its original conformation [127, 128]. The example system of pyridine with polyurethanes attains shape recovery of over 90 % following this stimulation [129].
Cellulose has been used as a reversible support to confer responsive reinforcement to polymers, in an example of tunicate biomimicry. Surface hydroxyl groups on cellulose nanofibers have strong tendency to associate with others, which is reversible with hydration. Capadona et al. demonstrated a switchable 40-fold change in the tensile modulus of their system and suggested applications in electrodes and medical devices [130]. Reversible hydrogen bonding between cellulose fibres provides another way to modulate the shape memory effect in polyurethanes, independent of thermal contributions [131].
The ability of the pine cone, wheat awn and other plants to undergo humidity-responsive actuation has provided inspiration for a simple biomimetic model system, consisting of paper (the active, cellulose containing component) glued onto a flexible polymer [18]. This mimic undergoes more complex actuation behaviour than the natural systems but is sufficiently robust to create a “flower” which can open and close in response to wetting over timescales of around an hour. Many related systems have been developed, for applications ranging from walls which open to provide venting in warm weather [13] to a synthetic leaf that wilts when tea is brewed [15], and a hygroscopic generator [132].
Chemical
Presence of chemical species is a ubiquitous natural trigger, whether this be ion concentration, pH change, or presence of a specific antigen. Volume-changes resulting from chemical triggers to hydrogels may reach 350-fold [133]. Polymeric thin films such as SU-8, a commonly used photoresist, may also be treated to show bending responses to solvent stimuli [134].
To create a polymer hydrogel that is sensitive to pH, acidic or basic functional groups must be added to the polymer backbone or side chains. As the pH of the aqueous medium contained within the hydrogel is altered, the polymer will release or accept protons which will trigger a response within the gel. For example, as the pH of an acidic gel is raised, the degree of ionisation of the polymer increases, which leads to electrostatic repulsion and swelling of the gel. A reduction in pH would lead to swelling in a basic polymer-based hydrogel. This swelling response of pH-sensitive hydrogels has been rigorously analysed by Brannon-Peppas in terms of Flory–Huggins and rubber elasticity theory, with added contributions from ionic interaction [135]. pH-responsive hydrogels are considered strong candidates for drug delivery due to their potential in sensing and targeting [12].
DNA reactive materials could be an interesting addition to the current growth in synthetic DNA construction and design. Hydrogels have been developed that selectively contract when exposed to specific antigens [136] and single strands of DNA [137], the latter with measurable difference in response to only one base change. These are suggested for use as DNA-sensing devices or DNA-triggered actuators. Double network shape memory hydrogels have been created with two artificial DNA sequences, one pH-sensitive and one double-stranded, conjoined to acrylamide. These gels turn quasi-liquid when exposed to selected pH levels which remove one network, but can recover their pre-programmed shapes using the secondary duplex DNA network [138, 139]. Microscale DNA-responsive hydrogel devices have been developed that are capable of bending in response to their chemical environment, and even selectively detecting, trapping and releasing bacterial cells [140].
Solvent-responsive polymers may be combined with flexible electrodes such as graphene to create self-rolling actuators and sensors for uses such as operation in confined spaces, as wearables and tools [141].
Applications shown for chemically responsive hydrogels include liquid microlenses composed of a ring of hydrogel enclosing water. This swells in response to pH, raising the meniscus within it, and has been formed into an insect-inspired compound eye, suggested for diagnostic or lab-on-chip application [142]. Eddington el al. demonstrated an internally self-regulating valve which maintained a constant pH due to the swelling and contracting of a pH-sensitive hydrogel blocking the inlet [120].
Dicker et al. propose a synthetic analogue to the sun-avoiding Cornish Mallow leaf, using a chemically responsive hydrogel rendered light-sensitive with the addition of photobase, with particular applications for passive control of solar panels [143]. Finally, cyclic motions are also possible: a Belousov–Zhabotinsky oscillating chemical reaction has been demonstrated in a hydrogel, creating a cyclic ‘walking’ motion reminiscent of self-sustaining responses seen in biology [144].
Heat
Thermal response is perhaps the best-known trigger for passive motion in the artificial world. Varying thermal coefficients are easy to observe, and bimetallic strip-based control systems have adopted this approach since the eighteenth century. Many commercial plastics such as polyesters and polyurethane are thermoplastics exhibiting the shape memory effect, due to their ease of processing. However, their use in post-manufacture shape-changing applications is currently novel.
A feature of soft matter is that its characteristic energy levels are on the order of the thermal fluctuations in the environment, and therefore, many examples of thermo-responsive self-shaping soft matter exist. The solvent-polymer interactions which mediate the hydrogel volume-change are temperature dependent, as is the mobility of SMP netpoints. LCE orientation changes and cis/trans switches also occur around room temperature thermal energy.
The balance between the entropic and enthalpic energy of polymers in hydrogels is necessarily temperature dependent, and for some hydrogels, there exists a point such that their polymers switch from a hydrophilic to a hydrophobic state. To minimise energy, they undergo a coil-globule transition, shielding hydrophobic sites and gaining in entropy from collapsing into a less stretched configuration. This decreases the space between them, ejecting solvent and losing volume. The temperature at which this occurs may be adjusted by the temperature of processing [145], blending [146] and microstructure [147], in addition to the inclusion of composites which may adjust temperature response through density changes [148], or Joule or surface plasmon resonance heating (see later sections on light, electricity and magnetism for these mechanisms).
The bending of a hydrogel bilayer rod with one thermo-responsive face was demonstrated by Hu and co-workers in 1995 [21] and modelled as a bimetallic strip. More modern work has explored different approaches to the creation of gradients or conjoined materials with different properties, as detailed in the final section.
There are many suggested applications for thermo-responsive materials, including power generation through body heat, self-opening curtains [149] and thermal control valves [150]. Kim et al. demonstrated a responsive hydrogel surface which could expose or reveal arbitrarily shaped functionalised regions in response to temperature, suggested for use in lab-on-chip diagnoses [151].
Light
Use of electromagnetic or radiation sensitive material opens up opportunities for remote activation and gradated stimulation compatible with existing control systems. Liquid crystal-based systems are well-known for their light response, triggering the transisomer switch referred to earlier. For LCEs, polymer systems and hydrogels, the addition of nanoparticle composites with tuned plasmon resonances have been shown to add photo-response by triggering heating [109, 152, 153].
The degree of response can be modulated by varying composite proportions. Zhang et al. [154] noted that the percentage of carbon nanotubes in a thermosensitive hydrogel material affects hydrogel response time to both optical (near IR) and thermal stimuli. They use these materials as heat-activated hinges attached to solid plates, and by varying the weight percentage of additives are able to adjust the response speed of petals opening on a model flower as a function of distance. Similar effects have been achieved by others using gold NP [155] and magnetite particles [109].
Materials with two modes of response to optical stimuli open up interesting possibilities for feedback loops. Camacho-Lopez et al. demonstrated a dye-doped LCE capable of spontaneously ‘swimming’ in a variety of surfactants: as the material is struck by light and expands, it dips below the surface, and in darkness shrinks and rises once more to complete the cycle [156]. The swimming action is directed away from the light source, which therefore is a potential source of directional control over a self-maintaining feedback loop, similar to those seen in many natural systems.
A similar bi-directional response from UV-responsive thin polydomain liquid crystal network films can be controlled by varying the polarisation direction of the light source. Oscillating the source polarisation direction therefore gives a method of optically controlling a cantilever [157]. A hydrogel micro-cantilever has also been shown to respond to UV illumination via expansion of the illuminated side and therefore bending, but this is so far irreversible, suggesting applications for light-triggered final assembly of microdevices rather than control [158]. The use of LCE sensitive to different wavelengths of light within the same system demonstrated the potential for complex behaviour achieved in situ with optical triggers [159].
It is also possible to use light to control optical systems, which suggests passive sensor applications. For example, soft liquid hydrogel microlenses can be remotely focused with IR light, and this is suggested for applications in endoscopy [160]—in contrast to the earlier-mentioned use of chemically responsive hydrogels which require direct stimuli but combine the functions of sensing and actuation. Koerner provides more information on light-triggered polymeric systems [161].
Electrical and magnetic
The electrical pulse of the action potential is a key stimulus to actuation and shaping in the natural world, where muscle contraction results from ion channels opened by voltages on the order of 10 mV [162]. Synthetically, multiple electroactive shape-changing polymers are known, of which none exhibits the high gain of the muscle interaction: indeed, many require kilovolts to match the modest 20 % contraction of muscle [163].
A broad range of electroactive polymers are described in Bar-Cohen’s canonical book on the subject [39] and have now been an active area of research for 30 years—however, no clear leader appears to be emerging in the field and we would refer an interested reader to the aforementioned book, reviews by Mirfakhai [163], Otero [164] and Baughman [165] for a deeper look at the subject. Early studies by Baughman showed the potential of carbon nanotube actuators [166], which have the advantages of low mass density, high mechanical strength and two directions of response depending on current direction. More recently, novel forms of carbon have been combined with conductive polymers, to improve the performance of said polymers while retaining their low cost and ease of processability [167, 168]. Flexible carbon-based materials may also be used as electrodes and in electrolytes, enabling the creation of multipurpose actuators that are also supercapacitors [169].
The addition of conductive elements to trigger heating, and therefore thermoresponse, can be applied to hydrogels, LCEs and SMPs. This essentially reduces down into the earlier section on thermal triggers, with additional potential for control and integration [86, 170]. Felton et al. demonstrated this approaching using Joule heating to trigger a sequence of plates connected by SMP hinges to fold into both an origami crane [171] and a walking robot [172].
Magnetic NP, commonly ferrites, may be added to hydrogels or SMPs to confer magnetoresponse upon these materials. This may act directly via the orientation of the magnetic domains in an external field, or through induced heating effects that then trigger a thermoresponse [173]. This methodology permits control of the shape memory effect by varying magnetic field strength and offers potential for ‘topping up’ the internal heat via induction effects, thus reducing the external temperature required to trigger [72]. Magnetic NPs may be blended into LCEs [173], SMPs [174] or hydrogels [170], or cross-linked into the polymer backbone [175].
The inclusion of magnetic NP enables magnetic fields to be used to guide microdevices including surgical microgrippers [176] and pollution-sensing fish [177].
Beyond isotropy: modification of structure
In order to achieve meaningful and complex material response, it is necessary to control the degree of sensitivity to stimuli, and also its direction. Non-trivial responses require some form of anisotropy in either the stimuli or the material. The former approach is easy in the case of electromagnetic fields or radiation, but less so for diffuse hygroscopic or chemical stimuli. Since these are the two most commonly found triggers in biological situations, evolved solutions tend to focus on manipulating the response through material structure, as seen in the earlier example of the wheat awn constrained in its swelling by fibre directionality. This also has the advantage of being independent and self-contained, although it carries the disadvantage of not being reconfigurable—often referred to as ‘programmable’ in the literature. Shape memory materials offer the prospect of resetting target and intermediate shapes, but require intervention to achieve this—holding a material curled, or stretching it, while it cools.
There are multiple ways to analyse and learn from natural material features, which have merit in different situations. For the purposes of this discussion, we have found it helpful to consider the distinction between how material properties are achieved, and the role they play for the organism overall. Using the filaree awn as an example, the variation in contraction direction is caused by fibre alignment. However, this varying contraction is used to create a spiralling structure which winds itself more tightly as it dries before breaking. So, a researcher looking to learn from the awn may choose to emulate orientation and alignment of constrictive elements, or the behaviour of a tightening spiral approaching breakpoint.
As seen with mimicry of the filaree awn [25], shape-change may be reproduced without using the same method seen in the natural example. Conversely, we will draw attention to some ways of reproducing the aligned or gradated structures that give rise to shape-changing properties in nature which have not yet been used to create synthetic shape-change. Together, we hope that this palette of options will spark ideas for how biological features may be emulated, and encourage researchers to keep an open mind regarding routes to the desired outcome.
Reproducing positioning and orientation
One-dimensional elements such as fibres and rods may restrict the movement of their surrounding matrix in a given direction, as noted above for plant cells. Alternatively, aligned one-dimensional elements may create one-dimension contraction, as the myofibrils do in muscle tissue [178], or expansion. We see that placement and orientational control at nano- and micro-length scales would be necessary to emulate these features.
Mesogenic elements in liquid crystals determine the direction of actuation [179]. Various methods of alignment are used to obtain single-crystal monodomains, which ensure these elements act together. Shearing surface force [44], electrical [180] and magnetic fields [61] are all well-known methods for aligning mesogens before initiating polymerisation to preserve this orientation. Photoalignment can create spatially varying responses in two dimensions, demonstrated both continuously [181, 182] and using a discrete voxel approach [60]. The development of local feature manipulation in LCEs is reviewed comprehensively by White et al. [37].
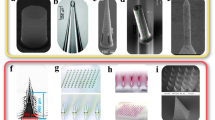
Techniques for positioning constraining fibres and plates have a long history in manufacture, primarily for reinforcing composites. Two-dimensional orientation dominates, although ultimately, as in nature, three-dimensional structures are required to respond to forces from all directions and so this is a natural final goal. Embedding of rigid units into polymers has been demonstrated with high-aspect ratio silicon nanocolumns (HARNS) around which a hydrogel was formed. The result was HARNS embedded in the hydrogel in specific orientations, which altered with respect to each other in accordance with the degree of hydration of hydrogel [126], suggested for use in microfluidics. Magnetic field effects have been used to align micrometre rods and platelets with superparamagnetic coating in various polymers [183], which has enabled twisting and bending motions via the application of local constraint (Fig. 7). There is potential for combination of this method with slip casting to fill moulded structures [184], but evidently magnetic field manipulation is restricted to strongly paramagnetic materials.

Pinecone biomimicry by Studart and Erb, using ultra-high magnetic response plates. a, b The natural principles of expansion and contraction in response to humidity. c (Above) diagrammatically the positioning of fibrous stiff cells (sclerenchyma) and short amorphous cells (sclereids) which give rise to the anisotropic movement of the pinecone, and (below) a schematic showing the synthetic reproduction of these properties with magnetic orientation of reinforcement. d, e The results; responsive synthetic pinecone scales produced with selective orientation within a gelatin matrix, in a similar bilayer structure, in both dry and wet states. Reproduced from Studart and Erb with permission from the Royal Society of Chemistry. [23] Copyright 2014
A more general approach has been demonstrated to position reinforcement fibres within a polymer matrix using ultrasound; [185] this allows arbitrary orientation in two dimensions and some degree of positional control, and is suitable for any material as long as it differs in density to the matrix. Future challenges involve extending this to three dimensions and accessing orientations which cross between layers.
The alignment of fibres in the direction of extrusion shows great potential when combined with 3D printing technologies. This has hitherto been a subject of research for stiff materials; for example, carbon nanofibers added to ABS printing were shown to align with the direction of extrusion. The bar samples produced showed decreased swelling and greater tensile strength, although at the cost of increased brittleness [186]. More complex extruded honeycomb structures rendered in resin with aligned fibres delivered a superior Young’s modulus for the lightweight samples, compared with other printing materials and natural balsa wood [187].
In softer materials, the addition of woven fibres has been successful in reinforcing hydrogels for strength [111] and directional confinement [188]. The alignment of constricting nanofibres within responsive electrospun polymer threads is a rapid way to generate hydrogels with ply angles analogous to those of classical composite design [189]. However, recent work by the Lewis group at Harvard’s Wyss Institute (Fig. 8) has taken this work into another elegant dimension, deriving mathematical models that allow the construction of arbitrary-curved shapes through the precise control of cellulose fibre direction, given by the path of an extrusion-based 3D printer [190].

Directional fibre orientation via extrusion, by Lewis et al., used to replicate complex orchid curvature and shape. a alignment during printer extrusion and the mesostructure which translates this into programmed curvature. b, c The cellulose alignment achieved during printing (scale bar 200 μm). d Multiple different shape-changes when immersed in water (scale bar 5 mm), in emulation of e, the dendrobium helix orchid. Adapted from Nature Materials with permission from Macmillan Publishers Ltd. [190] Copyright 2015
Extension of these orientating techniques to three dimensions would enable the creation of combined radial, longitudinal and helical fibres and structures; all key elements underlying the varied achievements of natural muscle, as exemplified by the hydrostatic skeleton [191].
Creating bending and gradated response
Considering the system from a different perspective, one can abstract the concept of simply joining materials with differing responses to the same stimulus. The bilayer motif is found in nature (for example, the pine cone and Venus fly trap), in the classic bimetallic strip, and from the very beginning of work in hydrogel actuators [21]. This is a prototypical example in many papers demonstrating newly discovered actuating materials. Stoychev et al. have explored the shapes formed by bilayers with varying dimensions as a result of diffusion and surface interactions, using a combination of finite element modelling and experimental work [192].
Lithographically patterning a bilayer structure introduces an extra dimension to bilayer fabrication, as shown by Bassik et al. [193]. Only those areas exposed to polymerising UV remain attached to the bottom layer, thus creating a surface with some bilayer bending regions and some monolayer flat regions. This is used to create an all-hydrogel version of a Venus flytrap. In another example of two-dimensional patterning, Andres et al. localise inkjet deposition of carbon nanotube composites within a polymer, creating folding regions with reduced hygroscopic swelling (as hinge points) [194].
More complicated curvature from a two-material system was demonstrated by Wu in 2013 [121], where hydrogels with thin, directed stripes of alternating chemical composition and coefficient of expansion were used to create complex deforming surfaces. The relatively small lateral modulation involved is directly reminiscent of plant motion, where, as in nature, small effects add up to create an overall larger movement.
A natural development from narrowly spaced alternating stripes is the transition to a continuously varying spatial response. This may be a gradient in composition; a natural example would be the combination of Type I and Type II muscles, whose different metabolic processes and response rates enable the many different behaviours required of skeletal muscle by changing the ratio of just two components [195]. Alternatively, it may be derived from structural variation or local amplification of a global stimulus.
The first case, of varying composition, has been realised in many different synthetic systems (see for example, Yu et al. with SMPs [196] and Maeda et al. with polymer gels [144]). A simple case is the variation of monomer concentration: gradients in monomer concentration throughout the injection forming of NIPAM sheets may be used to program the Gaussian curvature in a circular sample, allowing the spontaneous formation of domes and hyperbolic frills when heated [197]. Another approach is to introduce a second molecular species; for example, limited diffusion of a second polyacrylamide monomer into a pre-existing NIPAM gel is used to construct a bilayer strip and a flexible ‘hand’ gripping unit [21]. The gradient may also be created via post-treatment. For example, Zhao et al. expose a porous polymer network to a deprotonating acetone diffusion gradient, which causes variation in the degree of electrostatic complexation across the hydrogel and therefore its swelling response [119].
Structural modifications may result from altering the density of cross-links [22, 123] and the size of pores in hydrogels [198]. A more complex approach post-cured an SMP on a spatially varying thermal gradient, grading the glass temperature spatially and thus modulating the temperature at which different regions of the material regain their original shape [199]. Inert surface features such as a layer of micropillars can also direct the direction of deformation [46].
Finally, local amplification may derive from simple changes in colouration [200], or increased energy transmission induced by particles with a tailored surface plasmon resonance [155].
Form: a final boost to shape-change
Multifunctional materials exist to overcome the limitations of form. The sea cucumber not only needs a hard, defensive skin to resist ocean currents and predators, but also a soft, compliant dermis to take up shelter through narrow gaps in corals and to perform defensive evisceration [201]. Evidently, it can only have one skin, so the solution is to vary its stiffness, shifting between a tensile modulus of 5 and 50 MPa by modulating the interactions of collagen fibrils within the material [201]. This has been successfully emulated by Shanmuganathan et al. by the hydration-moderated interactions of cellulose nanowhiskers in low-density polymers [202].
Here, therefore, we see an example of material overcoming limitations of form. But the opposite situation can also be true: form may overcome the limitations of materials. Structural features, such as bistable shells [94], flexing keels [203] or collapsible chambers [92], expand the properties of materials through the structures they are formed into.
An example is an excellent final illustration of our journey through the materials, triggers and forms that can enable shape-change in soft materials. As discussed earlier, the variety of hydrogels available, combined with the many techniques for introducing anisotropy into their bulk, makes them an attractive material for volume-change-based actuation. However, the material experiences slow deployment times, as expansion is a diffusion-limited process [107], and the force exerted is typically small.
A solution to a similar problem is seen in the Venus flytrap, where the relatively weak forces exerted by water swelling release built-in strain, triggering a switch between two local minimal energy conformations [94]. Lee et al. emulated this with a combination of vasculature and pre-stress in the material to create the ‘jumping hydrogel’ (Fig. 9) [204]. Mesoscale channels created lithographically within a small hydrogel sample localise solvent exposure and therefore nanoscale swelling. The smaller scale also reduces diffusion length and therefore actuation time. Expansion and extension in a targeted small region were sufficient to flip the macroscale object into a second stable position, attain a maximum angular velocity an order of magnitude larger than the biological system, and release sufficient energy to propel it into the air. As noted by the authors, this design overcomes two known issues with hydrogels, namely their mechanical weakness and slow actuation, through incorporation in a wider system.

A biomimetic microgel, making use of microchannels and bistability, generates enough thrust to self-propel. The device is placed on a glass substrate (1). A solvent droplet fills the microfluidic network in the device by capillary action (2). Swelling results in bending of legs outwards (3–5). When bent, the device is ready to jump (6). Solvent in channels further evaporates, and snap-buckling takes place as the legs snap back to the original shape via de-swelling. This rapid motion produces enough thrust for the gel to jump out of the frame (7), landing outside the initial view field (8). Cross mark indicates the initial position of the device. Scale bar indicates 1 mm. Reproduced from Lee, Xia and Fang, with permission from the Royal Society of Chemistry. [204] Copyright 2010
Conclusion
A primary aim of this review was to examine the functionality and activation mechanisms of synthetic shape-changing materials used by the scientific community and benchmark our current knowledge of self-shaping materials against the strategies found in nature. As one might imagine, the conclusion is unavoidable; there will be no single material and manufacturing method which achieves the self-shaping the engineering and scientific community desires. Instead, we need to combine techniques to overcome shortcomings.
Key elements that have emerged in this review are the potential of nanoscopic features such as contracting fibres (as seen in muscle) and restrictive plates (as seen in plant tissues) to instigate and direct motion. These nanoelements must be united in a structured network, through embedding in a matrix or otherwise, potentially also including secondary mesoscale features. With current technology and materials, creating nanostructured materials in complex shapes requires either multiple steps (e.g. alignment followed by assembly), or dual development of materials and manufacturing (for example, alignment of fibre constructs during additive manufacture) [138, 140, 143].
From the top-down, we should look to create structures that minimise the force required to self-shape, and where possible target and combine stimuli to maximise the magnitude of the trigger delivered. The complementary use of different stimuli, such as the application of magnetic fields to reduce the temperature required for SMP relaxation [114], is a promising area, as is the potential of electrical or optical stimuli for remote, directional and modulated control.
Two themes are discernible from these examples: the potential for synergy between materials and form, and the need for features crossing multiple length scales—which require collaboration between manufacture and materials development. We therefore suggest simultaneous development of techniques for structuring materials, and the materials they will be used upon, as a fruitful direction. Diversification through the use of multiple techniques to create structures across complementary length scales, and responding to multiple stimuli, will combinatorially expand what is possible in this field.
Development of novel self-shaping materials can be expected to unlock applications and designs not yet considered, with corresponding changes in quality of life, material sustainability and environmental benefits. We consider that hierarchical material features, from nano- to macroscale, developed side-by-side with minimal-force structural designs, have the highest chance of blending nature’s responsive design space with the constructed world.
References
Nova A, Keten S, Pugno NM, Redaelli A, Buehler MJ (2010) Molecular and nanostructural mechanisms of deformation, strength and toughness of spider silk fibrils. Nano Lett 10:2626–2634. doi:10.1021/nl101341w
Rho J-Y, Kuhn-Spearing L, Zioupos P (1998) Mechanical properties and the hierarchical structure of bone. Med Eng Phys 20:92–102. doi:10.1016/S1350-4533(98)00007-1
Saranathan V, Osuji CO, Mochrie SGJ, Noh H, Narayanan S, Sandy A, Dufresne ER, Prum RO (2010) Structure, function, and self-assembly of single network gyroid (I4132) photonic crystals in butterfly wing scales. Proc Natl Acad Sci USA 107:11676–11681. doi:10.1073/pnas.0909616107
Allen JJ, Bell GRR, Kuzirian AM, Hanlon RT (2013) Cuttlefish skin papilla morphology suggests a muscular hydrostatic function for rapid changeability. J Morphol 274:645–656. doi:10.1002/jmor.20121
Lee H, Lee BP, Messersmith PB (2007) A reversible wet/dry adhesive inspired by mussels and geckos. Nature 448:338–341. doi:10.1038/nature05968
Mohd Jani J, Leary M, Subic A, Gibson MA (2014) A review of shape memory alloy research, applications and opportunities. Mater Des 56:1078–1113. doi:10.1016/j.matdes.2013.11.084
Irschik H (2002) A review on static and dynamic shape control of structures by piezoelectric actuation. Eng Struct 24:5–11. doi:10.1016/S0141-0296(01)00081-5
Doi M (2013) Soft matter physics. OUP, Oxford
Quake SR (2000) From micro- to nanofabrication with soft materials. Science 290:1536–1540. doi:10.1126/science.290.5496.1536
Bauer S, Bauer-Gogonea S, Graz I, Kaltenbrunner M, Keplinger C, Schwödiauer R (2014) 25th anniversary article: a soft future: from robots and sensor skin to energy harvesters. Adv Mater 26:149–161. doi:10.1002/adma.201303349
Rus D, Tolley MT (2015) Design, fabrication and control of soft robots. Nature 521:467–475. doi:10.1038/nature14543
Deligkaris K, Tadele TS, Olthuis W, van den Berg A (2010) Hydrogel-based devices for biomedical applications. Sens Actuators B 147:765–774. doi:10.1016/j.snb.2010.03.083
Holstov A, Bridgens B, Farmer G (2015) Hygromorphic materials for sustainable responsive architecture. Constr Build Mater 98:570–582. doi:10.1016/j.conbuildmat.2015.08.136
Zarzar LD, Kim P, Aizenberg J (2011) Bio-inspired design of submerged hydrogel-actuated polymer microstructures operating in response to pH. Adv Mater 23:1442–1446. doi:10.1002/adma.201004231
Yao L, Ou J, Cheng C-Y, Steiner H, Wang W, Wang G, Ishii H (2015) BioLogic. In: Proceedings of the 33rd annual ACM conference on human factors computing systems—CHI’15. ACM Press, New York, pp 1–10
Kim S, Laschi C, Trimmer B (2013) Soft robotics: a bioinspired evolution in robotics. Trends Biotechnol 31:287–294. doi:10.1016/j.tibtech.2013.03.002
Evangelista D, Hotton S, Dumais J (2011) The mechanics of explosive dispersal and self-burial in the seeds of the filaree, Erodium cicutarium (Geraniaceae). J Exp Biol 214:521–529. doi:10.1242/jeb.050567
Reyssat E, Mahadevan L (2009) Hygromorphs: from pine cones to biomimetic bilayers. J R Soc Interface 6:951–957. doi:10.1098/rsif.2009.0184
Rafsanjani A, Brulé V, Western TL, Pasini D (2015) Hydro-responsive curling of the resurrection plant Selaginella lepidophylla. Sci Rep 5:8064. doi:10.1038/srep08064
Timoshenko S (1925) Analysis of bi-metal thermostats. J Opt Soc Am 11:233–255. doi:10.1364/JOSA.11.000233
Hu Z, Zhang X, Li Y (1995) Synthesis and application of modulated polymer gels. Science 269:525–527. doi:10.1126/science.269.5223.525
Guvendiren M, Yang S, Burdick JA (2009) Swelling-induced surface patterns in hydrogels with gradient crosslinking density. Adv Funct Mater 19:3038–3045. doi:10.1002/adfm.200900622
Studart AR, Erb RM (2014) Bioinspired materials that self-shape through programmed microstructures. Soft Matter 10:1284–1294. doi:10.1039/c3sm51883c
Bar-On B, Sui X, Livanov K, Achrai B, Kalfon-Cohen E, Wiesel E, Wagner HD (2014) Structural origins of morphing in plant tissues. Appl Phys Lett 105:033703. doi:10.1063/1.4891191
Abraham Y, Tamburu C, Klein E, Dunlop JWC, Fratzl P, Raviv U, Elbaum R (2012) Tilted cellulose arrangement as a novel mechanism for hygroscopic coiling in the stork’s bill awn. J R Soc Interface 9:640–647. doi:10.1098/rsif.2011.0395
Aharoni H, Abraham Y, Elbaum R, Sharon E, Kupferman R (2012) Emergence of spontaneous twist and curvature in non-euclidean rods: application to erodium plant cells. Phys Rev Lett 108:238106. doi:10.1103/PhysRevLett.108.238106
Knight DP, Vollrath F (2002) Biological liquid crystal elastomers. Philos Trans R Soc Lond B 357:155–163. doi:10.1098/rstb.2001.1030
Behl M, Lendlein A (2007) Shape-memory polymers. Mater Today 10:20–28. doi:10.1016/S1369-7021(07)70047-0
Ahn S, Kasi RM, Kim S-C, Sharma N, Zhou Y (2008) Stimuli-responsive polymer gels. Soft Matter 4:1151–1157. doi:10.1039/b714376a
Geryak R, Tsukruk VV (2014) Reconfigurable and actuating structures from soft materials. Soft Matter 10:1246–1263. doi:10.1039/c3sm51768c
Meng H, Li G (2013) A review of stimuli-responsive shape memory polymer composites. Polymer (Guildf) 54:2199–2221. doi:10.1016/j.polymer.2013.02.023
Ionov L (2014) Polymeric actuators. Langmuir 31:5015–5024. doi:10.1021/la503407z
Lodish H, Kaiser C, Bretscher A, Amon A, Berk A, Krieger M, Polegh H, Scott M (2013) Myosin thick filaments and actin thin filaments in skeletal muscle slide past one another during contraction. In: Molecular cell biology, 7th edn. W. H. Freeman and Co, New York, p 801–802
Meyers MA, Chen P-Y, Lin AY-M, Seki Y (2008) Biological materials: structure and mechanical properties. Prog Mater Sci 53:1–206. doi:10.1016/j.pmatsci.2007.05.002
Wainwright PC, Kraklau DM, Bennett AF (1991) Kinematics of tongue projection in Chamaeleo oustaleti. J Exp Biol 159:109–133
Vogel S (2003) Achieving motility. In: Vogel S (ed) Comparative biomechanics—life’s physical world. Princeton University Press, Princeton, pp 453–582
White TJ, Broer DJ (2015) Programmable and adaptive mechanics with liquid crystal polymer networks and elastomers. Nat Mater 14:1087–1098. doi:10.1038/nmat4433
Shahinpoor M, Bar-Cohen Y, Simpson JO, Smith J (1998) Ionic polymer-metal composites (IPMCs) as biomimetic sensors, actuators and artificial muscles—a review. Smart Mater Struct 7:R15–R30. doi:10.1088/0964-1726/7/6/001
Bar-Cohen Y (2004) Electroactive polymer (EAP) actuators as artificial muscles: reality, potential, and challenges. Electroact Polym. doi:10.1117/3.547465
Pugal D, Jung K, Aabloo A, Kim KJ (2010) Ionic polymer-metal composite mechanoelectrical transduction: review and perspectives. Polym Int 59:279–289. doi:10.1002/pi.2759
Bahramzadeh Y, Shahinpoor M (2014) A review of ionic polymeric soft actuators and sensors. Soft Robot 1:38–52. doi:10.1089/soro.2013.0006
Brochu P, Pei Q (2010) Advances in dielectric elastomers for actuators and artificial muscles. Macromol Rapid Commun 31:10–36. doi:10.1002/marc.200900425
de Gennes P-G (1997) Un muscle artificiel semi-rapide. Comptes Rendus l’Académie des Sci—Ser IIB—Mech 324:343–348. doi:10.1016/S1251-8069(99)80044-X
Li M-H, Keller P (2006) Artificial muscles based on liquid crystal elastomers. Philos Trans A 364:2763–2777. doi:10.1098/rsta.2006.1853
Thomsen DL, Keller P, Naciri J, Pink R, Jeon H, Shenoy D, Ratna BR (2001) Liquid crystal elastomers with mechanical properties of a muscle. Macromolecules 34:5868–5875. doi:10.1021/ma001639q
Shahsavan H, Salili SM, Jákli A, Zhao B (2015) Smart muscle-driven self-cleaning of biomimetic microstructures from liquid crystal elastomers. Adv Mater 27:6828–6833. doi:10.1002/adma.201503203
Agrawal A, Chipara AC, Shamoo Y, Patra PK, Carey BJ, Ajayan PM, Chapman WG, Verduzco R (2013) Dynamic self-stiffening in liquid crystal elastomers. Nat Commun 4:1739. doi:10.1038/ncomms2772
Iwamoto H, Inoue K, Yagi N (2006) Evolution of long-range myofibrillar crystallinity in insect flight muscle as examined by X-ray cryomicrodiffraction. Proc Biol Sci 273:677–685. doi:10.1098/rspb.2005.3389
Seddon JM, Squires AM, Conn CE, Ces O, Heron AJ, Mulet X, Shearman GC, Templer RH (2006) Pressure-jump X-ray studies of liquid crystal transitions in lipids. Philos Trans A 364:2635–2655. doi:10.1098/rsta.2006.1844
Küpfer J, Finkelmann H (1991) Nematic liquid single crystal elastomers. Die Makromol Chemie Rapid Commun 12:717–726. doi:10.1002/marc.1991.030121211
Ohm C, Brehmer M, Zentel R (2010) Liquid crystalline elastomers as actuators and sensors. Adv Mater 22:3366–3387. doi:10.1002/adma.200904059
Hogan PM, Tajbakhsh AR, Terentjev EM (2002) UV manipulation of order and macroscopic shape in nematic elastomers. Phys Rev E 65:1–8. doi:10.1103/PhysRevE.65.041720
Tsutsumi O, Shiono T, Tomiki I, Galli G (1997) Photochemical phase transition behavior of nematic liquid crystals with azobenzene moieties as both mesogens and photosensitive chromophores. J Phys Chem B 101:1332–1337. doi:10.1021/jp961565d
Kempe MD, Scruggs NR, Verduzco R, Lal J, Kornfield JA (2004) Self-assembled liquid-crystalline gels designed from the bottom up. Nat Mater 3:177–182. doi:10.1038/nmat1074
Jiang H, Li C, Huang X (2013) Actuators based on liquid crystalline elastomer materials. Nanoscale 5:5225–5240. doi:10.1039/c3nr00037k
Chambers M, Finkelmann H, Remškar M, Sánchez-Ferrer A, Zalar B, Žumer S (2009) Liquid crystal elastomer–nanoparticle systems for actuation. J Mater Chem 19:1524–1531. doi:10.1039/B812423J
Urayama K (2007) Selected issues in liquid crystal elastomers and gels. Macromolecules 40:2277–2288. doi:10.1021/ma0623688
Sawa Y, Ye F, Urayama K, Takigawa T, Gimenez-Pinto V, Selinger RLB, Selinger JV (2011) Shape selection of twist-nematic-elastomer ribbons. Proc Natl Acad Sci USA 108:6364–6368. doi:10.1073/pnas.1017658108
de Haan LT, Schenning APHJ, Broer DJ (2014) Programmed morphing of liquid crystal networks. Polymer (Guildf) 55:5885–5896. doi:10.1016/j.polymer.2014.08.023
Ware TH, McConney ME, Wie JJ, Tondiglia VP, White TJ (2015) Actuating materials. Voxelated liquid crystal elastomers. Science 347:982–984. doi:10.1126/science.1261019
Buguin A, Li M-H, Silberzan P, Ladoux B, Keller P (2006) Micro-actuators: when artificial muscles made of nematic liquid crystal elastomers meet soft lithography. J Am Chem Soc 128:1088–1089. doi:10.1021/ja0575070
Alexander R (1991) Energy-saving mechanisms in walking and running. J Exp Biol 160:55–69
Kastelic J, Galeski A, Baer E (1978) The multicomposite structure of tendon. Connect Tissue Res 6:11–23. doi:10.3109/03008207809152283
Fratzl P (2003) Cellulose and collagen: from fibres to tissues. Curr Opin Colloid Interface Sci 8:32–39. doi:10.1016/S1359-0294(03)00011-6
Gatt R, Vella Wood M, Gatt A, Zarb F, Formosa C, Azzopardi KM, Casha A, Agius TP, Schembri-Wismayer P, Attard L, Chockalingam N, Grima JN (2015) Negative Poisson’s ratios in tendons: an unexpected mechanical response. Acta Biomater 24:201–208. doi:10.1016/j.actbio.2015.06.018
Bennett MB, Ker RF, Imery NJ, Alexander RM (1986) Mechanical properties of various mammalian tendons. J Zool 209:537–548. doi:10.1111/j.1469-7998.1986.tb03609.x
Leng J, Lan X, Liu Y, Du S (2011) Shape-memory polymers and their composites: stimulus methods and applications. Prog Mater Sci 56:1077–1135. doi:10.1016/j.pmatsci.2011.03.001
Xiao X, Hu J, Hu J, Zhu Y, Huang H, Lv J, Meng H, Li G, Anis A, Meng H, Zhou X, Mendez J, Zhu Y, Li J, Ma X, Tian H, Xue L, Dai S, Li Z, Jung YC, So HH, Cho JW, Chen S, Hu J, Yuen CW, Chen L, Huang WM, Yang B, An L, Li C, Chan YS, Lu H, Liu Y, Leng J, Du S, Dagnon KL, Huang H, Hu J, Zhu Y, Hu J, Zhu Y, Hu J, Dong ZE, Liu Y, Zimmermann EA, Fratzl P, Barth FG, Dawson C, Vincent JFV, Rocca AM, Elbaum R, Zaltzman L, Burgert I, Fratzl P, Meyers MA, McKittrick J, Chen PY, Fratzl P, Weinkamer R, Liu ZQ, Jiao D, Zhang ZF, Xu W, Ke G, Wu J, Wang X, Barba C, Ackbarow T, Chen X, Keten S, Buehler MJ, Speakman JB, Feughelman M, Feughelman M, Feughelman M, Fraser RDB, MacRae TP, Astbury WT, Street A, Astbury WT, Pauling L, Wortmann FJ, Rigby BJ, Phillips DG, Eaves JD, Schiessel H, Metzler R, Blumen A, Nonnenmacher TF, Williams G, Watts DC, Kumar B, Hu JL, Pan N, Behl M, Razzaq MY, Lendlein A, Lv H, Liu Y, Zhang D, Leng J, Du S, Du H, Zhang J, Liu Y, Kilpeläinen I, Akhtar W, Richard-Lacroix M, Pellerin C, Yao J, Liu Y, Yang S, Liu J, Ishida Y, Chabanne L, Antonietti M, Shalom M, Yan X, Ma M, Guo L, Anderson DG, Langer R, Nagia FA, EL-Mohamedy RSR, Liu C, Qin H, Mather PT (2016) Animal hairs as water-stimulated shape memory materials: mechanism and structural networks in molecular assemblies. Sci Rep 6:26393. doi:10.1038/srep26393
Liu ZQ, Jiao D, Zhang ZF (2015) Remarkable shape memory effect of a natural biopolymer in aqueous environment. Biomaterials 65:13–21. doi:10.1016/j.biomaterials.2015.06.032
Zhao Q, Qi HJ, Xie T (2015) Recent progress in shape memory polymer: new behavior, enabling materials, and mechanistic understanding. Prog Polym Sci 49–50:79–120. doi:10.1016/j.progpolymsci.2015.04.001
Lendlein A, Jiang H, Jünger O, Langer R (2005) Light-induced shape-memory polymers. Nature 434:879–882. doi:10.1038/nature03496
Kumar UN, Kratz K, Heuchel M, Behl M, Lendlein A (2011) Shape-memory nanocomposites with magnetically adjustable apparent switching temperatures. Adv Mater 23:4157–4162. doi:10.1002/adma.201102251
Wang CC, Huang WM, Ding Z, Zhao Y, Purnawali H (2012) Cooling-/water-responsive shape memory hybrids. Compos Sci Technol 72:1178–1182. doi:10.1016/j.compscitech.2012.03.027
Rousseau IA (2008) Challenges of shape memory polymers: a review of the progress toward overcoming SMP’s limitations. Polym Eng Sci 48:2075–2089. doi:10.1002/pen.21213
Momtaz M, Razavi-Nouri M, Barikani M (2014) Effect of block ratio and strain amplitude on thermal, structural, and shape memory properties of segmented polycaprolactone-based polyurethanes. J Mater Sci 49:7575–7584. doi:10.1007/s10853-014-8466-y
Messori M, Degli Esposti M, Paderni K, Pandini S, Passera S, Riccò T, Toselli M (2012) Chemical and thermomechanical tailoring of the shape memory effect in poly(ε-caprolactone)-based systems. J Mater Sci 48:424–440. doi:10.1007/s10853-012-6757-8
Zhuo H, Hu J, Chen S (2011) Study of the thermal properties of shape memory polyurethane nanofibrous nonwoven. J Mater Sci 46:3464–3469. doi:10.1007/s10853-011-5251-z
Auad ML, Contos VS, Nutt S, Aranguren MI, Marcovich NE (2008) Characterization of nanocellulose- reinforced shape memory polyurethanes. Polym Int 57:651–659. doi:10.1002/pi.2394
Miaudet P, Derré A, Maugey M, Zakri C, Piccione PM, Inoubli R, Poulin P (2007) Shape and temperature memory of nanocomposites with broadened glass transition. Science 318:1294–1296. doi:10.1126/science.1145593
Koerner H, Price G, Pearce NA, Alexander M, Vaia RA (2004) Remotely actuated polymer nanocomposites—stress-recovery of carbon-nanotube-filled thermoplastic elastomers. Nat Mater 3:115–120. doi:10.1038/nmat1059
Nishikawa M, Wakatsuki K, Takeda N (2010) Thermomechanical experiment and analysis on shape recovery properties of shape memory polymer influenced by fiber reinforcement. J Mater Sci 45:3957–3960. doi:10.1007/s10853-010-4545-x
Sun J-Y, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo Z (2012) Highly stretchable and tough hydrogels. Nature 489:133–136. doi:10.1038/nature11409
Ratna D, Karger-Kocsis J (2007) Recent advances in shape memory polymers and composites: a review. J Mater Sci 43:254–269. doi:10.1007/s10853-007-2176-7
Hager MD, Bode S, Weber C, Schubert US (2015) Shape memory polymers: past, present and future developments. Prog Polym Sci 49:3–33. doi:10.1016/j.progpolymsci.2015.04.002
Yan X, Wang F, Zheng B, Huang F (2012) Stimuli-responsive supramolecular polymeric materials. Chem Soc Rev 41:6042–6065. doi:10.1039/c2cs35091b
Liu Y, Lv H, Lan X, Leng J, Du S (2009) Review of electro-active shape-memory polymer composite. Compos Sci Technol 69:2064–2068. doi:10.1016/j.compscitech.2008.08.016
Stahlberg R (2009) The phytomimetic potential of three types of hydration motors that drive nastic plant movements. Mech Mater 41:1162–1171. doi:10.1016/j.mechmat.2009.05.003
Koller D (1990) Light-driven leaf movements. Plant Cell Environ 13:615–632. doi:10.1111/j.1365-3040.1990.tb01079.x
Luzar N, Gottsberger G (2001) Flower heliotropism and floral heating of five alpine plant species and the effect on flower visiting in Ranunculus montanus in the Austrian Alps. Arct Antarct Alp Res 33:93–99. doi:10.2307/1552282
Schwartz A, Koller D (1978) Phototropic response to vectorial light in leaves of Lavatera cretica L. Plant Physiol 61:924–928. doi:10.1104/pp.61.6.924
Whitaker DL, Webster LA, Edwards J (2007) The biomechanics of Cornus canadensis stamens are ideal for catapulting pollen vertically. Funct Ecol 21:219–225. doi:10.1111/j.1365-2435.2007.01249.x
Noblin X, Rojas NO, Westbrook J, Llorens C, Argentina M, Dumais J (2012) The fern sporangium: a unique catapult. Science 335:1322–1322. doi:10.1126/science.1215985
Llorens C, Argentina M, Rojas N, Westbrook J, Dumais J, Noblin X, Skotheim J, Mahadevan L, Dumais J, Forterre Y, Noblin X, Rojas N, Westbrook J, Llorens C, Argentina M, Dumais J, Borno R, Steinmeyer J, Maharbiz M, Martone P, Prantl K, Renner O, Ursprung A, King A, Ritman K, Milburn J, Haider K, Steudle E, Caupin F, Herbert E, Skotheim J, Mahadevan L, Epstein P, Plesset M (2016) The fern cavitation catapult: mechanism and design principles. J R Soc Interface 13:20150930. doi:10.1098/rsif.2015.0930
Forterre Y, Skotheim JM, Dumais J, Mahadevan L (2005) How the venus flytrap snaps. Nature 433:421–425. doi:10.1038/nature03185
Burgert I, Fratzl P (2009) Actuation systems in plants as prototypes for bioinspired devices. Philos Trans A 367:1541–1557. doi:10.1098/rsta.2009.0003
Zwieniecki MA, Melcher PJ, Michele Holbrook NM (2001) Hydrogel control of xylem hydraulic resistance in plants. Science 291:1059–1062. doi:10.1126/science.1057175
Guan Y, Zhang Y (2011) PNIPAM microgels for biomedical applications: from dispersed particles to 3D assemblies. Soft Matter 7:6375–6384. doi:10.1039/c0sm01541e
Guvendiren M, Lu HD, Burdick JA (2012) Shear-thinning hydrogels for biomedical applications. Soft Matter 8:260–272. doi:10.1039/C1SM06513K
Martin BD, Linhardt RJ, Dordick JS (1998) Highly swelling hydrogels from ordered galactose-based polyacrylates. Biomaterials 19:69–76. doi:10.1016/S0142-9612(97)00184-1
Osada Y, Matsuda A (1995) Shape memory in hydrogels. Nature 376:219. doi:10.1038/376219a0
Hennink WE, van Nostrum CF (2012) Novel crosslinking methods to design hydrogels. Adv Drug Deliv Rev 64:223–236. doi:10.1016/j.addr.2012.09.009
Chang C, Zhang L (2011) Cellulose-based hydrogels: present status and application prospects. Carbohydr Polym 84:40–53. doi:10.1016/j.carbpol.2010.12.023
Lam CX, Mo X, Teoh S, Hutmacher D (2002) Scaffold development using 3D printing with a starch-based polymer. Mater Sci Eng C 20:49–56. doi:10.1016/S0928-4931(02)00012-7
Koetting MC, Peters JT, Steichen SD, Peppas NA (2015) Stimulus-responsive hydrogels: theory, modern advances, and applications. Mater Sci Eng R Rep 93:1–49. doi:10.1016/j.mser.2015.04.001
Hardin BE, Snaith HJ, McGehee MD (2012) The renaissance of dye-sensitized solar cells. Nat Photonics 6:162–169. doi:10.1038/nphoton.2012.22
Calvert P (2009) Hydrogels for soft machines. Adv Mater 21:743–756. doi:10.1002/adma.200800534
Sato Matsuo E, Tanaka T (1988) Kinetics of discontinuous volume-phase transition of gels. J Chem Phys 89:1695. doi:10.1063/1.455115
Beebe D, Moore J, Bauer J, Yu Q, Liu R, Devadoss C, Jo B (2000) Functional hydrogel structures for autonomous flow control inside microfluidic channels. Nature 404:588–590. doi:10.1038/35007047
Lee E, Kim D, Kim H, Yoon J (2015) Photothermally driven fast responding photo-actuators fabricated with comb-type hydrogels and magnetite nanoparticles. Sci Rep 5:15124. doi:10.1038/srep15124
Ionov L (2014) Hydrogel-based actuators: possibilities and limitations. Mater Today 17:494–503. doi:10.1016/j.mattod.2014.07.002
Agrawal A, Rahbar N, Calvert PD (2013) Strong fiber-reinforced hydrogel. Acta Biomater 9:5313–5318. doi:10.1016/j.actbio.2012.10.011
Xia L-W, Xie R, Ju X-J, Wang W, Chen Q, Chu L-Y (2013) Nano-structured smart hydrogels with rapid response and high elasticity. Nat Commun 4:2226. doi:10.1038/ncomms3226
Schexnailder P, Schmidt G (2008) Nanocomposite polymer hydrogels. Colloid Polym Sci 287:1–11. doi:10.1007/s00396-008-1949-0
Mohr R, Kratz K, Weigel T, Lucka-Gabor M, Moneke M, Lendlein A (2006) Initiation of shape-memory effect by inductive heating of magnetic nanoparticles in thermoplastic polymers. Proc Natl Acad Sci USA 103:3540–3545. doi:10.1073/pnas.0600079103
Lee W-F, Tsao K-T (2009) Effect of silver nanoparticles content on the various properties of nanocomposite hydrogels by in situ polymerization. J Mater Sci 45:89–97. doi:10.1007/s10853-009-3896-7
Hebeish A, Farag S, Sharaf S, Shaheen TI (2014) Thermal responsive hydrogels based on semi interpenetrating network of poly(NIPAm) and cellulose nanowhiskers. Carbohydr Polym 102:159–166. doi:10.1016/j.carbpol.2013.10.054
Ionov L (2013) Biomimetic hydrogel-based actuating systems. Adv Funct Mater 23:4555–4570. doi:10.1002/adfm.201203692
Harris KD, Bastiaansen CWM, Broer DJ (2006) A glassy bending-mode polymeric actuator which deforms in response to solvent polarity. Macromol Rapid Commun 27:1323–1329. doi:10.1002/marc.200600342
Zhao Q, Dunlop JWC, Qiu X, Huang F, Zhang Z, Heyda J, Dzubiella J, Antonietti M, Yuan J (2014) An instant multi-responsive porous polymer actuator driven by solvent molecule sorption. Nat Commun 5:4293. doi:10.1038/ncomms5293
Eddington DT, Liu RH, Moore JS, Beebe DJ (2001) An organic self-regulating microfluidic system. Lab Chip 1:96–99. doi:10.1039/b108078d
Wu ZL, Moshe M, Greener J, Therien-Aubin H, Nie Z, Sharon E, Kumacheva E (2013) Three-dimensional shape transformations of hydrogel sheets induced by small-scale modulation of internal stresses. Nat Commun 4:1586. doi:10.1038/ncomms2549
Senff H, Richtering W (2000) Influence of cross-link density on rheological properties of temperature-sensitive microgel suspensions. Colloid Polym Sci 278:830–840. doi:10.1007/s003960000329
Palleau E, Morales D, Dickey MD, Velev OD (2013) Reversible patterning and actuation of hydrogels by electrically assisted ionoprinting. Nat Commun 4:2257. doi:10.1038/ncomms3257
Islam MR, Li X, Smyth K, Serpe MJ (2013) Polymer-based muscle expansion and contraction. Angew Chem Int Ed Engl 52:10330–10333. doi:10.1002/anie.201303475
Teramoto N, Shigehiro O, Ogawa Y, Maruyama Y, Shimasaki T, Shibata M (2014) Polymer foam-reinforced hydrogels inspired by plant body frameworks as high-performance soft matter. Polym J 46:592–597. doi:10.1038/pj.2014.41
Sidorenko A, Krupenkin T, Taylor A, Fratzl P, Aizenberg J (2007) Reversible switching of hydrogel-actuated nanostructures into complex micropatterns. Science 315:487–490. doi:10.1126/science.1135516
Huang WM, Yang B, An L, Li C, Chan YS (2005) Water-driven programmable polyurethane shape memory polymer: demonstration and mechanism. Appl Phys Lett 86:114105. doi:10.1063/1.1880448
Yang B, Huang WM, Li C, Lee CM, Li L (2004) On the effects of moisture in a polyurethane shape memory polymer. Smart Mater Struct 13:191–195. doi:10.1088/0964-1726/13/1/022
Chen S, Hu J, Zhuo H (2011) Study on the moisture absorption of pyridine containing polyurethane for moisture-responsive shape memory effects. J Mater Sci 46:6581–6588. doi:10.1007/s10853-011-5606-5
Capadona JR, Shanmuganathan K, Tyler DJ, Rowan SJ, Weder C (2008) Stimuli-responsive polymer nanocomposites inspired by the sea cucumber dermis. Science 319:1370–1374. doi:10.1126/science.1153307
Zhu Y, Hu J, Luo H, Young RJ, Deng L, Zhang S, Fan Y, Ye G (2012) Rapidly switchable water-sensitive shape-memory cellulose/elastomer nano-composites. Soft Matter 8:2509–2517. doi:10.1039/c2sm07035a
Ma M, Guo L, Anderson DG, Langer R (2013) Bio-inspired polymer composite actuator and generator driven by water gradients. Science 339:186–189. doi:10.1126/science.1230262
Tanaka T, Fillmore D, Sun S-T, Nishio I, Swislow G, Shah A (1980) Phase transitions in ionic gels. Phys Rev Lett 45:1636–1639. doi:10.1103/PhysRevLett.45.1636
Jamal M, Zarafshar AM, Gracias DH (2011) Differentially photo-crosslinked polymers enable self-assembling microfluidics. Nat Commun 2:527. doi:10.1038/ncomms1531
Brannon-Peppas L, Peppas NA (1991) Equilibrium swelling behavior of pH-sensitive hydrogels. Chem Eng Sci 46:715–722. doi:10.1016/0009-2509(91)80177-Z
Miyata T, Asami N, Uragami T (1999) A reversibly antigen-responsive hydrogel. Nature 399:766–769. doi:10.1038/21619
Gu H, Chao J, Xiao S-J, Seeman NC (2010) A proximity-based programmable DNA nanoscale assembly line. Nature 465:202–205. doi:10.1038/nature09026
Cheng E, Xing Y, Chen P, Yang Y, Sun Y, Zhou D, Xu L, Fan Q, Liu D (2009) A pH-triggered, fast-responding DNA hydrogel. Angew Chem Int Ed Engl 48:7660–7663. doi:10.1002/anie.200902538
Hu Y, Lu C-H, Guo W, Aleman-Garcia MA, Ren J, Willner I (2015) A shape memory acrylamide/DNA hydrogel exhibiting switchable dual pH-responsiveness. Adv Funct Mater 25:6867–6874. doi:10.1002/adfm.201503134
Kaehr B, Shear JB (2008) Multiphoton fabrication of chemically responsive protein hydrogels for microactuation. Proc Natl Acad Sci USA 105:8850–8854. doi:10.1073/pnas.0709571105
Deng T, Yoon C, Jin Q, Li M, Liu Z, Gracias DH (2015) Self-folding graphene-polymer bilayers. Appl Phys Lett 106:203108. doi:10.1063/1.4921530
Dong L, Agarwal AK, Beebe DJ, Jiang H (2006) Adaptive liquid microlenses activated by stimuli-responsive hydrogels. Nature 442:551–554. doi:10.1038/nature05024
Dicker MPM, Rossiter JM, Bond IP, Weaver PM (2014) Biomimetic photo-actuation: sensing, control and actuation in sun-tracking plants. Bioinspir Biomim 9:036015. doi:10.1088/1748-3182/9/3/036015
Maeda S, Hara Y, Yoshida R, Hashimoto S (2010) Active polymer gel actuators. Int J Mol Sci 11:52–66. doi:10.3390/ijms11010052
Rathjen CM, Park C-H, Goodrich PR, Walgenbach DD (1995) The effect of preparation temperature on some properties of a temperature-sensitive hydrogel. Polym Gels Netw 3:101–115. doi:10.1016/0966-7822(94)00030-B
Snowden MJ, Chowdhry BZ, Vincent B, Morris GE (1996) Colloidal copolymer microgels of N-isopropylacrylamide and acrylic acid: pH, ionic strength and temperature effects. J Chem Soc Faraday Trans 92:5013–5016. doi:10.1039/ft9969205013
Nguyen HH, Payré B, Fitremann J, Lauth-de Viguerie N, Marty J-D (2015) Thermoresponsive properties of PNIPAM-based hydrogels: effect of molecular architecture and embedded gold nanoparticles. Langmuir 31:4761–4768. doi:10.1021/acs.langmuir.5b00008
Kim H, Lee JA, Sim HJ, Lima MD, Baughman RH, Kim SJ (2016) Temperature-responsive tensile actuator based on multi-walled carbon nanotube yarn. Nano Micro Lett 8:254–259. doi:10.1007/s40820-016-0084-6
Yamamoto Y, Kanao K, Arie T, Akita S, Takei K (2015) Air ambient-operated pNIPAM-based flexible actuators stimulated by human body temperature and sunlight. ACS Appl Mater Interfaces 7:11002–11006. doi:10.1021/acsami.5b02544
Bakarich SE, Gorkin R, Panhuis M, Het IN, Spinks GM (2015) 4D printing with mechanically robust, thermally actuating hydrogels. Macromol Rapid Commun 36:1211–1217. doi:10.1002/marc.201500079
Kim J, Yoon J, Hayward RC (2010) Dynamic display of biomolecular patterns through an elastic creasing instability of stimuli-responsive hydrogels. Nat Mater 9:159–164. doi:10.1038/nmat2606
Yang L, Setyowati K, Li A, Gong S, Chen J (2008) Reversible infrared actuation of carbon nanotube-liquid crystalline elastomer nanocomposites. Adv Mater 20:2271–2275. doi:10.1002/adma.200702953
Zhang H, Xia H, Zhao Y (2012) Optically triggered and spatially controllable shape-memory polymer–gold nanoparticle composite materials. J Mater Chem 22:845–849. doi:10.1039/C1JM14615G
Zhang X, Pint CL, Lee MH, Schubert BE, Jamshidi A, Takei K, Ko H, Gillies A, Bardhan R, Urban JJ, Wu M, Fearing R, Javey A (2011) Optically- and thermally-responsive programmable materials based on carbon nanotube-hydrogel polymer composites. Nano Lett 11:3239–3244. doi:10.1021/nl201503e
Hribar KC, Metter RB, Ifkovits JL, Troxler T, Burdick JA (2009) Light-induced temperature transitions in biodegradable polymer and nanorod composites. Small 5:1830–1834. doi:10.1002/smll.200900395
Camacho-Lopez M, Finkelmann H, Palffy-Muhoray P, Shelley M (2004) Fast liquid-crystal elastomer swims into the dark. Nat Mater 3:307–310. doi:10.1038/nmat1118
Serak S, Tabiryan N, Vergara R, White TJ, Vaia RA, Bunning TJ (2010) Liquid crystalline polymer cantilever oscillators fueled by light. Soft Matter 6:779–783. doi:10.1039/B916831A
Xiong Z, Zheng M-L, Dong X-Z, Chen W-Q, Jin F, Zhao Z-S, Duan X-M (2011) Asymmetric microstructure of hydrogel: two-photon microfabrication and stimuli-responsive behavior. Soft Matter 7:10353–10359. doi:10.1039/c1sm06137b
van Oosten CL, Bastiaansen CWM, Broer DJ (2009) Printed artificial cilia from liquid-crystal network actuators modularly driven by light. Nat Mater 8:677–682. doi:10.1038/nmat2487
Zeng X, Jiang H (2008) Tunable liquid microlens actuated by infrared light-responsive hydrogel. Appl Phys Lett 93:151101. doi:10.1063/1.2996271
Koerner H, White TJ, Tabiryan NV, Bunning TJ, Vaia RA (2008) Photogenerating work from polymers. Mater Today 11:34–42. doi:10.1016/S1369-7021(08)70147-0
Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J (2000) Intracellular ion environment and membrane electric potential. In: Freeman WH (ed) Molecular cell biology, 4th edn. Scientific American Books, New York
Mirfakhrai T, Madden JDW, Baughman RH (2007) Polymer artificial muscles. Mater Today 10:30–38. doi:10.1016/S1369-7021(07)70048-2
Otero TF, Martinez JG, Arias-Pardilla J (2012) Biomimetic electrochemistry from conducting polymers. A review: artificial muscles, smart membranes, smart drug delivery and computer/neuron interfaces. Electrochim Acta 84:112–128. doi:10.1016/j.electacta.2012.03.097
Baughman RH (1996) Conducting polymer artificial muscles. Synth Met 78:339–353. doi:10.1016/0379-6779(96)80158-5
Baughman RH, Cui C, Zakhidov AA, Iqbal Z, Barisci JN, Spinks GM, Wallace GG, Mazzoldi A, De Rossi DD, De Rinzler AG, Jaschinski O, Roth S, Kertesz M, Baughman RH, Smela E, Inganäs O, Lundström I, Otero TF, Sansinena JM, Della Santa A, De Rossi D, Mazzoldi A, Gandhi MR, Murray P, Spinks GM, Wallace GG, Kaneto K, Kaneko M, Min Y, MacDiarmid AG, Liu J, Rinzler AG, Pietronero L, Strässler S, Kertesz M, Chan CT, Kamitakahara WA, Ho KM, Eklund PC, Baughman RH, Murthy NS, Eckhardt H, Kertesz M, Nixon DE, Perry GS, Murakami Y, Kishimoto T, Suematsu H, Fisher JE, Kim HJ, Cajipe VB, Kamitakahara WA, Zaresky JL, Eklund PC, Baron F, Flandrois S, Hauw C, Gaultier J, Flandrois S, Hauw C, Mathur RB, Spudich JA, Wong EW, Sheehan PE, Leber CM, Falvo MR, Thess A, Ye Y, Radin J-P, Yeager E, Gerischer H, McIntyre R, Scherson D, Storck W, Oren Y, Glatt I, Livnat A, Kafri O, Soffer A, Rao AM, Eklund PC, Bandow S, Thess A, Smalley RE, Gao G, Çagin T, Goddard WA, Treacy MMJ, Ebbesen TW, Gibson JM, Wong EW, Sheehan PE, Lieber CM, Zhang QM, Bharti V, Zhao X, Forster RJ (1999) Carbon nanotube actuators. Science 284:1340–1344. doi:10.1126/science.284.5418.1340
Liang J, Huang L, Li N, Huang Y, Wu Y, Fang S, Oh J, Kozlov M (2012) Electromechanical actuator with controllable motion, fast response rate, and high-frequency resonance based on graphene and polydiacetylene. ACS Nano 6:4508–4519. doi:10.1021/nn3006812
Zheng W, Razal JM, Whitten PG, Ovalle-Robles R, Wallace GG, Baughman RH, Spinks GM (2011) Artificial muscles based on polypyrrole/carbon nanotube laminates. Adv Mater 23:2966–2970. doi:10.1002/adma.201100512
Torop J, Palmre V, Arulepp M, Sugino T, Asaka K, Aabloo A (2011) Flexible supercapacitor-like actuator with carbide-derived carbon electrodes. Carbon N Y 49:3113–3119. doi:10.1016/j.carbon.2011.03.034
Satarkar NS, Biswal D, Hilt JZ (2010) Hydrogel nanocomposites: a review of applications as remote controlled biomaterials. Soft Matter 6:2364–2371. doi:10.1039/b925218p
Felton SM, Tolley MT, Shin B, Onal CD, Demaine ED, Rus D, Wood RJ (2013) Self-folding with shape memory composites. Soft Matter 9:7688–7694. doi:10.1039/c3sm51003d
Felton S, Tolley M, Demaine E, Rus D, Wood R (2014) Applied origami. A method for building self-folding machines. Science 345:644–646. doi:10.1126/science.1252610
Kaiser A, Winkler M, Krause S, Finkelmann H, Schmidt AM (2009) Magnetoactive liquid crystal elastomer nanocomposites. J Mater Chem 19:538–543. doi:10.1039/B813120C
Razzaq MY, Behl M, Lendlein A (2012) Memory-effects of magnetic nanocomposites. Nanoscale 4:6181–6195. doi:10.1039/c2nr31332d
Fuhrer R, Athanassiou EK, Luechinger NA, Stark WJ (2009) Crosslinking metal nanoparticles into the polymer backbone of hydrogels enables preparation of soft, magnetic field-driven actuators with muscle-like flexibility. Small 5:383–388. doi:10.1002/smll.200801091
Breger JC, Yoon C, Xiao R, Kwag HR, Wang MO, Fisher JP, Nguyen TD, Gracias DH (2015) Self-folding thermo-magnetically responsive soft microgrippers. ACS Appl Mater Interfaces 7:3398–3405. doi:10.1021/am508621s
Zhu W, Li J, Leong YJ, Rozen I, Qu X, Dong R, Wu Z, Gao W, Chung PH, Wang J, Chen S (2015) 3D-printed artificial microfish. Adv Mater 27:4411–4417. doi:10.1002/adma.201501372
Wang Z, Hang G, Wang Y, Li J, Du W (2008) Embedded SMA wire actuated biomimetic fin: a module for biomimetic underwater propulsion. Smart Mater Struct 17:025039. doi:10.1088/0964-1726/17/2/025039
Kondo M, Yu Y, Ikeda T (2006) How does the initial alignment of mesogens affect the photoinduced bending behavior of liquid-crystalline elastomers? Angew Chemie Int Ed 45:1378–1382. doi:10.1002/anie.200503684
Brehmer M, Zentel R, Wagenblast G, Siemensmeyer K (1994) Ferroelectric liquid-crystalline elastomers. Macromol Chem Phys 195:1891–1904. doi:10.1002/macp.1994.021950601
Ware TH, Perry ZP, Middleton CM, Iacono ST, White TJ (2015) Programmable liquid crystal elastomers prepared by thiol-ene photopolymerization. ACS Macro Lett 4:942–946. doi:10.1021/acsmacrolett.5b00511
de Haan LT, Sánchez-Somolinos C, Bastiaansen CMW, Schenning APHJ, Broer DJ (2012) Engineering of complex order and the macroscopic deformation of liquid crystal polymer networks. Angew Chemie 124:12637–12640. doi:10.1002/ange.201205964
Erb RM, Sander JS, Grisch R, Studart AR (2013) Self-shaping composites with programmable bioinspired microstructures. Nat Commun 4:1712. doi:10.1038/ncomms2666
Le Ferrand H, Bouville F, Niebel TP, Studart AR (2015) Magnetically assisted slip casting of bioinspired heterogeneous composites. Nat Mater 14:1172–1179. doi:10.1038/nmat4419
Llewellyn-Jones TM, Drinkwater BW, Trask RS (2016) 3D printed components with ultrasonically arranged microscale structure. Smart Mater Struct 25:02LT01. doi:10.1088/0964-1726/25/2/02LT01
Shofner ML, Lozano K, Rodríguez-Macías FJ, Barrera EV (2003) Nanofiber-reinforced polymers prepared by fused deposition modeling. J Appl Polym Sci 89:3081–3090. doi:10.1002/app.12496
Compton BG, Lewis JA (2014) 3D-printing of lightweight cellular composites. Adv Mater 26:6043–6043. doi:10.1002/adma.201470235
Etches JA, Bond IP (2011) Development of a self-actuating fibre reinforced ionic epoxy gel polymer composite. Smart Mater Struct 20:045020. doi:10.1088/0964-1726/20/4/045020
Liu L, Jiang S, Sun Y, Agarwal S (2016) Giving direction to motion and surface with ultra-fast speed using oriented hydrogel fibers. Adv Funct Mater 26:1021–1027. doi:10.1002/adfm.201503612
Sydney Gladman A, Matsumoto EA, Nuzzo RG, Mahadevan L, Lewis JA (2016) Biomimetic 4D printing. Nat Mater 15:413–418. doi:10.1038/nmat4544
Kier WM (2012) The diversity of hydrostatic skeletons. J Exp Biol 215:1247–1257. doi:10.1242/jeb.056549
Stoychev G, Zakharchenko S, Turcaud S, Dunlop JWC, Ionov L (2012) Shape-programmed folding of stimuli-responsive polymer bilayers. ACS Nano 6:3925–3934. doi:10.1021/nn300079f
Bassik N, Abebe BT, Laflin KE, Gracias DH (2010) Photolithographically patterned smart hydrogel based bilayer actuators. Polymer (Guildf) 51:6093–6098. doi:10.1016/j.polymer.2010.10.035
Andres CM, Zhu J, Shyu T, Flynn C, Kotov NA (2014) Shape-morphing nanocomposite origami. Langmuir 30:5378–5385. doi:10.1021/la404955s
Damron TA (2008) Skeletal muscle anatomy, physicology and mechanics. In: Tornetta P III, Einhorn TA (eds) Oncology and basic science, 2nd edn. Lippincott Williams & Wilkins, Philadelphia
Yu K, Ritchie A, Mao Y, Dunn ML, Qi HJ (2015) Controlled sequential shape changing components by 3D printing of shape memory polymer multimaterials. Procedia IUTAM 12:193–203. doi:10.1016/j.piutam.2014.12.021
Klein Y, Efrati E, Sharon E (2007) Shaping of elastic sheets by prescription of non-Euclidean metrics. Science 315:1116–1120. doi:10.1126/science.1135994
Luo R, Wu J, Dinh N-D, Chen C-H (2015) Gradient porous elastic hydrogels with shape-memory property and anisotropic responses for programmable locomotion. Adv Funct Mater 25:7272–7279. doi:10.1002/adfm.201503434
DiOrio AM, Luo X, Lee KM, Mather PT (2011) A functionally graded shape memory polymer. Soft Matter 7:68–74. doi:10.1039/C0SM00487A
Liu Y, Boyles JK, Genzer J, Dickey MD (2012) Self-folding of polymer sheets using local light absorption. Soft Matter 8:1764–1769. doi:10.1039/C1SM06564E
Motokawa T (1981) The stiffness change of the holothurian dermis caused by chemical and electrical stimulation. Comp Biochem Physiol Part C 70:41–48. doi:10.1016/0306-4492(81)90076-9
Shanmuganathan K, Capadona JR, Rowan SJ, Weder C (2010) Biomimetic mechanically adaptive nanocomposites. Prog Polym Sci 35:212–222. doi:10.1016/j.progpolymsci.2009.10.005
Harrington MJ, Razghandi K, Ditsch F, Guiducci L, Rueggeberg M, Dunlop JWC, Fratzl P, Neinhuis C, Burgert I (2011) Origami-like unfolding of hydro-actuated ice plant seed capsules. Nat Commun 2:337. doi:10.1038/ncomms1336
Lee H, Xia C, Fang NX (2010) First jump of microgel; actuation speed enhancement by elastic instability. Soft Matter 6:4342–4345. doi:10.1039/c0sm00092b
Li M-H, Keller P, Yang J, Albouy P-A (2004) An artificial muscle with lamellar structure based on a nematic triblock copolymer. Adv Mater 16:1922–1925. doi:10.1002/adma.200400658
Acknowledgements
KO and AS are funded through an Engineering and Physical Sciences Centre for Doctoral Training award to the Bristol Centre for Functional Nanomaterials, EPSRC Grant code EP/G036780/1. RST is an Engineering and Physical Sciences Research Council (EPSRC) Fellow funded under EPSRC ‘Engineering Fellowships for Growth’, Grant Number EP/M002489/1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Availability of research data
There is no underlying data associated with this paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Oliver, K., Seddon, A. & Trask, R.S. Morphing in nature and beyond: a review of natural and synthetic shape-changing materials and mechanisms. J Mater Sci 51, 10663–10689 (2016). https://doi.org/10.1007/s10853-016-0295-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-016-0295-8