
Abstract
Defect levels induced by defect-complexes in Ge play important roles in device fabrication, characterization, and processing. However, only a few defect levels induced by defect-complexes have been studied, hence limiting the knowledge of how to control the activities of numerous unknown defect-complexes in Ge. In this study, hybrid density functional theory calculations of defect-complexes involving oversize atom (indium) and n-type impurity atoms in Ge were performed. The formation energies, defect-complex stability, and electrical characteristics of induced defect levels in Ge were predicted. Under equilibrium conditions, the formation energy of the defect-complexes was predicted to be within the range of 5.90–11.38 eV. The defect-complexes formed by P and In atoms are the most stable defects with binding energy in the range of 3.31-3.33 eV. Defect levels acting as donors were induced in the band gap of the host Ge. Additionally, while shallow defect levels close to the conduction band were strongly induced by the interactions of Sb, P, and As interstitials with dopant (In), the double donors resulting from the interactions between P, As, N, and the host atoms including In atom are deep, leading to recombination centers. The results of this study could be applicable in device characterization, where the interaction of In atom and n-type impurities in Ge is essential. This report is important as it provides a theoretical understanding of the formation and control of donor-related defect-complexes in Ge.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Defect formation and engineering in semiconductors are important for controlling the electronic and electrical properties of their hosts [1,2,3,4,5]. Ge is one of the promising group IV semiconductors; it has a narrow band gap of 0.78 eV at 0 K [6], high resistivity, as well as high electron–hole mobility at low temperature (300 K) [7,8,9,10]. Ge is considered as one of the promising materials for the new generation development of integrated circuits, due to its unique transport properties [9, 11]. The ability of Ge to be directly grown on Si substrates makes it attractive for Si-integrated electronic and photonic device applications [7, 9, 11, 12].
Studies have shown that large n\(^{+}\) carrier concentrations are beneficial for fabricating ohmic contacts and producing low-parasitic-resistance n-MOSFETS [7, 13,14,15]. However, despite the technological applications of Ge as a semiconductor, there are difficulties in obtaining n\(^{+}\) carrier concentrations from donor doping. For instance, the possibility of a strong presence of an unstable oxide layer at the surface of Ge can restrict its application in electronic devices [12]. Another example is anomalous boron redistribution in the Ge melting phase acting as an electrically inactive pile-up. One recommended way to overcome this shortfall is by introducing oxygen as an external native oxide, preventing high boron activation in Ge [13]. However, doping Ge with donor atoms may be challenging due to the low solid solubility and high diffusion coefficient [16]. These properties tend to reduce the possibility of obtaining shallow doped regions in Ge [17]. Also, the combination of defect clusters involving donor and vacancy defects could enhances low activation energies [18]. Nevertheless, this shortcoming could be possibly corrected by the introduction of defect-complexes that may acts as donors in Ge. Firsthand information on how to control the impact of defect-complexes on the electronic properties, electrical conductivity, and the determination of the lifetime carriers of Ge could help enhance the quality of its device performance for future applications in MOSFETs [19,20,21,22].
A handful of well-known point defects in Ge that influences its electrical properties are interstitials, vacancies, defect-complexes, and substitutions [1, 2, 19, 23,24,25,26]. In the past, great achievements have been recorded toward the discovery, identification, and investigation of numerous point defects in Ge via the experimental techniques [27,28,29] or theoretical modeling [30,31,32,33]. For example, the impact of the self-interstitial [34], vacancies [35], di-interstitials [36], substitutional impurities [37, 38], vacancy complexes [23, 39], and defect-complexes [23, 39, 40] on the electronic, structural, and electrical properties of Ge have been reported.
Defect-complexes in semiconductors have been reported to induce electrically active defect levels in their hosts [3,4,5, 19, 41, 42] and numerous defect-complexes in Ge have been reported [23, 36, 43]. For instance, Al defect-complexes have shown to act as shallow donors in Ge, while exhibiting charged state controlled metastability characteristic [43]. The rare earth defect-complexes in Ge act as deep defect levels leading to recombination processes [5]. While the Tm defect-complex in Ge has been reported to induce shallow defect level [23], the Ce-substitution/Ge-interstitial, (Ce\(_\text{Ge}\)Ge\(_\text{i}\)) defect-complex in Ge exhibits a negative-U ordering and induces a deep donor level leading to recombination processes [5]. The Pr-substitution/Ge-interstitial Pr\(_\text{Ge}\)Ge\(_\text{i}\) defect-complex induces a shallow defect level near the valence band maximum. However, the Tm\(_\text{Ge}\)Ge\(_\text{i}\) and Er\(_\text{Ge}\)Ge\(_\text{i}\) defect-complexes are electrically inactive [5]. Ge di-interstitials induce defect levels close to the conduction band minimum (CBM) [36]. Recently, trivalent interstitial and substitution defect-complexes in Ge have been demonstrated to induce shallow defect levels [44]. For instance, Al\(_\text{i}\)Al\(_\text{Ge}\) induces a defect level approximately 0.02 eV from the CBM, while In\(_\text{i}\)In\(_\text{Ge}\) serves as a recombination center [44]. The electronic properties and defect levels induced by n/p-type defect-complexes in Ge have been studied [45]. The report discusses the electronic characteristics and defect levels caused by n/p-type defect-complexes in Ge. It further emphasizes the valuable insights gained from the unique response to introducing n/p-type interstitial and substitutional defect-complexes, highlighting their substantial influence on Ge properties. These insights are essential for guiding implantation procedures in Ge and Ge-based devices [45].
Despite the substantial breakthroughs in the study of point defects processes in Ge and other semiconductors [3,4,5, 7, 19, 41, 42], there are still numerous undiscovered point defects. As a result, their defect levels remain unknown. To the best of my knowledge, no theoretical information on the induced defect levels of the In and n-type impurities substitution–interstitial defect-complexes (In\(_\text{Ge}\)N\(_\text{i}\), In\(_\text{i}\)N\(_\text{Ge}\), In\(_\text{Ge}\)P\(_\text{i}\), In\(_\text{i}\)P\(_\text{Ge}\), In\(_\text{i}\)As\(_\text{Ge}\), In\(_\text{Ge}\)As\(_\text{i}\), In\(_\text{i}\)Sb\(_\text{Ge}\), and In\(_\text{Ge}\)Sb\(_\text{i}\)) in Ge have been published. Furthermore, the low solid solubility and high diffusion coefficient [16] in Ge tend to reduce the possibility of obtaining shallow doped regions [17]. This shortcoming could possibly be corrected by the introduction of defect-complexes formed by the interaction of donors with interstitial atoms. Hence, the motivation of this study is to investigate the influence of In and n-type impurities substitution–interstitial defect-complexes on the electronic and electrical properties of Ge, as well as to provide information on how to control donor-like defects for improved Ge-based technologies.
In this paper, the hybrid density functional theory was utilized to determine the formation and binding energies of substitutional and interstitial pairs of In and n-type impurities (N, P, As, and Sb) in Ge (In\(_\text{Ge}\)N\(_\text{i}\), In\(_\text{i}\)N\(_\text{Ge}\), In\(_\text{Ge}\)P\(_\text{i}\), In\(_\text{i}\)P\(_\text{Ge}\), In\(_\text{i}\)As\(_\text{Ge}\), In\(_\text{Ge}\)As\(_\text{i}\), In\(_\text{i}\)Sb\(_\text{Ge}\), and In\(_\text{Ge}\)Sb\(_\text{i}\)). The stability and electrically active defect levels induced by these complexes were predicted. The result demonstrates that substitutional and interstitial pairs of In and n-type impurities in Ge are stable with respect to their binding energies. Specifically, In\(_\text{Ge}\)P\(_\text{i}\) and In\(_\text{i}\)P\(_\text{Ge}\) are identified as the most energetically favorable defect-complexes. Furthermore, the studied defect-complexes exhibited donor behavior, with no traces of acceptor levels observed in Ge. The induced defect levels of In\(_\text{Ge}\)P\(_\text{i}\) and In\(_\text{Ge}\)As\(_\text{i}\) represent the shallowest donor defects and have the potential to donate electrons to the conduction band under thermal disturbances. The information presented in this report on the significance of the electrical properties of Ge at various defect levels resulting from the formation of defect-complexes is expected to be valuable in the process modeling of modern Ge-based devices for industrial applications.
This article is structured as follows: Sect. 2 provides detailed computational methods, including theoretical background. Results presentations and discussions on formation energy, stability, and induced defect levels in Ge are documented in Sect. 3. Finally, Sect. 4 presents a summary of the key results and conclusions of this research work.
2 Method
Density functional theory calculations using the Vienna ab-initio Simulation Package (VASP) were performed. The core and chemically active electrons were separated using the projector-augmented wave (PAW) method [46]. The hybrid functional with the generalized gradient approximation (GGA) functional of Perdew, Burke, and Ernzerhof (PBE) was used for exchange correlations. The hybrid approach combines the PBE and Hartree–Fock method. In this approach, a default mixing parameter of 25% and a screening parameter of 0.2 Å\(^{-1}\) were used to obtain an accurate Ge band gap of 0.78 eV, which is in agreement with Ref. [6]. To determine the appropriate supercell for charge defect modeling, an initial test for supercell convergence was conducted using a Ge self-interstitial, as reported in Ref. [47]. The results indicated that a 64-atom supercell is sufficient for modeling defects in Ge. To further demonstrate the selection of the supercell in this study, a simple calculation was performed introducing the +2 charge state of In\(_\text{Ge}\)P\(_\text{i}\) into supercells containing 64, 128, and 256 atoms. After minimizing the impact of defect–defect interactions, a 64-atom supercell was chosen as it proved suitable for modeling charge defects in Ge. For instance, the formation energies of the +2 charge state of In\(\text{Ge}\)P\(\text{i}\) in the 64, 128, and 256 atom supercells are 4.6571 eV, 4.6541 eV, and 4.6521 eV, respectively. Hence, the energy difference between the 64 and 216 atom supercells was less than 0.01 eV. For the \(2\times 2\times 2\) supercell relaxation, a Monkhorst–Pack k-point grid of \(4\times 4\times 4\) was used to integrate the Brillouin zone. The energy cutoff convergence criterion was set to 400 eV, the minimum total energy convergence criteria was set to 10\(^{-6}\), and the forces acting on each of the atoms were less than 0.001 eV/Å. Electronic properties calculations were performed with a denser k-point grid of \(16\times 16\times 16\).
Under thermodynamic equilibrium conditions, defects concentrations (C\(_{0}\) are related to the formation energy (\(E^{f}\)) through the Boltzmann constant (k\(_B\)) as follows:
While temperature (T) is measured in Kelvin, (n\(_0\)) represents the number of possible sites in a solid crystal where defect-complexes can occur per unit volume. The above equation suggests that an increase in the formation energy results in a decrease in the concentrations of defect-complexes, making the defect energetically less favorable. To predict the formation of defect-complexes in Ge, the total energy E(d, q) of the supercell containing the relaxed defect-complexes d in their charge state q was calculated. The formation energy of defect-complexes (\(E^f(d,q)\)) as a function of the electron Fermi energy (\(\varepsilon _F\)) is given as [48, 49].
The total energy of the relaxed supercell without a defect-complex is denoted as E(pure). The symbol \(\mu _{i}\) represents the chemical potential of the i-th type of atom, and \(\triangle (n)i\) is the difference in the number of individual atoms of type i between the pure and defective supercells. The chemical potentials for all atoms involved were determined by considering the total energy per atom. For this study, the chemical potentials used were \(-\)4.30 eV, \(-\)2.75 eV, \(-\)5.88 eV, \(-\)12.2 eV, \(-\)5.65 eV, and \(-\)4.03 eV for Ge, In, P, N, As, and Sb, respectively. When an impurity is added or removed from the host supercell, \(\triangle (n)\) assumes a negative or positive unity, respectively. In solid crystals, for any band gap \(E_\text{gap}\), the Fermi energy (\(\varepsilon _F\)) is measured from the valence band maximum (\(E_\text{V}\)) as
The \(E^q_{FNV}\) in Eq. 2 is the Freysoldt, Neugebauer and Van de Walle (FNV) correction term, which solves the problem of potential alignment between the charged defect and bulk at a point far from the defect. Also, it is used to remedy the charge corrections problem in a supercell of finite size [50, 51].
To predict the defect levels induced by the defect-complexes, it is essential to determine the charge state transition levels. A transition energy level (\(\epsilon (q/q_{0})\)) for a defect-complex is the Fermi energy at which its charge states (q and q\(_{0}\)) meet and have the same formation energy. The expression for \(\epsilon (q/q_{0})\) is provided in reference [3] as
To predict whether interstitial and substitutional defects can bind with the host atoms to form stable defect-complexes, the binding energy of these complexes were calculated. The binding energy (\(E_{b}\)) of a defect-complex is defined as the energy required to separate a cluster of defects into well-separated, non-interacting defects. The binding energy is calculated as [52]
were \(\sum \limits _{i} E^{f}{(isolated)}\) represents the sum of formation energies of the i-th isolated defects, and \(E^{f}{(defect-complex)}\) is the formation energy of the defect-complex. Equation 5 can be interpreted as the energy difference between the bonded structure and the isolated individual defects. If the binding energy of a defect-complex is positive, it indicates stability; otherwise, it suggests instability.
3 Results and discussion
3.1 Structural properties
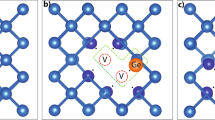
Figure 1 displays the relaxed geometric structure of the pristine Ge, In\(_\text{Ge}\)P\(_\text{i}\), and In\(_\text{Ge}\)Sb\(_\text{i}\). Table 1 lists the bond lengths of the nearest neighbor Ge, interstitial and substitution atoms in various defect-complexes. In Table 1, the interstitial and substitution atoms are denoted as X and Y, respectively.

The relaxed geometric structures of, a pristine Ge, b In\(_\text{Ge}\)P\(_\text{i}\) and, c In\(_\text{Ge}\)Sb\(_\text{i}\)
The X–Y bond lengths vary across different defect-complexes, ranging from 2.16 to 2.91 Å. Similar to the X–Ge bond lengths, the X–Y bond lengths also deviate from the pristine Ge-Ge bond length, depending on the specific defect configuration as listed in Table 1.
For the In\(_\text{Ge}\)N\(_\text{i}\) and In\(_\text{i}\)N\(_\text{Ge}\), the X–Y bond lengths are the same at 2.16 Å, which is 0.30 Å larger than the bond length of Ge-Ge. However, notable variations exist in other bond lengths within the In\(_\text{Ge}\)N\(_\text{i}\) and In\(_\text{i}\)N\(_\text{Ge}\) defect-complexes, such as the Y-Ge and X-Ge bonds. Furthermore, comparing defect-complexes across different group-V elements (P, As, Sb) reveals variations in bond lengths. These differences can be attributed to disparities in atomic radii and electronegativity. For example, defect-complexes involving In\(_\text{Ge}\)P\(_\text{i}\) and In\(_\text{i}\)P\(_\text{Ge}\), the X-Y bond lengths are slightly longer compared to those involving In\(_\text{Ge}\)N\(_\text{i}\) and In\(_\text{i}\)N\(_\text{Ge}\). However, the different between the bond length of the Ge-Ge and In-P for the In\(_\text{Ge}\)P\(_\text{i}\) and In\(_\text{i}\)P\(_\text{Ge}\) are 0.07 and 0.05 Å, respectively. It is imperative to note that the distortion exerted in the supercell due to external impurities when N is introduce either as interstitial or substitution is more that the bond length distortion experienced when P is introduced in Ge crystal either as interstitial or substitution atoms. This suggests potential differences in chemical bonding behavior between nitrogen and phosphorus atoms in the Ge lattice. When examining the bond length of X-Y, the difference in bond length between Ge-Ge and X-Y atoms is smaller when the impurities atoms is position on the Ge lattice, except for the In\(_\text{Ge}\)N\(_\text{i}\) and In\(_\text{i}\)N\(_\text{Ge}\), which have the same 0.30 Å.
The Y-Ge bond lengths vary across different defect-complexes, ranging from 1.89 to 2.58 Å. While the In\(_\text{Ge}\)N\(_\text{i}\) and In\(_\text{i}\)N\(_\text{Ge}\) induced more strain with a significant difference of 0.57 Å between the Ge–Ge and Ni–Ge bond lengths, the In\(_\text{Ge}\)P\(_\text{i}\) and In\(_\text{i}\)As\(_\text{Ge}\) induced less strain in the crystal structure. The Ni-Ge bond has the shortest length; however, it differs significantly from the equilibrium Ge–Ge bond length. Despite In\(_\text{Ge}\)Sb\(_\text{i}\) and In\(_\text{i}\)Sb\(_\text{Ge}\) having the longest bond lengths, the difference between Ge-Ge and Sb-Ge lengths is relatively smaller than that of the N–Ge. The X–Ge bond lengths vary across different defect-complexes, ranging from 2.07 to 2.88 Å. For the X–Ge bond length, it deviates significantly from the pristine Ge–Ge bond length. Specifically, the deviations for the N–Ge, P–Ge, As–Ge, and Sb–Ge bonds are 0.04, 0.11, 0.08, and 0.05, respectively. The extent of difference in bond length correlates with the degree of structural distortion experienced in the supercell. Comparing the bond lengths of the defect-complex with the Ge–Ge bond length provides insight into the extent of structural distortion or stability within the defect-complexes. Deviation from the pristine Ge-Ge bond length indicates alterations in the local atomic arrangement, potentially impacting the electronic and mechanical properties of the semiconductor material. These alterations can also affect the energy of formation of such defects in the host materials. Systems with substantial distortions in their atomic arrangement are likely to necessitate a significant amount of formation energy for their formation.
3.2 Formation energies of defect-complexes
The formation energy of impurities in a solid crystal system can offer insight into the likelihood of a specific defect formation. These formation energies have been utilized to ascertain whether the defect concentrations fall within the necessary range for a semiconductor. In this study, the formation energies of defect-complexes were calculated, as listed in Table 2. Under thermodynamic equilibrium conditions, the formation energy indicates that the most energetically favorable defect-complex is the In\(_\text{Ge}\)P\(_\text{i}\), with an energy of 5.84 eV. The relatively lower formation energy of In\(_\text{Ge}\)P\(_\text{i}\), compared to other impurities, suggests a potential solution to the challenges associated with achieving low defect concentrations when doping Ge with donor atoms. Additionally, In\(_\text{Ge}\)P\(_\text{i}\) may offer an alternative approach to addressing the challenges of low solubility and high diffusion coefficients associated with doping Ge with donor impurities [16]. Later in this article, it will be further demonstrated that the donor-like substitution–interstitial complexes formed by In atoms and n-type impurities in Ge could potentially yield shallow defect levels, less than 3 \(k_BT\).
The formation energy of In\(_\text{i}\)P\(_\text{Ge}\) is 0.06 eV higher than that of In\(_\text{Ge}\)P\(_\text{i}\). Conversely, In\(_\text{Ge}\)N\(_\text{i}\) and In\(_\text{i}\)N\(_\text{Ge}\) are the least energetically favorable defects, with formation energies approximately 5.00 eV higher than that of In\(_\text{Ge}\)P\(_\text{i}\). The difference in formation energies between In\(_\text{i}\)As\(_\text{Ge}\) and In\(_\text{Ge}\)As\(_\text{i}\) is small (0.01 eV), which is negligible within the electron-volt range. Insights into the defect engineering processes of In and p-type atom defect-complexes in Ge at zero Fermi energy, as reported in Ref [40], suggest a high potential for indium atoms to form stable defect-complexes. For instance, in Ref [40], the formation energies of In\(_\text{Ge}\)Al\(_\text{i}\) and In\(_\text{i}\)Al\(_\text{Ge}\) were reported as 6.16 eV and 5.44 eV, respectively. However, in this study, the formation energies of In\(_\text{Ge}\)P\(_\text{i}\) and In\(_\text{i}\)P\(_\text{Ge}\) are 5.84 eV and 5.90 eV, respectively. For defect-complexes formed by the interaction of In atom with gallium (Ga) or aluminum (Al), the formation energy results suggest that such defects are energetically favorable when In atom is located at the substitutional site. For example, the formation energies of In\(_\text{i}\)Al\(_\text{Ge}\) (In\(_\text{i}\)Al\(_\text{Ge}\)) and In\(_\text{Ge}\)Ga\(_\text{i}\) (In\(_\text{i}\)Ga\(_\text{Ge}\)) are 5.44 eV (6.16 eV) and 5.27 eV (6.06 eV), respectively [40]. The formation energies are strongly influenced by the type of atom, its radius, and the level of strain experienced by atomic bonds during defect formation. High strain resulting from lattice distortion leads to high formation energy, often observed in the case of oversized impurities. In this study, the amount of strain experienced by In\(_\text{Ge}\)N\(_\text{i}\) and In\(_\text{i}\)N\(_\text{Ge}\) is likely to be higher than that of other defect-complexes. Thus, careful consideration is needed before doping Ge with nitrogen to form such complexes. Oversize impurities are known to influence diffusivity, especially at high concentration levels. In this study, the influence of oversize impurities, such as In and Sb atoms, should be considered during defect processes. Below, is the outline of the order of formation energy under equilibrium conditions for the neutral charge state of defect-complexes formed by In and n-type atoms in Ge: In\(_\text{Ge}\)P\(_\text{i}<\) In\(_\text{i}\)P\(_\text{Ge}<\) In\(_\text{i}\)As\(_\text{Ge}<\) In\(_\text{Ge}\)As\(_\text{i}<\) In\(_\text{i}\)Sb\(_\text{Ge}<\) In\(_\text{Ge}\)Sb\(_\text{i}<\) In\(_\text{Ge}\)N\(_\text{i}\) In\(_\text{i}\)N\(_\text{Ge}\).
3.3 Binding energies of defect-complexes
Next, the stability of defect-complexes were examined by calculating their binding energies. In this study, when the binding energy is less than zero, it suggests that the defect-complexes may have difficulty binding, while a binding energy greater than zero indicates stability and favorable binding. As displayed in Table 2, all the defect-complexes have binding energies greater than zero and are stable, except for In\(_\text{Ge}\)N\(_\text{i}\), which has a negative binding energy, indicating instability. It is noteworthy to mention that despite In\(_\text{i}\)N\(_\text{Ge}\) having the highest formation energy, it remains stable in terms of its binding energy. The binding energy results show that the most stable defect-complexes are In\(_\text{Ge}\)As\(_\text{i}\) and In\(_\text{i}\)As\(_\text{Ge}\), with binding energies of 4.09 and 3.51 eV, respectively. Furthermore, the reported stable defect-complexes can only decompose into non-interacting defects at the expense of higher energy. For example, In\(_\text{Ge}\)As\(_\text{i}\) may decompose into non-interacting In\(_\text{Ge}\) and As\(_\text{i}\) with an energy cost.
3.4 Temperature-dependent band gap of Ge
Determining the temperature-dependent band gap of a material usually requires the application of theoretical models that illustrate how the band gap changes with temperature. Among the commonly employed theoretical models is the Varshni’s equation, which establishes a relationship between the band gap of a semiconductor and temperature [53, 54]. This equation is expressed as:
where E\(_\text{gap}\) (T) and E\(_\text{gap}\)(0) are the band gap at temperature T and at absolute zero temperature, respectively. The \(\alpha _\text{E}\), and \(\beta _\text{E}\) are material-dependent constants. In this study, we utilized Eq. (6) to derive the temperature dependence of Ge from 0 to 350 K, as depicted in Fig. 2. The values of \(\alpha _\text{E}\) and \(\beta _\text{E}\) were taken from Ref. [54]. At 300 K, the band gap of Ge is observed at 0.67 eV, which aligns with the findings of Ref. [54].

Temperature dependence of the band gap energies of Ge, determined using the Varshni’s equation. The horizontal red dotted line represents the band gap of Ge at 300K, measured at 0.67 eV
3.5 Defect levels induced by defect-complexes
Additional eigenstates may be created in semiconductors due to the presence of defects. These additional eigenstates can have a significant impact on the semiconductor’s electronic and optical properties [47]. The created allowed energy levels may lie within the conduction and valence bands [47]. The energy levels created by defects can be categorized as deep defect levels or shallow defect levels, depending on their position in the band gap [47]. Shallow levels are closer to the band edges; while, deep defect levels are farther away from the band edges. The specific energy and spatial location of these defect levels depend on the nature and type of defects present. The eigenstates that are slightly below the CBM, within an energy of approximately 3 \(k_BT\), can donate electrons to the conduction band, making them useful donors for electrical activities.
To provide information on defect levels and the electrical conductivity of defects in Ge, the defect levels induced by substitution–interstitial complexes formed by In and n-type atoms using Eq. (4) were calculated. As displayed in Table 3, In\(_\text{i}\)P\(_\text{Ge}\), In\(_\text{i}\)As\(_\text{Ge}\), and In\(_\text{Ge}\)Sb\(_\text{i}\) induce an electrically active defect level. In\(_\text{i}\)N\(_\text{Ge}\), In\(_\text{Ge}\)P\(_\text{i}\), and In\(_\text{Ge}\)As\(_\text{i}\) induce two distinct defect levels.
The (+1/0) defect level induced by In\(_\text{i}\)N\(_\text{Ge}\) could resonate between deep and shallow defect levels, depending on the temperature. For instance, at temperatures higher than 300 K, there is a high possibility of the (+1/0) level behaving as a shallow donor. At this moment, In\(_\text{i}\)N\(_\text{Ge}\) will donate electrons to the conduction band with energy at least lower than 3 \(k_BT\). As shown in Fig. 2 at 300 K, the 0.66 eV of the (+1/0) defect level of In\(_\text{i}\)N\(_\text{Ge}\) is positioned very close to the CBM; specifically, it is E\(_\text{C}\) - 0.01 eV. This proximity suggests that it could readily ionize by donating electrons to the conduction band. However, at low temperatures (below 300 K), the single (+1/0) energy level at E\(_\text{C}\) \(-\)0.12 eV induced by In\(_\text{i}\)N\(_\text{Ge}\) may be deep and could act as a recombination center.
The double (+2/+1) level at E\(_\text{V}\)+0.40 eV induced by In\(_\text{i}\)N\(_\text{Ge}\), as shown in Fig. 3a, is a deep defect level. Hence, it is postulated that In\(_\text{i}\)N\(_\text{Ge}\) exhibits temperature-dependent electrical characteristics. The single donor levels of In\(_\text{Ge}\)P\(_\text{i}\) and In\(_\text{Ge}\)As\(_\text{i}\) are shallow, positioned near the CBM. Specifically, the (+1/0) charge state transition level of In\(_\text{Ge}\)P\(_\text{i}\) is at E\(_\text{C}\) \(-\)0.08 (Fig. 3c), while that of In\(_\text{Ge}\)As\(_\text{i}\) is even closer to the CBM, at E\(_\text{C}\) \(-\)0.03 (Fig. 3e). This suggests that under equilibrium conditions, within an energy range of approximately 3 \(k_BT\), these defects may readily donate electrons to the conduction band.

Plot of the formation energy as a function of the Fermi level for the interstitial–substitutions defect-complexes in Ge. a In\(_\text{i}\)N\(_\text{Ge}\), b In\(_\text{i}\)P\(_\text{Ge}\), c In\(_\text{Ge}\)P\(_\text{i}\), d In\(_\text{i}\)As\(_\text{Ge}\), e In\(_\text{Ge}\)As\(_\text{i}\), f In\(_\text{Ge}\)Sb\(_\text{i}\) g In\(_\text{i}\)Sb\(_\text{Ge}\) and h the defect-complexes with their equilibrium most stable charge state configurations. The slope of each graph corresponds to the charge state
With the exception of In\(_\text{Ge}\)Sb\(_\text{i}\), all other double donor defect levels are relatively deep. The (+2/\(+\)1) defect level of In\(_\text{i}\)P\(_\text{Ge}\) is situated at E\(_\text{C}\) \(-\)0.13 eV (see Fig. 3b), which is 0.01 and 0.03 eV lower than the corresponding double donor levels of In\(_\text{i}\)As\(_\text{Ge}\) and In\(_\text{Ge}\)As\(_\text{i}\), respectively. The (+2/\(+\)1) defect levels of In\(_\text{i}\)As\(_\text{Ge}\) (Fig. 3d) and In\(_\text{Ge}\)As\(_\text{i}\) (Fig. 3e) are located at E\(_\text{V}\) \(-\)0.64 eV and E\(_\text{V}\) \(-\)0.62 eV, respectively. The shallow double charge state defect level of In\(_\text{Ge}\)Sb\(_\text{i}\) (E\(_\text{C}\) \(-\)0.09 eV), as depicted in Fig. 3g, is a significant defect that is less likely to cause electron recombination and can readily donate electrons to the conduction band. In contrast, In\(_\text{i}\)Sb\(_\text{Ge}\) did not induce any defect levels in the band gap of Ge. However, the +2 charge state dominates the Fermi energy range from 0 to 0.78 eV. The feasibility of inducing defect levels would only be possible above the CBM. Therefore, any attempts to alter the electronic properties of Ge in this configuration (In\(_\text{i}\)Sb\(_\text{Ge}\)) within the specified energy range may not yield encouraging results, as the dominant charge state remains +2. The substitution–interstitial complexes formed by In atom with n-type impurities function solely as donor defects, with no presence of acceptor levels. The shallow double and single charge state defect levels induced by these defect-complexes are advantageous for enhancing electrical conductivity performance. They mitigate electron recombination, facilitate electron donation to the conduction band, and consequently augment the conductivity of the host Ge material.
As mentioned in the introduction, doping Ge with donor atoms can be challenging due to the low solid solubility and high diffusion coefficient of impurities, which can reduce the possibility of obtaining shallow doped regions in Ge. However, in this report, it is shown that there is a possibility of doping Ge with donors by forming defect-complexes, which are electrically active defects. This paper provides insight into how these defect-complexes may be accessed to modulate the electrical conductivity and structural properties of Ge. This approach could potentially enhance the quality of Ge-based devices for future applications in MOSFETs. In future studies, it will be important to focus on the diffusion and migration properties of indium substitution and interstitial defect-complexes in Ge to gain a more comprehensive understanding of their impact in Ge-based devices.
4 Conclusion
Results of hybrid density functional theory study of defect-complexes formed by indium and n-type impurity atoms in Ge are presented. The study includes an analysis of the structural behavior of these substitution–interstitial defect-complexes, their formation energy, binding energy, stability, and thermodynamic charge state transition levels. The defect-complexes investigated include In\(_\text{Ge}\)N\(_\text{i}\), In\(_\text{i}\)N\(_\text{Ge}\), In\(_\text{Ge}\)P\(_\text{i}\), In\(_\text{i}\)P\(_\text{Ge}\), In\(_\text{i}\)As\(_\text{Ge}\), In\(_\text{Ge}\)As\(_\text{i}\), In\(_\text{i}\)Sb\(_\text{Ge}\), and In\(_\text{Ge}\)Sb\(_\text{i}\). The predicted results indicate that In\(_\text{i}\)P\(_\text{Ge}\) is the most energetically favorable defect-complex under equilibrium conditions, with a formation energy of 5.84 eV. Conversely, In\(_\text{i}\)N\(_\text{Ge}\) is the least favorable defect-complex. However, apart from In\(_\text{Ge}\)N\(_\text{i}\), all other defect-complexes are stable. This suggests the possibility of substitution atoms forming strong bonds with interstitial atoms in the host Ge. The most stable defect-complex is In\(_\text{Ge}\)As\(_\text{i}\), with a binding energy of 4.09 eV. The substitution–interstitial defect-complexes formed by In and n-type impurity atoms in Ge induced defect levels that primarily act as donors. There is no evidence of acceptor defect levels induced in the band gap of Ge by the investigated defect-complexes. The single donor defect levels induced by In\(_\text{i}\)N\(_\text{Ge}\), In\(_\text{Ge}\)P\(_\text{i}\), and In\(_\text{Ge}\)As\(_\text{i}\) are shallow levels located close to the conduction band. In contrast, the double (+2/+1) donor defect levels induced by these same complexes are deep and may act as recombination centers. This study highlights the behavior of substitution–interstitial defect-complexes formed by In and n-type impurity atoms in Ge. Additionally, it offers insights into controlling donor-like defects during device fabrication and defect processes. Furthermore, the research opens up a new scientific area related to oversize impurity atoms for defect engineering in Ge, with potential applications in modern generation MOSFETs. It also provides a frontier insight into the experimental synthesis of interstitial and substitution defect-complexes of In and n-type impurity atoms in Ge.
Data availability
Not Applicable
References
Auret, F., van Rensburg, P.J., Hayes, M., Nel, J., Coelho, S., Meyer, W., Decoster, S., Matias, V., Vantomme, A., Smeets, D.: Nuclear instruments and methods in physics research section B: beam interactions with materials and atoms. Nucl. Instrum. Methods Phys. Res. 257(1–2), 169–171 (2007)
Omotoso, E., Paradzah, A.T., Igumbor, E., Taleatu, B., Meyer, W.E., Auret, F.D.: Determination of capture barrier energy of the e-center in palladium Schottky barrier diodes of antimony-doped germanium by varying the pulse width. Mater. Res. Express 7(2), 025901 (2020)
Igumbor, E., Dongho-Nguimdo, G., Mapasha, R., Omotoso, E., Meyer, W.: Stability, electronic and defect levels induced by substitution of al and p pair in 4h-sic. J. Phys. Chem. Solids 142, 109448 (2020)
Khirunenko, L.I., Sosnin, M.G., Duvanskii, A.V., Abrosimov, N.V., Riemann, H.: Electronic absorption of interstitial boron-related defects in silicon. Phys. Status Solidi (a) 214(7), 1700245 (2017)
Igumbor, E., Omotoso, E., Tunhuma, S.M., Danga, H.T., Meyer, W.E.: Rare earth substitutional impurities in germanium: a hybrid density functional theory study. Nucl. Instrum. Methods Phys. Res., Sect. B 409, 31–35 (2017)
Morin, F.J., Maita, J.P.: Conductivity and Hall effect in the intrinsic range of germanium. Phys. Rev. 94(6), 1525–1529 (1954)
Claeys, C., Simoen, E.: Germanium-Based Technologies: From Materials to Devices. Elsevier, Amsterdam (2011)
Ruan, D.B., Chang-Liao, K.S., Hong, Z.Q., Huang, J., Yi, S.H., Liu, G.T., Chiu, P.C., Li, Y.L.: Radiation effects and reliability characteristics of Ge pMOSFETS. Microelectron. Eng. 216, 111034 (2019)
Yadav, D., Nair, D.R.: Impact of source to drain tunneling on the ballistic performance of Si, Ge, GaSb, and GeSn nanowire p-MOSFETs. IEEE J. Electron Devices Soc. 8, 308–315 (2020)
Liu, H., Pasanen, T.P., Fung, T.H., Isometsä, J., Leiviskä, O., Vähänissi, V., Savin, H.: Comparison of sinx-based surface passivation between germanium and silicon. Phys. status solidi (a) 220(2), 2200690 (2023)
Wen, W.C., Nagatomi, Y., Akamine, H., Yamamoto, K., Wang, D., Nakashima, H.: Interface trap and border trap characterization for Al2O3/GeOx/Ge gate stacks and influence of these traps on mobility of ge p-mosfet. AIP Adv. 10(6), 065119 (2020)
Pastor, D., Gandhi, H.H., Monmeyran, C.P., Akey, A.J., Milazzo, R., Cai, Y., Napolitani, E., Gwilliam, R.M., Crowe, I.F., Michel, J., Kimerling, L.: High level active n+ doping of strained germanium through co-implantation and nanosecond pulsed laser melting. J. Appl. Phys. 123(16), 165101 (2018)
Milazzo, R., Impellizzeri, G., Piccinotti, D., La Magna, A., Fortunato, G., De Salvador, D., Carnera, A., Portavoce, A., Mangelinck, D., Privitera, V., Napolitani, E.: Impurity and defect interactions during laser thermal annealing in Ge. J. Appl. Phys. 119(4), 045702 (2016)
Sumathi, R.R., Gybin, A., Gradwohl, K.P., Palleti, P.C., Pietsch, M., Irmscher, K., Dropka, N., Juda, U.: Development of large-diameter and very high purity Ge crystal growth technology for devices. Cryst. Res. Technol. 58(5), 2200286 (2023)
Sgourou, E.N., Daskalopulu, A., Tsoukalas, L.H., Stamoulis, G., Vovk, R.V., Chroneos, A.: Seventy-five years since the point-contact transistor: germanium revisited. Appl. Sci. 12(23), 11993 (2022)
Patel, N.S., Monmeyran, C., Agarwal, A., Kimerling, L.C.: Point defect states in Sb-doped germanium. J. Appl. Phys. 118(15), 155702 (2015)
Chroneos, A.: Dopant-vacancy cluster formation in germanium. J. Appl. Phys. 107, 076102 (2010)
Carraro, C., Milazzo, R., Sgarbossa, F., Fontana, D., Maggioni, G., Raniero, W., Scarpa, D., Baldassarre, L., Ortolani, M., Andrighetto, A., Napoli, D.: N-type heavy doping with ultralow resistivity in Ge by Sb deposition and pulsed laser melting. Appl. Surf. Sci. 509, 145229 (2020)
Igumbor, E., Mapasha, R.E., Meyer, W.E.: Ab initio study of aluminium impurity and interstitial-substitutional complexes in Ge using a hybrid functional (HSE). J. Electron. Mater. 46(7), 3880–3887 (2017)
Śpiewak, P., Vanhellemont, J., Sueoka, K., Kurzydłowski, K., Romandic, I.: First principles calculations of the formation energy and deep levels associated with the neutral and charged vacancy in germanium. J. Appl. Phys. 103(8), 086103 (2008)
Winter, E., Schreiber, W., Schygulla, P., Souza, P., Janz, S., Lackner, D., Ohlmann, J.: Iii-v material growth on electrochemically porosified ge substrates. J. Cryst. Growth 602, 126980 (2023)
Saraswat, K.C.: Germanium: back to the future. 75th Anniversary of the Transistor. pp. 415–429 (2023)
Igumbor, E., Mapasha, R.E., Andrew, R., Meyer, W.E.: A first principle hybrid functional calculation of TmGe3+-VGe defect complexes in germanium. Comput. Cond. Mater. 8, 31–35 (2016)
Chroneos, A., Uberuaga, B.P., Grimes, R.W.: Carbon, dopant, and vacancy interactions in germanium. J. Appl. Phys. 102(8), 083707 (2007)
Nyamhere, C., Das, M., Auret, F.D., Chawanda, A.: A study of electron induced defects in n-type germanium by deep level transient spectroscopy (DLTS). Phys. Status Solidi C 5(2), 623–625 (2008)
Prucnal, S., Liu, F., Voelskow, M., Vines, L., Rebohle, L., Lang, D., Berencén, Y., Andric, S., Boettger, R., Helm, M., Zhou, S.: Ultra-doped n-type germanium thin films for sensing in the mid-infrared. Sci. Rep. 6(1), 1–8 (2016)
Danga, H., Tunhuma, S., Auret, F., Igumbor, E., Omotoso, E., Meyer, W.: The study of low temperature irradiation induced defects in p-Si using deep-level transient spectroscopy. Nucl. Instrum. Methods Phys. Res., Sect. B 442, 28–30 (2019)
Simoen, E., Vanhellemont, J.: On the diffusion and activation of ion-implanted n-type dopants in germanium. J. Appl. Phys. 106(10), 103516 (2009)
Gora, V.E., Auret, F.D., Danga, H.T., Tunhuma, S.M., Nyamhere, C., Igumbor, E., Chawanda, A.: Barrier height inhomogeneities on Pd/n-4h-SiC Schottky diodes in a wide temperature range. Mater. Sci. Eng., B 247, 114370 (2019)
Vanhellemont, J., Śpiewak, P., Sueoka, K.: On the solubility and diffusivity of the intrinsic point defects in germanium. J. Appl. Phys. 101(3), 036103 (2007)
Deák, P., Aradi, B., Frauenheim, T., Janzén, E., Gali, A.: Accurate defect levels obtained from the HSE06 range-separated hybrid functional. Phys. Rev. B 81, 153203 (2010)
Kuisma, M., Ojanen, J., Enkovaara, J., Rantala, T.: Kohn-sham potential with discontinuity for band gap materials. Phys. Rev. B 82(11), 115106 (2010)
Igumbor, E., Omotoso, E., Meyer, W.E.: Nano Hybrids and Composites, vol. 16, pp. 47–51. Trans Tech Publ, Stafa-Zurich (2017)
Carvalho, A., Jones, R., Janke, C., Goss, J.P., Briddon, P.R., Coutinho, J., Öberg, S.: Electronic and dynamical properties of the silicon trivacancy. Phys. Rev. Lett. 99, 175502 (2007)
Janke, C., Jones, R., Öberg, S., Briddon, P.R.: Supercell and cluster density functional calculations of the thermal stability of the divacancy in germanium. Phys. Rev. B 75, 195–208 (2007)
Igumbor, E., Ouma, C., Webb, G., Meyer, W.: Ab-initio study of germanium di-interstitial using a hybrid functional (HSE). Phys. B 480, 191–195 (2016)
Coutinho, J., Öberg, S., Torres, V.J.B., Barroso, M., Jones, R., Briddon, P.R.: Donor-vacancy complexes in Ge: cluster and supercell calculations. Phys. Rev. B 73, 235213 (2006)
Igumbor, E., Meyer, W.E.: A hybrid functional calculation of Tm3+ defects in germanium (Ge). Mater. Sci. Semicond. Process. 43, 129–133 (2016)
Chroneos, A., Grimes, R.W., Tsamis, C.: Atomic scale simulations of arsenic-vacancy complexes in germanium and silicon. Mater. Sci. Semicond. Process. 9(4–5), 536–540 (2006)
Igumbor, E., Dongho-Nguimdo, G., Mapasha, R.E., Meyer, W.E.: Electronic properties and defect levels induced by group iii substitution-interstitial complexes in Ge. J. Mater. Sci. 54(15), 10798–10808 (2019)
Csóré, A., Von Bardeleben, H., Cantin, J., Gali, A.: Characterization and formation of NV centers in 3C, 4H, and 6H SiC: an ab initio study. Phys. Rev. B 96(8), 085204 (2017)
Dolgolenko, A., Varentsov, M., Gaidar, G.: Energy-level position of bistable (cics) 0 defect in the b configuration in the forbidden band of n-Si. Phys. Status solidi (b) 241(13), 2914–2922 (2004)
Igumbor, E., Mapasha, R.E. and Meyer, W.E.: Ab-Initio Study of Aluminium Impurity and Interstitial-Substitutional Complexes in Ge Using a Hybrid Functional (HSE). J. Electron. Mater., 46, pp. 3880–3887
Igumbor, E., Dongho-Nguimdo, M., Mapasha, E., Kalimuthu, R., Raji, A., Meyer, W.: Trivalent atom defect-complex induced defect levels in germanium for enhanced ge-based device performance. J. Electron. Mater. 53, 1903 (2024)
Igumbor, E., Olaniyan, O., Dongho-Nguimdo, G.M., Mapasha, R., Ahmad, S., Omotoso, E., Meyer, W.E.: Electronic properties and defect levels induced by n/p-type defect-complexes in ge. Mater. Sci. Semicond. Process. 150, 106906 (2022)
Blochl, P.E.: Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994)
Igumbor, E.: Hybrid functional study of point defects in germanium. Ph.D. thesis, University of Pretoria (2017)
Zhang, S.B., Northrup, J.E.: Chemical potential dependence of defect formation energies in GaAs: Application to Ga self-diffusion. Phys. Rev. Lett. 67(17), 2339–2342 (1991)
Freysoldt, C., Grabowski, B., Hickel, T., Neugebauer, J., Kresse, G., Janotti, A., de Walle, V., C. G.: First-principles calculations for point defects in solids. Rev. Mod. Phys. 86, 253–305 (2014)
Freysoldt, C., Neugebauer, J., Van de Walle, C.G.: Fully ab initio finite-size corrections for charged-defect supercell calculations. Phys. Rev. Lett. 102, 016402 (2009)
Freysoldt, C., Neugebauer, J., Van de Walle, C.G.: Electrostatic interactions between charged defects in supercells. Phys. Status solidi (b) 248(5), 1067–1076 (2011)
Zollo, G., Lee, Y.J., Nieminen, R.M.: Properties of intrinsic di-interstitials in GaAs. J. Phys. Cond. Mater. 16(49), 8991 (2004)
Yacobi, B.G.: Semiconductor Materials: An Introduction to Basic Principles. Springer, Berlin (2003)
Geng, P., Li, W., Zhang, X., Zhang, X., Deng, Y., Kou, H.: A novel theoretical model for the temperature dependence of band gap energy in semiconductors. J. Phys. D Appl. Phys. 50(40), 40LT02 (2017)
Acknowledgements
Emmanuel Igumbor is grateful to the University of Johannesburg for funding and the Center for High Performance Computing (CHPC) Cape Town for providing computational resources.
Funding
Open access funding provided by University of Johannesburg. The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
Emmanuel Igumbor contributed in the following conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, validation, visualization, writing—original draft, and writing—review and editing
Corresponding author
Ethics declarations
Conflict of interest
The author declares that they have no Conflict of interest.
Ethical approval
Not applicable
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Igumbor, E. Donor-induced electrically charged defect levels: examining the role of indium and n-type defect-complexes in germanium. J Comput Electron (2024). https://doi.org/10.1007/s10825-024-02179-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10825-024-02179-0